

Abstract
Glioblastoma multiforme is the most frequent, aggressive and fatal type of brain tumor. Glioblastomas are characterized by their infiltrating nature, high proliferation rate and resistance to chemotherapy and radiation. Recently, oncologic therapy experienced a rapid evolution towards “targeted therapy,” which is the employment of drugs directed against particular targets that play essential roles in proliferation, survival and invasiveness of cancer cells. A number of molecules involved in signal transduction pathways are used as molecular targets for the treatment of various tumors. In fact, inhibitors of these molecules have already entered the clinic or are undergoing clinical trials. Cellular receptors are clear examples of such targets and in the case of glioblastoma multiforme, some of these receptors and their ligands have become relevant. In this review, the importance of glioblastoma multiforme in signaling pathways initiated by extracellular tyrosine kinase receptors such as EGFR, PDGFR and IGF-1R will be discussed. We will describe their ligands, family members, structure, activation mechanism, downstream molecules, as well as the interaction among these pathways. Lastly, we will provide an up-to-date review of the current targeted therapies in cancer, in particular glioblastoma that employ inhibitors of these pathways and their benefits.
Keywords: RTK, EGFR, PDGFR, IGF-1R, glioblastoma multiforme
1. Introduction
Cancer is a very heterogeneous disease that represents a complex social and health problem. Cancer is the first cause of death worldwide, causing 8 million annual deaths worldwide [1]. Among all types of cancer, brain tumors represent 2% of all cancers and 90% of tumors of the central nervous system [2]. Within brain tumors, gliomas have high incidence in adults and represent 70% of all brain tumors [3]. Glioblastoma multiforme (GBM) is the most frequent subtype of glioma (about 55%) and represents 20% of the brain tumors [4]. GBM is a grade IV glioma and the most deadly—it is the most difficult type of brain tumor to treat. Current therapy includes surgery and radiation [5]. These tumors, however, are highly resistant to chemotherapy. The first line chemotherapeutic treatment at present is the alkylating agent temozolomide [6,7]. Therapeutic options for GBM are very limited and there is therefore a great need for better treatments. Transmembrane tyrosine kinase receptors play an important role in neoplasia. Once these receptors are activated, they are capable of transducing signals that result in cell proliferation and cell survival. One therapeutic strategy to combat cancer is hampering with these signaling cascades. Among these signaling amplification cascades, the Ras/Raf/Mek/Erk and he PI3K/Akt/mTOR pathway are known to play important and decisive roles.
2. Signaling Pathways and Cancer Treatment
Signal transduction is the process by which a cell converts an extracellular stimulus into an intracellular signal. This process is initiated upon binding of extracellular signaling molecules to cellular receptors. Cell proliferation and cell death are highly regulated processes in which signal transduction pathways intervene. Tumor cells possess abnormalities in molecules involved in these signaling pathways and, as a result, develop an uncontrolled proliferation and defects in apoptotic mechanisms. For this reason, signaling pathways play a pivotal role in the search for molecular targets that will be employed in cancer treatment. Cellular transmembrane receptors are clear examples of such targets. In this sense, there are several inhibitors against cellular receptors undergoing clinical trials or being utilized for the treatment of various types of cancer (Table 1).
Table 1.
Anti-tumor drugs targeted against cellular tyrosine kinase receptors.
| Drug | Company | Target | Indication |
|---|---|---|---|
| Gleevec® (Imatinib) | Novartis Pharma | PDGFR | Chronic myeloid leukemia (CML) and gastrointenstinal stromal tumor (GIST) |
| Erbitux® (Cetuximab/C225) | Merck | EGFR | Colorectal cancer and head and neck squamous cell tumors |
| Tykerb® (Lapatinib) | GlaxoSmithKline | ErbB-2/EGFR | ErbB-2 positive, advanced breast cancer, previously treated with anthracyclines, taxanes or Herceptin® |
| Iressa® (Gefitinib) | AstraZeneca | EGFR | Non-small cell lung cancer (NSCLC) |
| Tarceva® (Erlotinib) | Roche | EGFR | Non-small cell lung cancer (NSCLC) |
| Vectibix® (Panitumumab) | AMGEN | EGFR | Metastasic colorectal cancer |
| Herceptin® (Trastuzumab) | Merck | ErbB-2 | ErbB-2 positive metastasic breast cancer |
| Votrient® (Pazopanib) | GlaxoSmithKline | PDGFR VEGFR, c-KIT |
Advanced renal carcinoma |
3. Transmembrane Receptor Tyrosine Kinases
Cellular receptors with tyrosine kinase enzymatic activity catalyze the transfer of phosphate groups and ATP towards hydroxyl groups present in the tyrosine residues of proteins. These membrane-bound receptors are activated by growth factors, cytokines and hormones. Upon activation by ligand binding, they can transduce signals and rule important processes such as proliferation, apoptosis, motility, angiogenesis, or cell differentiation [8]. There are about 58 known tyrosine kinase receptors in humans, classified into 20 subtypes or families, according to structural aspects and the ligands that activate them [9]. Some examples include the epidermal growth factor receptor (EGFR), the vascular endothelial growth factor receptor (VEGFR), or the platelet-derived growth factor receptor (PDGFR) family. All receptor tyrosine kinases (RTK) are transmembrane proteins, whose amino-terminal end is extracellular (transmembrane proteins type I). Their general structure is comprised of an extracellular ligand-binding domain (ectodomain), a small hydrophobic transmembrane domain and a cytoplasmic domain, which contains a conserved region with tyrosine kinase activity. This region consists of two lobules (N-terminal and C-terminal) that form a hinge where the ATP needed for the catalytic reactions is located [10]. Activation of RTK takes place upon ligand binding at the extracellular level. This binding induces oligomerization of receptor monomers, usually dimerization. In this phenomenon, juxtaposition of the tyrosine-kinase domains of both receptors stabilizes the kinase active state [11]. Upon kinase activation, each monomer phosphorylates tyrosine residues in the cytoplasmic tail of the opposite monomer (trans-phosphorylation). Then, these phosphorylated residues are recognized by cytoplasmic proteins containing Src homology-2 (SH2) or phosphotyrosine-binding (PTB) domains, triggering different signaling cascades. Cytoplasmic proteins with SH2 or PTB domains can be effectors, proteins with enzymatic activity, or adaptors, proteins that mediate the activation of enzymes lacking these recognition sites. Some examples of signaling molecules are: phosphoinositide 3-kinase (PI3K), phospholipase C (PLC), growth factor receptor-binding protein (Grb), or the kinase Src, The main signaling pathways activated by RTK are: PI3K/Akt, Ras/Raf/ERK1/2 and signal transduction and activator of transcription (STAT) pathways (Figure 1).
Figure 1.

Main signal transduction pathways initiated by RTK.
The PI3K/Akt pathway participates in apoptosis, migration and cell invasion control [12]. This signaling cascade is initiated by PI3K activation due to RTK phosphorylation. PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) producing phosphatidylinositol 3,4,5-triphosphate (PIP3), which mediates the activation of the serine/threonine kinase Akt (also known as protein kinase B). PIP3 induces Akt anchorage to the cytosolic side of the plasma membrane, where the phosphoinositide-dependent protein kinase 1 (PDK1) and the phosphoinositide-dependent protein kinase 2 (PDK2) activate Akt by phosphorylating threonine 308 and serine 473 residues, respectively. The once elusive PDK2, however, has been recently identified as mammalian target of rapamycin (mTOR) in a rapamycin-insensitive complex with rictor and Sin1 [13]. Upon phosphorylation, Akt is able to phosphorylate a plethora of substrates involved in cell cycle regulation, apoptosis, protein synthesis, glucose metabolism, and so forth [12,14]. A frequent alteration found in glioblastoma that affects this signaling pathway is mutation or genetic loss of the tumor suppressor gene PTEN (Phosphatase and Tensin homologue deleted on chromosome ten), which encodes a dual-specificity protein phosphatase that catalyzes PIP3 dephosphorylation [15]. Therefore, PTEN is a key negative regulator of the PI3K/Akt pathway. About 20% to 40% of glioblastomas present PTEN mutational inactivation [16] and about 35% of glioblastomas suffer genetic loss due to promoter methylation [17].
The Ras/Raf/ERK1/2 pathway is the main mitogenic route initiated by RTK. This signaling pathway is triggered upon binding of the adaptor molecule Grb2 to phosphorylated tyrosines located in receptor cytoplasmic tails. This binding produces a conformational change in Sos, which recruits and activates the GTP hydrolase (GTPase) Ras. Subsequently, Ras activates the serine/threonine kinase Raf, which activates MEK 1/2 until finally MEK 1/2 phosphorylates and activates extracellular signal-regulated kinase 1/2 (ERK1/2), which in turn, can phosphorylate more than a hundred proteins with distinct functions [18]. Among these targets, we can find transcription factors involved in cell proliferation (c-Myc, c-Jun, c-Fos, Elk1, Ets-1, p62) [19], proteins involved in cell migration [20], or proteins that regulate GAP junctions [21]. This signaling pathway is frequently altered in glioblastoma. According to “The Cancer Genome Atlas”, 86% of glioblastomas present at least one alteration that affects the Ras/Raf/ERK 1/2 pathway.
The JAK/STAT pathway is initiated upon ligand binding to RTK, which activates the kinase function of members of the Janus family of tyrosine kinases (JAK), which in turn, are autophosphorylated. STAT proteins then bind to the receptor phospho-tyrosine residues through their SH2 domains, where they become phosphorylated by JAK. Once phosphorylated, STAT factors dimerize, translocate to the nucleus and induce expression of anti-apoptotic and cell cycle regulatory proteins [22]. Thus, the JAK/STAT pathway represents the link between extracellular signals and transcriptional responses within the nucleus. STATs may also be directly phosphorylated by RTK such as EGFR and PDGFR and by non-receptor tyrosine kinases such as c-src. In addition, several MAPK can phosphorylate STAT at a serine near its C-terminus, increasing its transcriptional activity. Signal-transducing adapter molecules (STAM) aid transcriptional activation of specific genes such as MYC [23]. There are three classes of negative regulators: Suppressors of cytokine signaling (SOCS), which directly bind to and inactivate JAKs [24], protein inhibitors of activated Stats (PIAS), which bind phosphorylated STAT dimers, preventing DNA recognition [25] and protein phosphatases, which inactivate RTK [26].
3.1. Epidermal Growth Factor Receptor (EGFR)
EGFR (ErbB1/HER1) is membrane-bound receptor with tyrosine kinase activity that is expressed in a whole variety of tissues and takes part in processes such as proliferation, differentiation, motility or survival [27]. EGFR belongs to the family of ErbB receptors together with ErbB-2 (Neu/HER-2) [28], ErbB-3 (HER-3) [29] and ErbB-4 (HER-4) [30]. EGFR was identified in 1976 by Carpenter and Cohen [31], several years after the isolation of the epidermal growth factor (EGF) [32]. The discovery some years later that EGFR had tyrosine kinase activity was an upheaval in growth factor and cancer biology [33,34]. Moreover, it was found afterwards that the avian erythroblastic leukemia viral (ErbB) oncogene encodes a truncated EGFR form [35], which suggests that EGFR plays a role in tumorigenesis and can be used as a molecular target for cancer therapy.
3.1.1. Structure and Activation Mechanism
The family of erbB receptors is made up of a 620 amino acid extracellular ligand-binding domain that contains four cysteine-rich regions, a small hydrophobic transmembrane-spanning domain with an alpha-helix structure and a cytoplasmic domain of about 550 amino acids formed by a region with tyrosine kinase activity (270 amino acids), flanked by a juxtamembrane region (45 amino acids) and a tyrosine-rich carboxy-terminal end (230 amin oacids).
ErbB receptor family activation is triggered upon ligand binding to the extracellular domain [36]. In the absence of stimulus, the receptor molecule is held in an autoinhibitory conformational state in which subdomains II and IV are interacting between themselves. Ligand binds to subdomains I and III, which produces conformational changes that promote receptor dimerization [37]. Ligand binding alters the relative subdomain positioning so that the subdomain II dimerization arm of one receptor reaches the other receptor molecule [38]. This dimerization process requires the binding of two ligand molecules onto two receptor molecules [39]. When the dimerization takes places between equal family members is called homodimerization; if it occurs between different family members is named heterodimerization [40]. As a consequence of dimerization driven by ligand binding, cytoplasmic tyrosine-kinase regions are juxtaposed (C-terminal end of one kinase placed close together to N-terminal end of the other kinase) and undergo allosteric activation [41]. Once phosphorylated, these kinases phosphorylate tyrosine residues in the C-terminal end of the other molecule, in a process known as transphosphorylation [36]. After ligand-driven receptor activation and subsequent signaling-cascade triggering, receptor molecules are endocytosed by the cell in order to either be proteolytically degraded in the lysosomes or be recycled back to the plasma membrane [42].
Although ErbB receptor activation has always been related to the dimerization process, it the presence of ErbB family receptors in the plasma membrane comprised by preformed inactive homo or heterodimers has been reported by some authors [43].
ErbB-3 receptor differs from the rest of the family members because it lacks some residues in its kinase domain that are essential for catalysis. Therefore, even though this receptor is able to bind ATP, it lacks enzymatic activity. However, in spite of everything, the erbB-3 receptor is able to transduce signals by its ability to form heterodimers with other members of the erbB family of receptors [44,45].
3.1.2. ErbB Ligands
ErbB receptors are recognized by different structurally related growth factors. There are about ten known ligands, which consist of 55 amino acids, characterized by three disulfide bonds and a loop-rich structure. The best known ligands are: epidermal growth factor (EGF), amphiregulin (AR), transforming growth factor alpha (TGFα), epiregulin (ERG), betacellulin (BTC), heparin-binding epidermal growth factor (HB-EGF) and neuregulins (NRG) 1 through 4. These growth factors are synthesized as precursor polypeptides that contain several soluble ligand units. After synthesis, these growth factor precursors are anchored to the plasma membrane exposing to the extracellular medium the ligand units, which are sequentially released through proteolytic processing by ADAM family metalloproteinases [46,47] and different protein kinase C (PKC) isoforms [48]. Interestingly, when these precursor forms are anchored to the plasma membrane, they are able to transduce signals upon binding to erbB receptors present in adjacent cells, in a process known as juxtacrine signaling [49].
Ligands show distinct affinity for the different erbB receptors. In this sense, some receptors share ligands and some ligands bind exclusively specific receptors. EGF, AR and TGFα bind EGFR, HRG, BTC and HB-EGF bind EGFR and erbB-4 [50], NRG1 and NRG2 bind erbB-3 and erbB-4 [51,52], and NRG3 and NRG4 bind only erbB-4 [53] (Table 2).
Table 2.
ErbB family members and their ligands.
| RECEPTOR | Locus | Protein size | Kinase activity | Ligands |
|---|---|---|---|---|
| EGFR | 7p13-q22 | 170 KDa | yes | EGF, TGFα, AR, HB-EGF, ERG and BTC |
| ErbB-2 | 17q21 | 185 KDa | yes | -- |
| ErbB-3 | 12q13 | 190 KDa | no | NRG 1-4 |
| ErbB-4 | 2q33 | 180 KDa | yes | NRG 1-4, HB-EGF, ERG and BTC |
Thus far, no ligands able to bind erbB-2 have being identified. It is known that erbB-2 activation is secondary to the activation of other ErbB family members [54]. In this respect, it has been shown that erbB-2 is always constitutively available for dimerization, since it is present in an active conformation at all times [55].
The prevalence of homodimer and heterodimer formation follows a hierarchical order determined by the abundance and presence of both receptors and ligands [56,57,58]. The distinct homodimer and heterodimer formation produces a great variety of cell signaling, depending upon the ligand and receptor combination. Homodimers cause a less durable signaling than heterodimers [59]. ErbB-2 is the most frequent partner found in heterodimers and its presence increases ligand-binding affinity [60,61]. Heterodimers containing ErbB-2 are less frequently endocytosed and degraded [62]. ErbB-2 and erbB-3 heterodimer is considered the most potent [45,60] and may be oncogenic, as it plays a pivotal role in signaling of erbB-2-overexpressing tumors [63]. The receptor combination can determine the type and magnitude of the initiated signaling cascade. For example, erbB-3 preferentially triggers the PI3K pathway due to six tyrosine residues binding PI3K [64], whereas EGFR and erbB-2 bind PI3K through adaptor proteins. On the other hand, erbB-4 activates less cytoplasmic proteins than the other receptors [65].
3.1.3. EGFR and Cancer
EGFR overexpression and activation is considered an etiological factor in several types of cancer, such as head and neck squamous cell cancer, non-small cell lung cancer (NSCLC), colorectal cancer, breast cancer and some tumors of the central nervous system. In particular, 50% of GBM present EGFR hyperactivation or amplification [66]. In glioblastoma, EGFR gene amplification leads to receptor overexpression and rise of mutant forms. Moreover, the truncated EGFR form known as EGFRvIII or variant III appears frequently in GBM. This mutant form is present in about 20%–30% of glioblastomas [67,68,69], and has also been found in medulloblastomas, breast, ovary and lung cancer [70], but not in healthy tissues. EGFRvIII mutant results from a deletion comprising a region between exon 2 and exon 7 and therefore lacks the ligand-binding domain [71]. Since this mutant EGFR variant cannot bind ligands, it is constitutively active [72] and highly tumorigenic [73]. There are other EGFR mutants present in glioblastoma with different deletions affecting either the extracellular or cytoplasmic domain [74]. Other variants such as TDM/18-26 present tandem duplications [75,76]. In addition, activating mutations have been found in lung carcinoma, which are associated with prognosis and therapy response [77]. Ligand-dependent activation of wild-type EGFR and constitutive activation of EGFR mutants function in a different manner. EGFR and its ligands EGF and TGFα are expressed during brain development in a spatial and timely fashion [78]. EGFR stimulation by its ligands occurs in an autocrine, paracrine or juxtracrine manner, triggering signaling pathways involved in proliferation, survival, migration, invasion and angiogenesis of primary brain cells, including tumor-initiating cells [79]. The constitutively activated EGFRvIII form, however, acts in the absence of ligand. This variant exhibits lower autophosphorylation levels than the ligand-driven wild-type receptor [80] and its rate of receptor internalization via endocytosis and lysosomal degradation is lower than the degradation rate of its wild-type counterpart [81]. Similarly, the TDM/18-26 mutant exhibits a lower internalization rate than the wild-type receptor, as it is poorly downregulated upon ligand binding and its constitutive phosphorylation accounts for its oncogenic potential [82]. Therefore, the signaling cascade initiated by receptor mutant forms is more persistent and, hence, more tumorigenic than the signaling initiated by ligand-driven wild-type receptor molecules. Moreover, wild-type EGFR and EGFRvIII can heterodimerize and contribute to the malignant development of brain cells.
It is important to note that the RTK c-Met and its ligand hepatocyte growth factor (HGF)/Scatter factor are overexpressed in gliomas and that their expression levels correlate with tumor grade [83]. In addition, it has been shown that c-Met plays a role in cell proliferation, motility, migration, invasion, angiogenesis and survival in several types of cancer [84]. EGFR and c-Met trigger the same signal transduction pathways and may elicit similar responses. In fact, the cross-talk between both receptors may affect the duration and strength of the response [85] and contribute to a malignant phenotype. Several reports have shown the coexistence of EGFR and c-Met in solid malignancies. In particular, EGFR and c-Met are coexpressed in GBM, and dysregulated EGFR signaling in glioblastoma increases HGF binding to c-Met promoting cell invasion [86]. A recent report has shown that the cross talk between EGFR and c-Met induces proliferation, invasion and migration in glioblastoma cells and therefore contributes to tumorigenesis [87]. Additionally, the highly oncogenic EGFRvIII mutant activates c-Met and EGFR wild-type can antagonize c-Met activation [88].
Consistent with the EGFR amplification observed in EGFRvIII-bearing glioblastomas, EGFR was highly expressed in classical neurospheres derived from these tumors, but barely detectable in mesenchymal or preneural neurospheres that harbor EGFR wild-type gene. However, c-Met oncogene was expressed in mesenchymal and preneural neurospheres, but absent in EGFR overexpressing neurospheres, indicating that EGFR amplification and c-Met overexpression are mutually exclusive. These results suggest that c-Met is a functional marker of glioblastoma stem cells [89].
Translocations are frequent in human gliomas. For example, recurrent translocations in glioma cells fuse the EGFR coding sequence to several partners, EGFR-SEPT14 being the most common functional gene fusion in human glioblastoma [90].
Upon ligand binding, EGFR translocates to the nucleus, where it can modulate gene transcription [91] by interacting with STAT3 [92]; it can also promote radioresistance [93,94,95] and chemoresistance [96]. It is interesting to mention that EGFR can also translocate to the mitochondria. In fact, EGFR colocalizes with FAK in GBM samples from patients [97]. Likewise, upon HGF stimulation, c-Met translocates to the nucleus, where it triggers calcium signals [98].
3.2. Platelet-Derived Growth Factor Receptor (PDGFR)
The platelet-derived growth factor receptor (PDGFR) family constitutes the subfamily III of RTK [99] and is formed by PDGFRα and PDGFRβ [100]. PDGF receptors are related to cell migration, proliferation and survival processes [101]. PDGFRα and β play fundamental roles during embryogenesis [102]. PDGFRα is involved in the development of organs and structures such as lungs, skin, gonads, central nervous system and skeleton. PDGFRβ takes part in early hematopoiesis and blood vessel formation [103]. PDGFRα is expressed in mesenchymal cells, especially in oligodendrocyte progenitors and mesenchymal progenitors of lung, skin and intestine. PDGFRβ is mainly expressed in vascular smooth muscle and pericytes [104].
3.2.1. Structure and Activation Mechanism
PDGFRα and PDGFRβ are membrane-bound glycoproteins formed by an extracellular ligand-binding domain, composed of five immunoglobulin-like subdomains, a transmembrane domain and a cytoplasmic domain with tyrosine kinase activity (Figure 2). Like other RTK, PDGFR is activated upon ligand binding to the first three immunoglobulin-like domains in the ectodomain [105], triggering oligomerization of two receptor molecules. Dimerization can take place between two α or two β molecules, resulting in αα or ββ homodimers, respectively, or between α and β isoforms, giving rise to αβ heterodimers [106]. This dimerization event juxtaposes the cytoplasmic domains of both receptor molecules, leading to tyrosine kinase activation and trans-phosphorylation of tyrosine residues located in the transmembrane and cytoplasmic domains of the receptor [107]. These phosphorylated residues are recognized by PTB or SH2 domain-containing signal transduction molecules, such as phospholipase C γ (PLCγ) [108], Ras, PI3K or Src [109]. Activated PDGF receptors trigger PI3K/Akt [110], PLCγ [111] and Ras/Raf/Erk 1/2 pathways [112].
Figure 2.
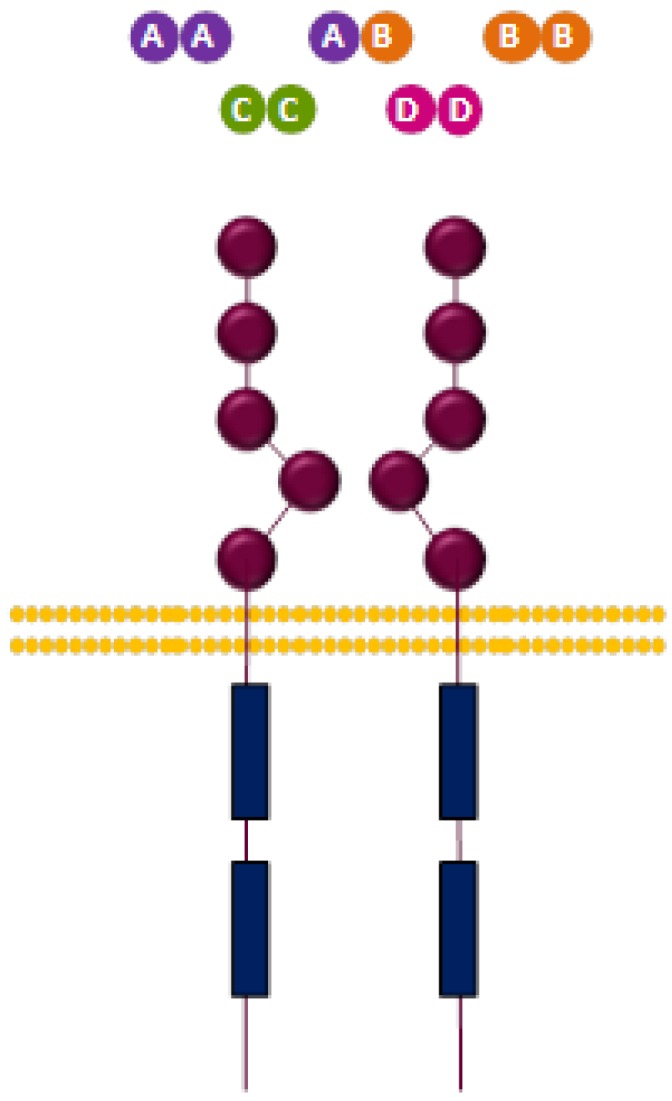
Structure of PDGFR and ligands.
PDGFRα and PDGFRβ activate different signaling molecules. For instance, PLCγ binds PDGFRα with higher affinity than PDGFRβ; Crk adaptor protein binds PDGFRα but not PDGFRβ; conversely, GAP binds PDGFRβ, but nor PDGFRα [113]. Moreover, PDGFRβ mobilizes calcium ions more efficiently than PDGFRα [114]. It has been shown in mice than the substitution of the PDGFRβ cytoplasmic domain for the PDGFRα domain leads to phenotypic differences [115], which indicates that both isoforms trigger different signaling mechanisms.
3.2.2. PDGFR Ligands
PDGFR ligands are platelet-derived growth factors (PDGF). The first PDGF was isolated in 1979 [116]. Nowadays, there are four known PDGFR ligands: PDGF-A, PDGF-B, PDGF-C and PDGF-D. These growth factors are comprised of 100 amino acids and are codified by four different genes. PDGF family requires proteolytic processing for its activation. They are linked by disulfide bonds, forming functional homodimers and heterodimers. The PDGF family can be classified in two subfamilies according to structural differences and proteolytic processing. PDGF-A and PDGF-B constitute one subfamily and are secreted as functional forms after amino-terminal end intracellular cleavage. On the other hand, PDGF-C and PDGF-D are not intracellularly processed, but rather activated after secretion by cleavage of the CUB motif, present on the amino-terminal end [117].
PDGFs act mainly in a paracrine fashion. However, it has been observed that in some tumors they can trigger autocrine signaling, although PDGFRs do not physiologically function in an autocrine manner. These growth factors play a pivotal role during development, but lack physiological functions in the adult [104].
PDGFR ligands are differentially expressed in the organism. In this sense, PDGF-B is expressed mainly in vascular endothelial cells, megakariocytes and neurons; PDGF-A and PDGF-C in epithelial, muscle and neuronal progenitor cells; and PDGF-D in fibroblasts and smooth muscle cells [118].
Different PDGFs combinations have been described, which are: PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD. These dimers present differential affinity for PDGFRs and generate distinct PDGFR dimer configurations. In this regard, PDGFR-α can be activated by PDGF-AA, PDGF-AB, PDGF-BB or PDGF-CC [100], whereas PDGFR-β can be stimulated by PDGF-BB or PDGF-DD. In addition, PDGFR-α/β heterodimer can be activated upon binding of PDGF-AB, PDGF-BB or PDGF-CC [119] (Table 3).
Table 3.
PDGFR family members and their ligands. Different PDGF combinations.
| Ligands | Dimers |
|---|---|
| AA, BB, CC, AB | PDGFR-α/α |
| BB, DD | PDGFR-β/β |
| BB, CC, AB | PDGFR-α/β |
3.2.3. PDGFR and Cancer
PDGFR family as well as receptor ligands have been associated with tumorigenesis, tumor survival and poor prognosis [104]. PDGFR and PDGF transforming ability has been shown in several reports, thereby indicating that PDGF-B expression or overexpression in mice brain cells results in oligodendrogliomas and oligoastrocytomas [120] and tumors resembling glioblastomas [121]. It is now thought that gliomas could be originated from neural stem cells present in the adult brain [122] and that PDGFR could be involved in this oncogenic process. This hypothesis is sustained by two findings. First, adult neural stem cells express PDGFRα and proliferate in response to PDGF originating glioma-like hyperplasias [123]. Second, adult neural precursors, which have undergone spontaneous malignant transformation, show PDGFRα-deficient inactivation and cell proliferation inhibition after PDGFRα silencing [124].
Overexpression of PDGFR and/or their ligands have been documented in several human cancers [125]. In the case of gliomas, PDGFRα overexpression is related to tumor malignancy [126]. In addition, PDGFRα is related to tumor aggressiveness and poor prognosis in breast and prostate cancer. In this sense, PDGFs expression [127] and stromal PDGFRβ expression is associated with unfavorable clinicopathological parameters and shortened survival in breast cancer [128]. In prostate cancer, metastatic potential of tumor cells is related to PDGFRα expression [129] and PDGF-D overexpression is known to be involved in epithelial-mesenchymal transition [130].
Autocrine stimulation due to PDGFR and PDGF coexpression contributes to proliferation of glioblastoma and other gliomas [131,132], breast tumors [133] or leukemia [134]. Moreover, in some tumors, PDGF paracrine signaling with stroma tumor cells that express PDGFR (pericytes, fibroblasts and endothelial cells), which is involved in tumor progression [135], has been described.
In contrast, PDGFR mutations are not very frequent in cancer cells. Activating PDGFRα mutations have been found in a small percentage of glioblastomas [136,137] and gastrointenstinal stromal tumors (GIST) [138]. The mutation found in 40% of glioblastomas with amplified PDGFRA [137] consists of a deletion of exons 8 and 9 coding for an extracellular domain fragment [139]. This mutant receptor variant, which is known as PDGFRAΔ8,9, exhibits high tyrosine kinase activity and transforming potential [136,137]. On the other hand, there are five different translocation described for PDGFRB in leukemia cells, which lead to constitutively active receptor production [140].
Translocations that disrupt and constitutively activate the PDGFRB gene have been described in CML patients. These patients respond well to Imatinib mesylate therapy [141]. Reciprocal translocation within o near the chromosome 4q12 locus of the PDGFRA and KIT genes have been reported in leukemias that give rise to a fusion tyrosine kinase. It has been described as a gain-of-function protein, resulting from an interstitial deletion rather than reciprocal translocation that fuses the PDGFRA gene with a FIP1-like-1 (FIP1L1) gene. The gene product is a tyrosine kinase with transforming ability [142].
3.3. Insulin-Like Growth Factor 1 Receptor (IGF-1R)
Insulin-like growth factor 1 receptor (IGF-1R) belongs to the subfamily II of RTK, together with the insulin receptor and insulin-like growth factor 2 receptor (IGF-2R), also known as mannose 6- phosphate receptor. IGF-1R is ubiqitously expressed in several human tissues and is involved in mitogenic signaling processes, malignant transformation, protection against apoptosis and cell differentiation [143,144]. IGF-1R plays a role in organ development [145] and is essential for brain development [146].
3.3.1. Structure and Activation Mechanism
IGF-1R is made up of two α extracellular ligand-binding subunits and β subunits forming the membrane-spanning region and the cytoplasmic domain containing the tyrosine kinase activity region (residues 931-1337) [143]. The α subunits are composed of 706 amino acids, whereas the β subunits are composed of 626 amino acids. The α subunits are associated between themselves and linked to the β subunits by disulfide bonds (Figure 3). The ectodomain subunits are composed of two leucine-rich (L1 and L2), one cysteine-rich (CR) and three fibronectine III-like regions (FnIII-1, FnIII-2 and FnIII-3). The tyrosine kinase region (residues 973-1229) has three tyrosine residues essential for receptor activation (Y1131, Y1135 e Y1136) [147], and is flanked by two regulatory regions: the juxtamembrane region and the carboxy-terminal end [147,148].
Figure 3.
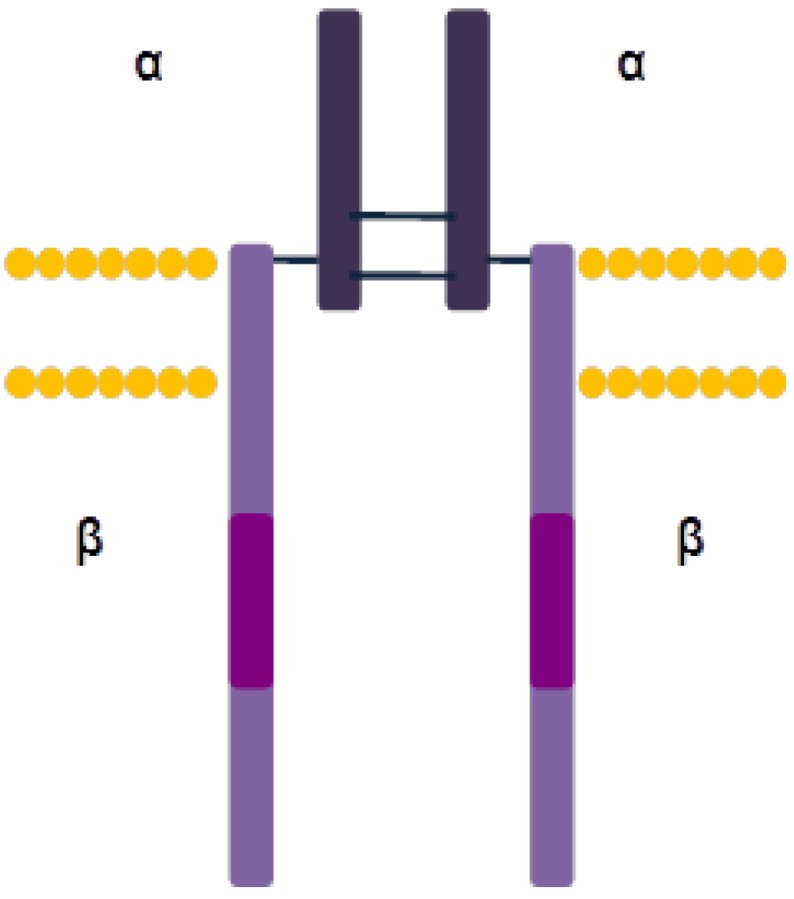
IGF-1R structure.
IGF-1R is synthesized as a single polypeptide chain that is proteolytically processed in the Golgi apparatus, giving rise to the α and β subunits, which are linked by disulfide bonds [143]. IGF-1R differs from the majority of the RTK because it is presented as a dimer in the absence of ligand stimulus. Nonactivated IGF-1R is found in an autoinhibitory conformation in which the ATP-binding site and active center are inaccessible [149]. Ligand binding to IGF-1R takes place through the cysteine-rich region in the ectodomain and leads to allosteric interaction between the two monomers. This interaction produces phosphorylation of the three tyrosine residues within the kinase domain activation loop (Tyr1131, Tyr1135 and Tyr1136) and receptor activation [150]. Tyr 1131 and Tyr1135 phosphorylation destabilizes the autoinhibitory confirmation, whereas Tyr1136 phosphorylation stabilizes the catalytically active conformation [149]. Then, tyrosine kinase of each monomer phosphorylates tyrosines located on juxtamembrane domain and carboxy-terminal end of the other monomer, which recruit SH2 or PTB domain-containing signaling molecules, such as 14-3-3, Shc o IRS-1-4 [144]. These signaling molecules initiate the PI3K/Akt or the Ras/Raf/Mek/Erk pathway. After signaling, the receptor is endocytosed and degraded in the lysosomes [151].
Insulin receptor (IR) shares significant structural and functional similarity with IGF-1R. The differences reside mainly in the ligand-recognition sites [152,153]. There are two isoforms presents in mammals: IR-A and IR-B, which result from mRNA splicing and differ in one exon. The IR-A isoform lacks exon 11, which codes for a 12-amino-acid fragment (residues 717–728) of the α subunit carboxy-terminal end [154]. IR-A plays a role during embryogenesis and fetal development. IR-B is expressed in liver, muscle, adipose tissue and kidney, where it regulates homeostasis glucose [155].
IGF-2R is momomeric; it lacks tyrosine kinase activity and is not involved in signal transduction [156]. Rather, IGF-2R in involved in lysosomal-enzyme trafficking towards lysosomes [157].
3.3.2. IGF-1R Ligands
The ligands of this receptor family are insulin, insulin growth factor I (IGF-I) and insulin growth factor II (IGF-II). These ligands are 70-amino-acid polypeptides with a similar tridimensional structure. Insulin binds IRs with high affinity and IGF-1R with low affinity. IGFs, especially IGF-I, binds IGF-1R with high affinity and IRs with low affinity [158,159]. However, the only ligand that binds IGF-2R is IGF-II, which is concomitantly degraded [156] (Table 4). HybridIR-IGF-1R dimers in cells that express both receptors have been described [160,161,162]. However, data is contradictory regarding hybrid receptor-ligand affinity. Some results indicate that insulin and IGF-II bind IR-A-IGF-1R hybrid, and that IGF-I, on the other hand, binds IR-B-IGF-1R hybrid [158]. More recently, it has been shown that IGF-I e IGF-II ligands bind hybrids formed by either IR-A or IR-B with affinity, whereas insulin binds with low affinity [159].
Table 4.
Insulin receptor family members and its ligands.
| RECEPTOR | Locus | Structure | Kinase activity | Ligands |
|---|---|---|---|---|
| IR-A | 19p13.2 | dimer | yes | Insulin, IGF-I, IGF-II. |
| IR-B | 19p13.2 | dimer | yes | Insulin, IGF-I |
| IGF-1R | 15q26 | dimer | yes | IGF-I, IGF-II, Insulin |
| IGF-2R | 6q26-27 | monomer | no | IGF-II, lyosomal enzymes |
IGFs are regulated by insulin-like growth factor-binding proteins (IGFBP). These molecules are cytoplasmic proteins that bind IGF-I and IGF-II with affinity preventing their degradation. However, it is not clear whether they stimulate or inhibit IGFs, since they negatively modulate these growth factors’ interaction with their receptors [163].
3.3.3. IGF-1R and Cancer
IGF-1R has been associated with malignant cell transformation and tumor progression. Moreover, IGF-1R expression is linked to oncogenesis, as it has been shown that it is positively regulated by several oncogenes (mutant TP53, c-MYB, etc.), and that tumor-suppressor gene loss (TP53, BRCA1 or WT1) leads to its overexpression [164]. The first studies showing a relationship between IGF-1R and malignant transformation indicate that mouse embryo fibroblasts lacking IGF-1R were resistant to transformation driven by several oncogenes, and that this malignant transformation was established upon IGF-1R re-expression [165,166]. In another more recent study, IGF-1R was overexpressed in RIP1-Tag2 mice and, consequently, pancreatic tumor development was accelerated [167]. Other studies show that expression of a constitutively active IGF-1R form in mice provoked spontaneous development of salivary and mammary gland carcinomas [168]. Moreover, transgenic mice overexpressing IGF-1R induced lung tumorigenesis [169].
IGF-1R is overexpressed in melanoma, colon, pancreatic, prostate, breast and renal cell carcinoma [170,171,172,173,174]. It has been shown that inhibition of IGF-1R expression or activity produces antiproliferative effects in glioblastoma [175,176,177,178]. However, no activating mutations have been described, IGF-1R overexpression being the most common alteration found in tumors. However, apart from a small percentage of breast tumors [179], this overexpression is not due to gene amplification [180].
In addition, IGF-1R ligands are related to tumorigenesis. High IGF-1R levels in serum are associated with an elevated risk to develop colon, breast or prostate cancer. It has also been shown that IGF-I overexpression in the basal epidermis layer and in the prostatic epithelium leads to spontaneous tumor formation in mice [181,182]. IGF-1R ligands expression, which is restricted to brain during fetal development, reappears in glioblastoma [183,184]. This suggests that IGFs’ expression contributes to the development of these tumors. Additionally, it has been observed that IGF-I induces in vitro proliferation and cell migration in GBM [185].
IGF-1R represents a potential target for cancer treatment since is not essential for normal cell survival [186]. There are about 30 molecules targeted against IGF-1R in different clinical trial phases [187], while different approaches to interfere with IGF-1R-initiated cell signaling are being evaluated. These strategies include: antibodies which block receptor-ligand binding, tyrosine kinase inhibitors, antisense oligonucleotides or interference RNA to silence receptor expression, etc. [187]. IGF-1 and IGF-II represent potential targets, as well. However, only neutralizing antibodies have been developed thus far [188,189]. An additional strategy is based on IGFBP induction in order to inhibit IGF binding to IGF-1R [190,191].
Like other RTKs, IGF-1R can translocate to the nucleus where it interacts with chromatin, suggesting a role in transcriptional regulation [192]. IGF-1 induces IGF-1R modification by small ubiquitin-like modifier protein-1 (SUMO-1) and its translocation to the nucleus [193].
4. Current Targeted Therapies
4.1. EGFR
EGF was the first growth factor discovered [32], followed by EGFR isolation [31]. In fact, there was no precedent that cellular receptors had tyrosine kinase activity until it was demonstrated that EGFR activation led to phosphorylation of cytoplasmic proteins tyrosine residues [34]. In addition, dimerization as an activation mechanism was also observed for the first time in EGFR [194,195,196]. Moreover, several studies have shown that EGFR inhibition has a pivotal role as an anti-tumor agent [197] and, for this reason, several drugs targeted against EGFR are currently employed for the treatment of head and neck, colorectal, lung and pancreatic cancer [198].
The main prognostic factors in GBM are: patient’s age, cognitive state, mutational status of the isocitrate dehydrogenase 1 (IDH1) gene and promoter methylation of the O6- methyl guanine methyl transferase (MGMT) gene [199]. The prognostic value of EGFR amplification and/or overexpression in GBM remains controversial, since some studies support that that EGFR is a poor prognosis factor [200], and others that there is no relationship between EGFR levels and glioma evolution [201].
At present, the only response therapy marker utilized is MGMT promoter methylation status, as it defines the degree of response to alkylating agents such as Temozolomide (Temodar®/Temodal®) [202]. The use of small-molecule inhibitors (Erlotinib, Gefintib, Labatinib, etc.) and monoclonal antibodies targeted against EGFR (Cetuximab and Panitututmab) is a common therapeutic strategy in several solid tumors, including gliomas. Small-molecule tyrosine kinase inhibitors are especially attractive, as they are able to cross the blood–brain barrier [203]. However, there is only one study that associates the degree of response to the EGFR inhibitor Erlotinib with high EGFR expression levels [204]. We have shown that the response to EGFR inhibitors is not necessarily related to the EGFR levels in glioblastoma cell lines [205]. In this respect, EGFR tyrosine kinase inhibitors (AG1478, Gefitinib, Erlotinib and Lapatinib) were able to abrogate GBM cell line growth, whereas Cetuximab had no effect. Furthermore, small-molecule EGFR inhibitors were able to prevent phosphorylation of erbB-3 and erbB-4, whereas Cetuximab only hindered EGFR phosphorylation, suggesting that EGFR tyrosine kinase inhibitors may mediate their anti-proliferative effects through other erbB family members. Therefore, we show that it is important to characterize patients not only measuring EGFR status (gene amplification, presence of mutations or expression levels), but also monitoring erbB-2, erbB-3 and erbB4 levels, since all these receptors interact among themselves and can induce a response.
Several clinical trials using EGFR inhibitors such as Erlotinib are being conducted in GBM [206] as monotherapy in GBM, or in combination with other agents. In addition, the new inhibitor GW572016 (Lapatinib, Tykerb®) has entered clinical trials of patients with recurrent GBM [207,208]. In addition, we and others have shown in a preclinical setting that mutant EGFR forms such as the EGFRvIII or the TDM/18-26 variant are also abrogated by EGFR inhibitors [205,209]. Moreover, it has been shown that coexpression of EGFFRvIII and functional PTEN can be responsive markers to Gefitinib and Erlotinib response [210].
4.2. PDGFR
Alterations in signaling initiated by PDGFR have been found in several types of tumors [104]. In particular, it has been shown that PDGF and PDGFR signaling is an important event for both the onset and transformation from astrocytoma to GBM [211]. In particular, PDGFRα overexpression has been detected in all subtypes of gliomas, especially in glioblastoma [212]. Moreover, PDGFR mRNA has been detected in several human GBM cell lines; whereas no expression has been observed in normal fetal and adult brain tissues [132]. Interestingly, both PDGFRs and their ligands are coexpressed in astrocytic gliomas and different GBM cell lines [131,213,214]. These findings indicate that autocrine signaling triggered by PDGF is an important event in glioma proliferation and malignant transformation.
There are several antitumor agents targeted against PDGFR. These include: Imatinib (Gleevec®), which is employed for the treatment of chronic myeloid leukemia (CML) or GIST, Sorafenib (Nexavar®) for advanced renal carcinoma, Nilotinib (Tasigna®) for CML, or Sunitinib (Sutent®) for kidney cell carcinoma or Imatinib-resistant GIST. All these drugs are multi-targeted tyrosine kinase inhibitors and can inhibit other RTK apart from PDGFR, such as VEGFR FGFR, Bcr-Abl (c-Abl) and stem cell factor receptor (c-Kit). It has been shown that Imatinib inhibits GBM cell proliferation and induces cell cycle arrest in the G1 phase of the cell cycle [215]. Another study shows that Imatinib inhibits cell migration and acts synergistically with temozolomide and hydroxyurea [216]. In a phase II study of Sunitinib in patients with high-grade glioma, no correlation was found between VEGFR2, PDGFR-α, and KIT levels and response to treatment [217]. Other clinical trials with several PDGFR inhibitors are currently ongoing and show promising results. [218].
4.3. IGF-1R
IGF-1R is a potential target for cancer therapeutics since it plays a fundamental role in the progression of several types of cancer [170,171,172,173,174,186,219], and is involved in resistance to cancer therapy [220]. IGF-1R ligands expression, restricted to brain during fetal development, reappears in glioblastoma [183,184]. Therefore, signaling initiated by IGF-1R is important for glioblastoma progression and development. In fact, several studies show that inhibition of IGF-1R expression exerts antiproliferative effects in glioblastoma [175,176,177,178].
IGF-1R can modify sensitivity to several chemotherapeutic agents [221]. For example, PI3K/Akt activation induced by IGF mediates resistance to EGFR blockade in glioblastoma [222]. However, biomarkers of response to IGF-1R inhibitors are unknown as yet. In this respect, it has been shown that insulin receptor substrate-1 (IRS-1) levels are associated to the IGF-1R inhibitor NVP-AEW541 response [223], independently of the IGF-1R expression levels [224].
Multiple approaches are being employed to abolish IGF-1R signaling in vivo and in vitro. These approaches include dominant negative and kinase defective mutants, anti-sense oligonucleotides, small interference RNA (siRNA), small molecule tyrosine kinase inhibitors, blocking antibodies and IGF binding proteins (IGFBP) [225]. IGFBPs can modulate IGFs activity. They have been shown to induce apoptosis and regulate cell survival in the absence of ligand [226]. Therefore, treatment with recombinant IGFBPs represents a different approach for cancer therapeutics. So far, several small molecule inhibitors targeted against IGF-1R have entered clinical trials: OSI-906 (OSI Pharmaceuticals), XL-228 (Exelixis) and INSM-18 (Insmed), while others are in preclinical phase: A-928605 (Abbot), BMS-536924 (Bristol-Myers Squibb), BMS-554417 (Bristol-Myers Squibb), picropodophyllin (PPP) (Karolinska Cancer Institute and Biovitrum), NVP-ADW742 (Novartis Pharma) and NVP- AEW541 (Novartis Pharma) [221]. However, high homology between IGFR and IR represents a specificity caveat for drug design [143]. Conversely, antibodies against IGF-1R do not bind IR. There are several IGF-1R blocking antibodies currently in clinical early development: AVE-1642 (Sanofi-Aventis), SCH-717454 (Schering-Plough), CP-751,871(Pfizer), IMC-A12 (ImClone Systems), BIIB022 (Biogen Idec), MK-0646 (Merck), R1507 (Roche) and AMG 479 (Amgen) [221].
Interestingly, the ciclolignan PPP shows high specificity for IGFR-1R due to its binding to the substrate site, instead of to the ATP site [227], and for this reason it does not affect IR [227,228]. PPP reduces dramatically both subcutaneous and intracerebral xenografts, indicating that this inhibitor is able to cross the blood–brain barrier [229]. A small number of clinical trials in patients with glioma, using IGF-1R inhibitors including PPP as single agents, or in combination with other chemotherapeutic drugs, are currently ongoing [230].
4.4. MicroRNAs in Glioblastoma
MicroRNAs (miRNAs) have gained recent interest as potential therapeutic tools. These short noncoding RNA molecules modulate the expression of numerous target genes, and can act either as oncogenes or tumor suppressor genes. They represent an additional regulation step of endogenous gene transcription and translation. A novel therapeutic option is the delivery of miRNAs to modulate the expression of target mRNAs [231]. Mature miRNAs are one strand RNA non-coding molecules (19–25 nucleotides) processed from longer RNA molecules (70 nucleotides). Once a miRNA is processed, the RNA-induced silencing complex (RISC) binds to the mature miRNA, which, in turn, binds the 3’untranslated region of target mRNAs in two ways: (1) upon binding to imperfect complementary sites, blocking gene expression at the translation level; or (2) upon binding perfectly complementary sites, acting as a siRNA, subsequently inducing mRNA cleavage [232]. In regards to GBM, a significant amount of work has been carried out in deciphering alterations of miRNA expression, their impact in glioma development and patient prognosis and the potential of miRNA-based technology [233]. miRNAs play roles in tumor-initiating brain cells, cell cycle, proliferation, apoptosis, invasion and angiogenesis. A review by Moller and coauthors [231] has revised over a hundred articles and has identified 253 upregulated, 95 downregulated, and 17 unclear miRNAs with respect to expression levels in GBM. In regards to RTK, miR-7, whose expression is downregulated in GBM, has been shown to inhibit EGFR, IRS-1 and IRS-1 and, consequently, the PI3K/Akt pathway [234]. miR-7 is involved in differentiation, invasion and proliferation. Expression of miR-128 is also repressed in GBM. This miRNA involved in proliferation, self-renewal and tumor growth can modulate differentiation by targeting EGFR and PDGFRα and may be a candidate glioma tumor suppressor [235]. miR-503 expression is repressed in GBM and its upregulation inhibits IGF-1R expression, suggesting that miR-503 is a candidate tumor suppressor, as well [236]. Conversely, miRNAs’ expression can be regulated by RTK signaling. It has been recently demonstrated that PDGF-BB induces miR-146b, which consequently downregulates EGFR [237], thereby having an impact on migration and invasion [238]. The most-studied miRNA is miR-21, whose expression is upregulated in GBM and has oncogenic potential. It downregulates PTEN and the EGFR pathway in a PTEN-independent manner [239]. This anti-apoptotic miRNA is involved in chemoresistance, invasion, proliferation and tumor growth [240]. Therefore, apart from their roles in gene regulation, miRNAs have a significant potential in diagnosis, prognosis and novel therapeutics in glioblastoma.
4.5. Failure of Current Therapies
The standard current care for GBM patients consists of radiotherapy plus adjuvant DNA-alkylating agents, such as temozolomide, nitrosourea agents or cisplatin. These therapies, however, have a poor median survival time of less than 15 months after diagnosis [241]. The failure of such treatments is due to overexpression of several oncogenic proteins and development of resistance mechanisms. The initiation and progression of medulloblastomas and glioblastomas are associated with genetic alterations in PTEN, TP53, p16INK4A and p19ARF, among others. These alterations result in constitutive and sustained activation of signaling pathways initiated by RTK, such as EGFR. In turn, EGFR amplifications and mutations found in 30%–60% of GBM [200] contribute to the onset and progression of primary brain tumors. Moreover, activation of the PI3K/Akt pathway can be detected in about 77%–87% of tumor tissues of GBM patients [242]. Other pathways that are also frequently deregulated and constitutively activated in brain tumor cells are: Sonic Hedgehog, Wnt/β-catenin, c-Met/HGF, PDGFRs, IGF-1R, VEGFR, c-Kit, Notch, etc. [79]. The activation of such pathways promotes the sustained growth of brain tumor-initiating, stem or progenitor cells. These cells, which express typical stem cell-like markers such as CD133, Nestin, CD44, Nanog, Klf4 and/or Oct-3/4, have the ability of forming spheres in vitro and tumors in animal models [243]. These cancer stem cells, which have long-term self-renewal and replicative immortality in vitro and tumor-initiating potential in vivo, hold intrinsic radio- and chemoresistance, which is a major cause of failure to treatment. In this respect, it has been shown that EGFR inhibition induces c-Met activation in tumor-initiating cells in an EGFR-driven GBM mouse model [244].
Despite aggressive treatments, recurrence takes place in 90% of GBM patients. Another cause of failure to response treatment is the low bioability of anticancer drugs delivered to brain tumor cells due to restrictions imposed by the brain–blood barrier.
Resistance may be limited to a single agent or associated with cross-resistance to different drugs with or without structural similarity to the primary agent [245]. This pleiotropic phenomenon known as multidrug resistance (MDR) [246] could be one of the causes of the poor outcome present in GBM. Although several mechanisms could be involved in the acquisition of this phenotype, the role of P-glycoprotein (Pgp), a member of the ATP-binding cassette (ABC) transporter family, has been well established [247,248,249]. Pgp, encoded by the gene MDR1, was first identified as a consequence of its overexpression in multidrug-resistant tumor cells, where it mediates the ATP-dependent efflux of a variety of chemotherapeutic agents [250]. Expression of Pgp and several multidrug-resistant proteins (MRPs) have been found in glioblastoma cell lines [251]. Additionally, Pgp is overexpressed in endothelial cells of capillaries that form the brain–blood barrier [252,253]. Moreover, MRP-1 and MGMT expression can be significant prognostic factors for the overall survival of GBM patients [254]. Therefore, overexpression of members of the ABC transporter family involved in drug efflux may play a role in intrinsic resistance to chemotherapeutic drugs in glioblastoma.
In addition, c-Met may contribute to acquired resistance to small-molecule EGFR inhibitors such as Gefitinib by erbB3 engagement and reactivation of the PI3K/Akt pathway [255], even when EGFR antagonists are combined with c-Met inhibitors [256].
It has been shown that Cetuximab resistance may also be due to c-Met/HGF activation [257] and that resistance to anti-EGFR therapy is mediated by IGF-1R activation [222]. Moreover, IGFBP-3 and IGF-BP4 downregulation can mediate acquired resistance to EGFR inhibitors by engagement of the IGF-1R pathway [258]. Acquired resistance has also been developed to the anti-erbB2 monoclonal antibody Trastuzumab (Herceptin®) in HER2+ breast cancer patients, which seems to be mediated by IGF-1R [259].
In the cases presented so far, resistance was caused by activation of alternative pathways, but the drug target itself remained sensitive to the treatment. Another mechanism of acquired resistance is genetic alteration of the molecular target [260]. This phenomenon was initially observed in Imatinib-resistant CML and GIST, where the respective targets, Bcr-Abl and c-Kit acquired a mutation in a “gatekeeper” residue of the ATP-binding pocket [261,262]. Likewise, the T790M mutation in the EGFR gene rose after chronic exposure to Gefitinib in NSCLC, conferring resistance to this inhibitor [263].
To summarize, several factors are involved in failure to targeted therapy: the presence of initiating-tumor cells, the low bioability of anti-tumor drugs that need to cross the blood–brain barrier, genetic alterations, oncogene activation in brain tumor cells, intrinsic resistance due to overexpression of drug transporters and acquired resistance to RTK inhibitors.
In order to overcome resistance to small molecule tyrosine kinase inhibitors, the use of oncogene-addicted cell lines highly sensitive to these agents could aid to identify possible escape mechanisms [260]. In addition, the development of small-molecule second-generation compounds which do not require the “gatekeeper” residue and the development of multi-targeted kinase inhibitors are useful approaches to override resistance mechanisms [264]. Combination of RTK antagonists with inhibitors targeting different signaling pathways that share common downstream mediators (e.g., EGFR and IGF-1R) or with other drugs involved in survival or angiogenesis could also be helpful strategies [265].
5. Conclusions
We have described in this review the signaling pathways initiated by three tyrosine kinase receptor families: EGFR, PDGFR and IGF-1R. We have explained their structure, family members, ligands, and mechanisms of action. More importantly, we have illustrated the role that these receptors and their ligands play in cancer in general, especially in glioblastoma. Lastly, we have explained the current cancer therapies employing inhibitors targeted against these receptors, with special emphasis on glioblastoma, the role of miRNAs, and possible causes of failure to current therapies, such as oncogene activation, presence of tumor-initiating cells, and chemoresistance.
Acknowledgments
We would like to thank our lab members for helpful comments. This article has been funded by Instituto de Salud Carlos III grant FIS PI041084 to I. Martínez-Lacaci, by a grant from the Fundación de Investigación Médica Mutua Madrileña to I. Martínez-Lacaci and by Conselleria de Sanidad GVA grant AP-089/10 to M. Saceda.
Author Contributions
Estefanía Carrasco-García wrote part of the manuscript, searched for references and prepared the figures. Miguel Saceda designed the structure of the review and wrote part of the manuscript. Isabel Martínez-Lacaci wrote part of the manuscript and corrected the English grammar. All co-authors proofread the manuscript and gave helpful suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Ferlay J., Shin H.R., Bray F., Forman D., Mathers C., Parkin D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Bondy M.L., Scheurer M.E., Malmer B., Barnholtz-Sloan J.S., Davis F.G., Il'yasova D., Kruchko C., McCarthy B.J., Rajaraman P., Schwartzbaum J.A., et al. Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer. 2008;113:1953–1968. doi: 10.1002/cncr.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohgaki H. Epidemiology of brain tumors. Methods Mol. Biol. 2009;472:323–342. doi: 10.1007/978-1-60327-492-0_14. [DOI] [PubMed] [Google Scholar]
- 4.Gurney J.G., Kadan-Lottick N. Brain and other central nervous system tumors: Rates, trends, and epidemiology. Curr. Opin. Oncol. 2001;13:160–166. doi: 10.1097/00001622-200105000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R., Hegi M.E., Van den Bent M.J., Mason W.P., Weller M., Mirimanoff R.O., Cairncross J.G. Changing paradigms--an update on the multidisciplinary management of malignant glioma. Oncologist. 2006;11:165–180. doi: 10.1634/theoncologist.11-2-165. [DOI] [PubMed] [Google Scholar]
- 6.Villano J.L., Seery T.E., Bressler L.R. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother. Pharmacol. 2009;64:647–655. doi: 10.1007/s00280-009-1050-5. [DOI] [PubMed] [Google Scholar]
- 7.Koukourakis G.V., Kouloulias V., Zacharias G., Papadimitriou C., Pantelakos P., Maravelis G., Fotineas A., Beli I., Chaldeopoulos D., Kouvaris J. Temozolomide with radiation therapy in high grade brain gliomas: pharmaceuticals considerations and efficacy; a review article. Molecules. 2009;14:1561–1577. doi: 10.3390/molecules14041561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/S0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 9.Robinson D.R., Wu Y.M., Lin S.F. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 10.Hubbard S.R., Till J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 11.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/S0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 12.Fayard E., Xue G., Parcellier A., Bozulic L., Hemmings B.A. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- 13.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 14.Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 15.Maehama T., Dixon J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 16.Wang S.I., Puc J., Li J., Bruce J.N., Cairns P., Sidransky D., Parsons R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183–4186. [PubMed] [Google Scholar]
- 17.Baeza N., Weller M., Yonekawa Y., Kleihues P., Ohgaki H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 2003;106:479–485. doi: 10.1007/s00401-003-0748-4. [DOI] [PubMed] [Google Scholar]
- 18.Yoon S., Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 19.Pages G., Lenormand P., L'Allemain G., Chambard J.C., Meloche S., Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. USA. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klemke R.L., Cai S., Giannini A.L., Gallagher P.J., de Lanerolle P., Cheresh D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warn-Cramer B.J., Cottrell G.T., Burt J.M., Lau A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998;273:9188–9196. doi: 10.1074/jbc.273.15.9188. [DOI] [PubMed] [Google Scholar]
- 22.Lord J.D., McIntosh B.C., Greenberg P.D., Nelson B.H. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J. Immunol. 2000;164:2533–2541. doi: 10.4049/jimmunol.164.5.2533. [DOI] [PubMed] [Google Scholar]
- 23.Rawlings J.S., Rosler K.M., Harrison D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 24.Krebs D.L., Hilton D.J. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 25.Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006;16:196–202. doi: 10.1038/sj.cr.7310027. [DOI] [PubMed] [Google Scholar]
- 26.Aaronson D.S., Horvath C.M. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 27.Sibilia M., Kroismayr R., Lichtenberger B.M., Natarajan A., Hecking M., Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007;75:770–787. doi: 10.1111/j.1432-0436.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama T., Sudo C., Ogawara H., Toyoshima K., Yamamoto T. The product of the human c-erbB-2 gene: A 185-kilodalton glycoprotein with tyrosine kinase activity. Science. 1986;232:1644–1646. doi: 10.1126/science.3012781. [DOI] [PubMed] [Google Scholar]
- 29.Kraus M.H., Issing W., Miki T., Popescu N.C., Aaronson S.A. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: evidence for overexpression in a subset of human mammary tumors. Proc. Natl. Acad. Sci. USA. 1989;86:9193–9197. doi: 10.1073/pnas.86.23.9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plowman G.D., Culouscou J.M., Whitney G.S., Green J.M., Carlton G.W., Foy L., Neubauer M.G., Shoyab M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl. Acad. Sci. USA. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carpenter G., Cohen S. Human epidermal growth factor and the proliferation of human fibroblasts. J. Cell Physiol. 1976;88:227–237. doi: 10.1002/jcp.1040880212. [DOI] [PubMed] [Google Scholar]
- 32.Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962;237:1555–1562. [PubMed] [Google Scholar]
- 33.Chinkers M., Cohen S. Purified EGF receptor-kinase interacts specifically with antibodies to Rous sarcoma virus transforming protein. Nature. 1981;290:516–519. doi: 10.1038/290516a0. [DOI] [PubMed] [Google Scholar]
- 34.Ushiro H., Cohen S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J. Biol. Chem. 1980;255:8363–8365. [PubMed] [Google Scholar]
- 35.Downward J., Yarden Y., Mayes E., Scrace G., Totty N., Stockwell P., Ullrich A., Schlessinger J., Waterfield M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521–527. doi: 10.1038/307521a0. [DOI] [PubMed] [Google Scholar]
- 36.Yarden Y., Schlessinger J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987;26:1434–1442. doi: 10.1021/bi00379a034. [DOI] [PubMed] [Google Scholar]
- 37.Lax I., Bellot F., Howk R., Ullrich A., Givol D., Schlessinger J. Functional analysis of the ligand binding site of EGF-receptor utilizing chimeric chicken/human receptor molecules. EMBO J. 1989;8:421–427. doi: 10.1002/j.1460-2075.1989.tb03393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogiso H., Ishitani R., Nureki O., Fukai S., Yamanaka M., Kim J.H., Saito K., Sakamoto A., Inoue M., Shirouzu M., et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/S0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 39.Lemmon M.A., Bu Z., Ladbury J.E., Zhou M., Pinchasi D., Lax I., Engelman D.M., Schlessinger J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997;16:281–294. doi: 10.1093/emboj/16.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemmon M.A., Schlessinger J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem. Sci. 1994;19:459–463. doi: 10.1016/0968-0004(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Gureasko J., Shen K., Cole P.A., Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 42.Wiley H.S., Herbst J.J., Walsh B.J., Lauffenburger D.A., Rosenfeld M.G., Gill G.N. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 1991;266:11083–11094. [PubMed] [Google Scholar]
- 43.Moriki T., Maruyama H., Maruyama I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001;311:1011–1026. doi: 10.1006/jmbi.2001.4923. [DOI] [PubMed] [Google Scholar]
- 44.Guy P.M., Platko J.V., Cantley L.C., Cerione R.A., Carraway K.L., III Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. USA. 1994;91:8132–8136. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pinkas-Kramarski R., Soussan L., Waterman H., Levkowitz G., Alroy I., Klapper L., Lavi S., Seger R., Ratzkin B.J., Sela M., et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996;15:2452–2467. [PMC free article] [PubMed] [Google Scholar]
- 46.Lee D.C., Rose T.M., Webb N.R., Todaro G.J. Cloning and sequence analysis of a cDNA for rat transforming growth factor-alpha. Nature. 1985;313:489–491. doi: 10.1038/313489a0. [DOI] [PubMed] [Google Scholar]
- 47.Pandiella A., Massague J. Cleavage of the membrane precursor for transforming growth factor alpha is a regulated process. Proc. Natl. Acad. Sci. USA. 1991;88:1726–1730. doi: 10.1073/pnas.88.5.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrlich A., Klinman E., Fu J., Sadegh C., Lodish H. Ectodomain cleavage of the EGF ligands HB-EGF, neuregulin1-beta, and TGF-alpha is specifically triggered by different stimuli and involves different PKC isoenzymes. FASEB J. 2008;22:4281–4295. doi: 10.1096/fj.08-113852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anklesaria P., Teixido J., Laiho M., Pierce J.H., Greenberger J.S., Massague J. Cell-cell adhesion mediated by binding of membrane-anchored transforming growth factor alpha to epidermal growth factor receptors promotes cell proliferation. Proc. Natl. Acad. Sci. USA. 1990;87:3289–3293. doi: 10.1073/pnas.87.9.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riese D.J., Bermingham Y., van Raaij T.M., Buckley S., Plowman G.D., Stern D.F. Betacellulin activates the epidermal growth factor receptor and erbB-4, and induces cellular response patterns distinct from those stimulated by epidermal growth factor or neuregulin-beta. Oncogene. 1996;12:345–353. [PubMed] [Google Scholar]
- 51.Carraway K.L., III, Weber J.L., Unger M.J., Ledesma J., Yu N., Gassmann M., Lai C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature. 1997;387:512–516. doi: 10.1038/387512a0. [DOI] [PubMed] [Google Scholar]
- 52.Chang H., Riese D.J., Gilbert W., Stern D.F., McMahan U.J. Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature. 1997;387:509–512. doi: 10.1038/387509a0. [DOI] [PubMed] [Google Scholar]
- 53.Zhang D., Sliwkowski M.X., Mark M., Frantz G., Akita R., Sun Y., Hillan K., Crowley C., Brush J., Godowski P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA. 1997;94:9562–9567. doi: 10.1073/pnas.94.18.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klapper L.N., Glathe S., Vaisman N., Hynes N.E., Andrews G.C., Sela M., Yarden Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA. 1999;96:4995–5000. doi: 10.1073/pnas.96.9.4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garrett T.P., McKern N.M., Lou M., Elleman T.C., Adams T.E., Lovrecz G.O., Kofler M., Jorissen R.N., Nice E.C., Burgess A.W., et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell. 2003;11:495–505. doi: 10.1016/S1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 56.Earp H.S., Dawson T.L., Li X., Yu H. Heterodimerization and functional interaction between EGF receptor family members: a new signaling paradigm with implications for breast cancer research. Breast Cancer Res. Treat. 1995;35:115–132. doi: 10.1007/BF00694752. [DOI] [PubMed] [Google Scholar]
- 57.Riese D.J., Stern D.F. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 58.Gamett D.C., Pearson G., Cerione R.A., Friedberg I. Secondary dimerization between members of the epidermal growth factor receptor family. J. Biol. Chem. 1997;272:12052–12056. doi: 10.1074/jbc.272.18.12052. [DOI] [PubMed] [Google Scholar]
- 59.Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 60.Tzahar E., Waterman H., Chen X., Levkowitz G., Karunagaran D., Lavi S., Ratzkin B.J., Yarden Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol. 1996;16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graus-Porta D., Beerli R.R., Daly J.M., Hynes N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z., Zhang L., Yeung T.K., Chen X. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell. 1999;10:1621–1636. doi: 10.1091/mbc.10.5.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holbro T., Beerli R.R., Maurer F., Koziczak M., Barbas C.F., Hynes N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fedi P., Pierce J.H., di Fiore P.P., Kraus M.H. Efficient coupling with phosphatidylinositol 3-kinase, but not phospholipase C gamma or GTPase-activating protein, distinguishes ErbB-3 signaling from that of other ErbB/EGFR family members. Mol. Cell Biol. 1994;14:492–500. doi: 10.1128/mcb.14.1.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonner J.A., Harari P.M., Giralt J., Azarnia N., Shin D.M., Cohen R.B., Jones C.U., Sur R., Raben D., Jassem J., et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 66.Watanabe K., Tachibana O., Sata K., Yonekawa Y., Kleihues P., Ohgaki H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996;6:217–223. doi: 10.1111/j.1750-3639.1996.tb00848.x. [DOI] [PubMed] [Google Scholar]
- 67.Kuan C.T., Wikstrand C.J., Bigner D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr. Relat Cancer. 2001;8:83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- 68.Aldape K.D., Ballman K., Furth A., Buckner J.C., Giannini C., Burger P.C., Scheithauer B.W., Jenkins R.B., James C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004;63:700–707. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 69.Gan H.K., Kaye A.H., Luwor R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Moscatello D.K., Holgado-Madruga M., Godwin A.K., Ramirez G., Gunn G., Zoltick P.W., Biegel J.A., Hayes R.L., Wong A.J. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55:5536–5539. [PubMed] [Google Scholar]
- 71.Sugawa N., Ekstrand A.J., James C.D., Collins V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ekstrand A.J., Longo N., Hamid M.L., Olson J.J., Liu L., Collins V.P., James C.D. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9:2313–2320. [PubMed] [Google Scholar]
- 73.Nishikawa R., Ji X.D., Harmon R.C., Lazar C.S., Gill G.N., Cavenee W.K., Huang H.J. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malden L.T., Novak U., Kaye A.H., Burgess A.W. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res. 1988;48:2711–2714. [PubMed] [Google Scholar]
- 75.Panneerselvam K., Kanakaraj P., Raj S., Das M., Bishayee S. Characterization of a novel epidermal-growth-factor-receptor-related 200-kDa tyrosine kinase in tumor cells. Eur. J. Biochem. 1995;230:951–957. doi: 10.1111/j.1432-1033.1995.tb20641.x. [DOI] [PubMed] [Google Scholar]
- 76.Steck P.A., Lee P., Hung M.C., Yung W.K. Expression of an altered epidermal growth factor receptor by human glioblastoma cells. Cancer Res. 1988;48:5433–5439. [PubMed] [Google Scholar]
- 77.Da Cunha S.G., Shepherd F.A., Tsao M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011;6:49–69. doi: 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- 78.Seroogy K.B., Gall C.M., Lee D.C., Kornblum H.I. Proliferative zones of postnatal rat brain express epidermal growth factor receptor mRNA. Brain Res. 1995;670:157–164. doi: 10.1016/0006-8993(94)01300-7. [DOI] [PubMed] [Google Scholar]
- 79.Mimeault M., Batra S.K. Complex oncogenic signaling networks regulate brain tumor-initiating cells and their progenies: pivotal roles of wild-type EGFR, EGFRvIII mutant and hedgehog cascades and novel multitargeted therapies. Brain Pathol. 2011;21:479–500. doi: 10.1111/j.1750-3639.2011.00505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang H.S., Nagane M., Klingbeil C.K., Lin H., Nishikawa R., Ji X.D., Huang C.M., Gill G.N., Wiley H.S., Cavenee W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 81.Grandal M.V., Zandi R., Pedersen M.W., Willumsen B.M., van D.B., Poulsen H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis. 2007;28:1408–1417. doi: 10.1093/carcin/bgm058. [DOI] [PubMed] [Google Scholar]
- 82.Ciesielski M.J., Fenstermaker R.A. Oncogenic epidermal growth factor receptor mutants with tandem duplication: gene structure and effects on receptor function. Oncogene. 2000;19:810–820. doi: 10.1038/sj.onc.1203409. [DOI] [PubMed] [Google Scholar]
- 83.Lamszus K., Laterra J., Westphal M., Rosen E.M. Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. Int. J. Dev. Neurosci. 1999;17:517–530. doi: 10.1016/S0736-5748(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 84.Gentile A., Trusolino L., Comoglio P.M. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27:85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- 85.Trusolino L., Bertotti A., Comoglio P.M. MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 86.Koochekpour S., Jeffers M., Rulong S., Taylor G., Klineberg E., Hudson E.A., Resau J.H., Vande Woude G.F. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997;57:5391–5398. [PubMed] [Google Scholar]
- 87.Velpula K.K., Dasari V.R., Asuthkar S., Gorantla B., Tsung A.J. EGFR and c-Met Cross Talk in Glioblastoma and Its Regulation by Human Cord Blood Stem Cells. Transl. Oncol. 2012;5:379–392. doi: 10.1593/tlo.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li L., Puliyappadamba V.T., Chakraborty S., Rehman A., Vemireddy V., Saha D., Souza R.F., Hatanpaa K.J., Koduru P., Burma S., et al. EGFR wild type antagonizes EGFRvIII-mediated activation of Met in glioblastoma. Oncogene. 2013 doi: 10.1038/onc.2013.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De B.F., Casanova E., Medico E., Pellegatta S., Orzan F., Albano R., Luraghi P., Reato G., D'Ambrosio A., Porrati P., et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Res. 2012;72:4537–4550. doi: 10.1158/0008-5472.CAN-11-3490. [DOI] [PubMed] [Google Scholar]
- 90.Frattini V., Trifonov V., Chan J.M., Castano A., Lia M., Abate F., Keir S.T., Ji A.X., Zoppoli P., Niola F., et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin S.Y., Makino K., Xia W., Matin A., Wen Y., Kwong K.Y., Bourguignon L., Hung M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 92.Lo H.W., Hsu S.C., Ali-Seyed M., Gunduz M., Xia W., Wei Y., Bartholomeusz G., Shih J.Y., Hung M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 93.Dittmann K., Mayer C., Kehlbach R., Rodemann H.P. The radioprotector Bowman-Birk proteinase inhibitor stimulates DNA repair via epidermal growth factor receptor phosphorylation and nuclear transport. Radiother. Oncol. 2008;86:375–382. doi: 10.1016/j.radonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 94.Dittmann K., Mayer C., Kehlbach R., Rodemann H.P. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol. Cancer. 2008;7 doi: 10.1186/1476-4598-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dittmann K.H., Mayer C., Ohneseit P.A., Raju U., Andratschke N.H., Milas L., Rodemann H.P. Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA-repair: a COX-2 independent mechanism. Int. J. Radiat. Oncol. Biol. Phys. 2008;70:203–212. doi: 10.1016/j.ijrobp.2007.08.065. [DOI] [PubMed] [Google Scholar]
- 96.Li C., Iida M., Dunn E.F., Ghia A.J., Wheeler D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28:3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dasari V.R., Velpula K.K., Alapati K., Gujrati M., Tsung A.J. Cord blood stem cells inhibit epidermal growth factor receptor translocation to mitochondria in glioblastoma. PLoS. One. 2012;7:e31884. doi: 10.1371/journal.pone.0031884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Gomes D.A., Rodrigues M.A., Leite M.F., Gomez M.V., Varnai P., Balla T., Bennett A.M., Nathanson M.H. c-Met must translocate to the nucleus to initiate calcium signals. J. Biol. Chem. 2008;283:4344–4351. doi: 10.1074/jbc.M706550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heldin C.H., Westermark B. Platelet-derived growth factor: Mechanism of action and possible in vivo function. Cell Regul. 1990;1:555–566. doi: 10.1091/mbc.1.8.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Matsui T., Heidaran M., Miki T., Popescu N., La Rochelle W., Kraus M., Pierce J., Aaronson S. Isolation of a novel receptor cDNA establishes the existence of two PDGF receptor genes. Science. 1989;243:800–804. doi: 10.1126/science.2536956. [DOI] [PubMed] [Google Scholar]
- 101.Ferns G.A., Sprugel K.H., Seifert R.A., Bowen-Pope D.F., Kelly J.D., Murray M., Raines E.W., Ross R. Relative platelet-derived growth factor receptor subunit expression determines cell migration to different dimeric forms of PDGF. Growth Factors. 1990;3:315–324. doi: 10.3109/08977199009003674. [DOI] [PubMed] [Google Scholar]
- 102.Betsholtz C., Karlsson L., Lindahl P. Developmental roles of platelet-derived growth factors. Bioessays. 2001;23:494–507. doi: 10.1002/bies.1069. [DOI] [PubMed] [Google Scholar]
- 103.Betsholtz C., Lindblom P., Bjarnegard M., Enge M., Gerhardt H., Lindahl P. Role of platelet-derived growth factor in mesangium development and vasculopathies: lessons from platelet-derived growth factor and platelet-derived growth factor receptor mutations in mice. Curr. Opin. Nephrol. Hypertens. 2004;13:45–52. doi: 10.1097/00041552-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 104.Andrae J., Gallini R., Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Heidaran M.A., Pierce J.H., Jensen R.A., Matsui T., Aaronson S.A. Chimeric alpha- and beta-platelet-derived growth factor (PDGF) receptors define three immunoglobulin-like domains of the alpha-PDGF receptor that determine PDGF-AA binding specificity. J. Biol. Chem. 1990;265:18741–18744. [PubMed] [Google Scholar]
- 106.Kanakaraj P., Raj S., Khan S.A., Bishayee S. Ligand-induced interaction between alpha- and beta-type platelet-derived growth factor (PDGF) receptors: Role of receptor heterodimers in kinase activation. Biochemistry. 1991;30:1761–1767. doi: 10.1021/bi00221a005. [DOI] [PubMed] [Google Scholar]
- 107.Kelly J.D., Haldeman B.A., Grant F.J., Murray M.J., Seifert R.A., Bowen-Pope D.F., Cooper J.A., Kazlauskas A. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 1991;266:8987–8992. [PubMed] [Google Scholar]
- 108.Kumjian D.A., Wahl M.I., Rhee S.G., Daniel T.O. Platelet-derived growth factor (PDGF) binding promotes physical association of PDGF receptor with phospholipase C. Proc. Natl. Acad. Sci. USA. 1989;86:8232–8236. doi: 10.1073/pnas.86.21.8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tallquist M., Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 110.Escobedo J.A., Kaplan D.R., Kavanaugh W.M., Turck C.W., Williams L.T. A phosphatidylinositol-3 kinase binds to platelet-derived growth factor receptors through a specific receptor sequence containing phosphotyrosine. Mol. Cell Biol. 1991;11:1125–1132. doi: 10.1128/mcb.11.2.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ronnstrand L., Mori S., Arridsson A.K., Eriksson A., Wernstedt C., Hellman U., Claesson-Welsh L., Heldin C.H. Identification of two C-terminal autophosphorylation sites in the PDGF beta-receptor: involvement in the interaction with phospholipase C-gamma. EMBO J. 1992;11:3911–3919. doi: 10.1002/j.1460-2075.1992.tb05484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marra F., Pinzani M., DeFranco R., Laffi G., Gentilini P. Involvement of phosphatidylinositol 3-kinase in the activation of extracellular signal-regulated kinase by PDGF in hepatic stellate cells. FEBS Lett. 1995;376:141–145. doi: 10.1016/0014-5793(95)01261-0. [DOI] [PubMed] [Google Scholar]
- 113.Heidaran M.A., Beeler J.F., Yu J.C., Ishibashi T., LaRochelle W.J., Pierce J.H., Aaronson S.A. Differences in substrate specificities of alpha and beta platelet-derived growth factor (PDGF) receptors. Correlation with their ability to mediate PDGF transforming functions. J. Biol. Chem. 1993;268:9287–9295. [PubMed] [Google Scholar]
- 114.Diliberto P.A., Gordon G.W., Yu C.L., Earp H.S., Herman B. Platelet-derived growth factor (PDGF) alpha receptor activation modulates the calcium mobilizing activity of the PDGF beta receptor in Balb/c3T3 fibroblasts. J. Biol. Chem. 1992;267:11888–11897. [PubMed] [Google Scholar]
- 115.Klinghoffer R.A., Mueting-Nelsen P.F., Faerman A., Shani M., Soriano P. The two PDGF receptors maintain conserved signaling in vivo despite divergent embryological functions. Mol. Cell. 2001;7:343–354. doi: 10.1016/S1097-2765(01)00182-4. [DOI] [PubMed] [Google Scholar]
- 116.Antoniades H.N., Scher C.D., Stiles C.D. Purification of human platelet-derived growth factor. Proc. Natl. Acad. Sci. USA. 1979;76:1809–1813. doi: 10.1073/pnas.76.4.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li X., Ponten A., Aase K., Karlsson L., Abramsson A., Uutela M., Backstrom G., Hellstrom M., Bostrom H., Li H., et al. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat. Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- 118.Heldin C.H., Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 119.Eriksson A., Rorsman C., Ernlund A., Claesson-Welsh L., Heldin C.H. Ligand-induced homo- and hetero-dimerization of platelet-derived growth factor alpha- and beta-receptors in intact cells. Growth Factors. 1992;6:1–14. doi: 10.3109/08977199209008867. [DOI] [PubMed] [Google Scholar]
- 120.Dai C., Celestino J.C., Okada Y., Louis D.N., Fuller G.N., Holland E.C. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Uhrbom L., Hesselager G., Nister M., Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–5279. [PubMed] [Google Scholar]
- 122.Schiffer D., Annovazzi L., Caldera V., Mellai M. On the origin and growth of gliomas. Anticancer Res. 2010;30:1977–1998. [PubMed] [Google Scholar]
- 123.Jackson E.L., Garcia-Verdugo J.M., Gil-Perotin S., Roy M., Quinones-Hinojosa A., VandenBerg S., Alvarez-Buylla A. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–199. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 124.Siebzehnrubl F.A., Jeske I., Muller D., Buslei R., Coras R., Hahnen E., Huttner H.B., Corbeil D., Kaesbauer J., Appl T., et al. Spontaneous in vitro transformation of adult neural precursors into stem-like cancer cells. Brain Pathol. 2009;19:399–408. doi: 10.1111/j.1750-3639.2008.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Antoniades H.N., Galanopoulos T., Neville-Golden J., O'Hara C.J. Malignant epithelial cells in primary human lung carcinomas coexpress in vivo platelet-derived growth factor (PDGF) and PDGF receptor mRNAs and their protein products. Proc. Natl. Acad. Sci. USA. 1992;89:3942–3946. doi: 10.1073/pnas.89.9.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hermanson M., Funa K., Koopmann J., Maintz D., Waha A., Westermark B., Heldin C.H., Wiestler O.D., Louis D.N., von Deimling A., et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor alpha receptor expression in human malignant gliomas. Cancer Res. 1996;56:164–171. [PubMed] [Google Scholar]
- 127.Seymour L., Dajee D., Bezwoda W.R. Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res. Treat. 1993;26:247–252. doi: 10.1007/BF00665802. [DOI] [PubMed] [Google Scholar]
- 128.Paulsson J., Sjoblom T., Micke P., Ponten F., Landberg G., Heldin C.H., Bergh J., Brennan D.J., Jirstrom K., Ostman A. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am. J. Pathol. 2009;175:334–341. doi: 10.2353/ajpath.2009.081030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dolloff N.G., Shulby S.S., Nelson A.V., Stearns M.E., Johannes G.J., Thomas J.D., Meucci O., Fatatis A. Bone-metastatic potential of human prostate cancer cells correlates with Akt/PKB activation by alpha platelet-derived growth factor receptor. Oncogene. 2005;24:6848–6854. doi: 10.1038/sj.onc.1208815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kong D., Wang Z., Sarkar S.H., Li Y., Banerjee S., Saliganan A., Kim H.R., Cher M.L., Sarkar F.H. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells. 2008;26:1425–1435. doi: 10.1634/stemcells.2007-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nister M., Libermann T.A., Betsholtz C., Pettersson M., Claesson-Welsh L., Heldin C.H., Schlessinger J., Westermark B. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res. 1988;48:3910–3918. [PubMed] [Google Scholar]
- 132.Lokker N.A., Sullivan C.M., Hollenbach S.J., Israel M.A., Giese N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729–3735. [PubMed] [Google Scholar]
- 133.Jechlinger M., Sommer A., Moriggl R., Seither P., Kraut N., Capodiecci P., Donovan M., Cordon-Cardo C., Beug H., Grunert S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 2006;116:1561–1570. doi: 10.1172/JCI24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ho C.L., Hsu L.F., Phyliky R.L., Li C.Y. Autocrine expression of platelet-derived growth factor B in B cell chronic lymphocytic leukemia. Acta Haematol. 2005;114:133–140. doi: 10.1159/000087886. [DOI] [PubMed] [Google Scholar]
- 135.Furuhashi M., Sjoblom T., Abramsson A., Ellingsen J., Micke P., Li H., Bergsten-Folestad E., Eriksson U., Heuchel R., Betsholtz C., et al. Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Res. 2004;64:2725–2733. doi: 10.1158/0008-5472.CAN-03-1489. [DOI] [PubMed] [Google Scholar]
- 136.Clarke I.D., Dirks P.B. A human brain tumor-derived PDGFR-alpha deletion mutant is transforming. Oncogene. 2003;22:722–733. doi: 10.1038/sj.onc.1206160. [DOI] [PubMed] [Google Scholar]
- 137.Ozawa T., Brennan C.W., Wang L., Squatrito M., Sasayama T., Nakada M., Huse J.T., Pedraza A., Utsuki S., Yasui Y., et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010;24:2205–2218. doi: 10.1101/gad.1972310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Heinrich M.C., Corless C.L., Demetri G.D., Blanke C.D., von M.M., Joensuu H., McGreevey L.S., Chen C.J., van den Abbeele A.D., Druker B.J., et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 139.Kumabe T., Sohma Y., Kayama T., Yoshimoto T., Yamamoto T. Amplification of alpha-platelet-derived growth factor receptor gene lacking an exon coding for a portion of the extracellular region in a primary brain tumor of glial origin. Oncogene. 1992;7:627–633. [PubMed] [Google Scholar]
- 140.Cross N.C., Reiter A. Tyrosine kinase fusion genes in chronic myeloproliferative diseases. Leukemia. 2002;16:1207–1212. doi: 10.1038/sj.leu.2402556. [DOI] [PubMed] [Google Scholar]
- 141.Baxter E.J., Kulkarni S., Vizmanos J.L., Jaju R., Martinelli G., Testoni N., Hughes G., Salamanchuk Z., Calasanz M.J., Lahortiga I., et al. Novel translocations that disrupt the platelet-derived growth factor receptor beta (PDGFRB) gene in BCR-ABL-negative chronic myeloproliferative disorders. Br. J. Haematol. 2003;120:251–256. doi: 10.1046/j.1365-2141.2003.04051.x. [DOI] [PubMed] [Google Scholar]
- 142.Gotlib J., Cools J., Malone J.M., III, Schrier S.L., Gilliland D.G., Coutre S.E. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: Implications for diagnosis, classification, and management. Blood. 2004;103:2879–2891. doi: 10.1182/blood-2003-06-1824. [DOI] [PubMed] [Google Scholar]
- 143.Ullrich A., Gray A., Tam A.W., Yang-Feng T., Tsubokawa M., Collins C., Henzel W., Le Bon T., Kathuria S., Chen E. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986;5:2503–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baserga R., Hongo A., Rubini M., Prisco M., Valentinis B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim. Biophys. Acta. 1997;1332:F105–F126. doi: 10.1016/s0304-419x(97)00007-3. [DOI] [PubMed] [Google Scholar]
- 145.Liu J.P., Baker J., Perkins A.S., Robertson E.J., Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 146.De la Monte S.M., Tong M., Bowling N., Moskal P. si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: relevance to fetal alcohol spectrum disorder. Mol. Brain. 2011;4:13. doi: 10.1186/1756-6606-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hernandez-Sanchez C., Blakesley V., Kalebic T., Helman L., LeRoith D. The role of the tyrosine kinase domain of the insulin-like growth factor-I receptor in intracellular signaling, cellular proliferation, and tumorigenesis. J. Biol. Chem. 1995;270:29176–29181. doi: 10.1074/jbc.270.49.29176. [DOI] [PubMed] [Google Scholar]
- 148.Hubbard S.R. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004;5:464–471. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- 149.Favelyukis S., Till J.H., Hubbard S.R., Miller W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001;8:1058–1063. doi: 10.1038/nsb721. [DOI] [PubMed] [Google Scholar]
- 150.Kato H., Faria T.N., Stannard B., Roberts C.T., Jr., LeRoith D. Essential role of tyrosine residues 1131, 1135, and 1136 of the insulin-like growth factor-I (IGF-I) receptor in IGF-I action. Mol. Endocrinol. 1994;8:40–50. doi: 10.1210/mend.8.1.7512194. [DOI] [PubMed] [Google Scholar]
- 151.Furlanetto R.W. Receptor-mediated endocytosis and lysosomal processing of insulin-like growth factor I by mitogenically responsive cells. Endocrinology. 1988;122:2044–2053. doi: 10.1210/endo-122-5-2044. [DOI] [PubMed] [Google Scholar]
- 152.Lou M., Garrett T.P., McKern N.M., Hoyne P.A., Epa V.C., Bentley J.D., Lovrecz G.O., Cosgrove L.J., Frenkel M.J., Ward C.W. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. USA. 2006;103:12429–12434. doi: 10.1073/pnas.0605395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lawrence M.C., McKern N.M., Ward C.W. Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol. 2007;17:699–705. doi: 10.1016/j.sbi.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 154.Seino S., Bell G.I. Alternative splicing of human insulin receptor messenger RNA. Biochem. Biophys. Res. Commun. 1989;159:312–316. doi: 10.1016/0006-291X(89)92439-X. [DOI] [PubMed] [Google Scholar]
- 155.Belfiore A., Frasca F., Pandini G., Sciacca L., Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009;30:586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 156.Pavelic K., Bukovic D., Pavelic J. The role of insulin-like growth factor 2 and its receptors in human tumors. Mol. Med. 2002;8:771–780. [PMC free article] [PubMed] [Google Scholar]
- 157.Ludwig T., Munier-Lehmann H., Bauer U., Hollinshead M., Ovitt C., Lobel P., Hoflack B. Differential sorting of lysosomal enzymes in mannose 6-phosphate receptor-deficient fibroblasts. EMBO J. 1994;13:3430–3437. doi: 10.1002/j.1460-2075.1994.tb06648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pandini G., Frasca F., Mineo R., Sciacca L., Vigneri R., Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002;277:39684–39695. doi: 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- 159.Benyoucef S., Surinya K.H., Hadaschik D., Siddle K. Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007;403:603–613. doi: 10.1042/BJ20061709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Moxham C.P., Duronio V., Jacobs S. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J. Biol. Chem. 1989;264:13238–13244. [PubMed] [Google Scholar]
- 161.Soos M.A., Siddle K. Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem. J. 1989;263:553–563. doi: 10.1042/bj2630553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Treadway J.L., Morrison B.D., Goldfine I.D., Pessin J.E. Assembly of insulin/insulin-like growth factor-1 hybrid receptors in vitro. J. Biol. Chem. 1989;264:21450–21453. [PubMed] [Google Scholar]
- 163.Baxter R.C. Insulin-like growth factor (IGF)-binding proteins: Interactions with IGFs and intrinsic bioactivities. Am. J. Physiol Endocrinol. Metab. 2000;278:E967–E976. doi: 10.1152/ajpendo.2000.278.6.E967. [DOI] [PubMed] [Google Scholar]
- 164.Werner H., Shalita-Chesner M., Abramovitch S., Idelman G., Shaharabani-Gargir L., Glaser T. Regulation of the insulin-like growth factor-I receptor gene by oncogenes and antioncogenes: implications in human cancer. Mol. Genet. Metab. 2000;71:315–320. doi: 10.1006/mgme.2000.3044. [DOI] [PubMed] [Google Scholar]
- 165.Sell C., Rubini M., Rubin R., Liu J.P., Efstratiadis A., Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc. Natl. Acad. Sci. USA. 1993;90:11217–11221. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sell C., Dumenil G., Deveaud C., Miura M., Coppola D., DeAngelis T., Rubin R., Efstratiadis A., Baserga R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell Biol. 1994;14:3604–3612. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lopez T., Hanahan D. Elevated levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2002;1:339–353. doi: 10.1016/S1535-6108(02)00055-7. [DOI] [PubMed] [Google Scholar]
- 168.Carboni J.M., Lee A.V., Hadsell D.L., Rowley B.R., Lee F.Y., Bol D.K., Camuso A.E., Gottardis M., Greer A.F., Ho C.P., et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005;65:3781–3787. doi: 10.1158/0008-5472.CAN-04-4602. [DOI] [PubMed] [Google Scholar]
- 169.Linnerth N.M., Siwicky M.D., Campbell C.I., Watson K.L., Petrik J.J., Whitsett J.A., Moorehead R.A. Type I insulin-like growth factor receptor induces pulmonary tumorigenesis. Neoplasia. 2009;11:672–682. doi: 10.1593/neo.09310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.All-Ericsson C., Girnita L., Seregard S., Bartolazzi A., Jager M.J., Larsson O. Insulin-like growth factor-1 receptor in uveal melanoma: A predictor for metastatic disease and a potential therapeutic target. Invest Ophthalmol. Vis. Sci. 2002;43:1–8. [PubMed] [Google Scholar]
- 171.Hellawell G.O., Turner G.D., Davies D.R., Poulsom R., Brewster S.F., Macaulay V.M. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 2002;62:2942–2950. [PubMed] [Google Scholar]
- 172.Weber M.M., Fottner C., Liu S.B., Jung M.C., Engelhardt D., Baretton G.B. Overexpression of the insulin-like growth factor I receptor in human colon carcinomas. Cancer. 2002;95:2086–2095. doi: 10.1002/cncr.10945. [DOI] [PubMed] [Google Scholar]
- 173.Bergmann U., Funatomi H., Yokoyama M., Beger H.G., Korc M. Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 1995;55:2007–2011. [PubMed] [Google Scholar]
- 174.Sichani M.M., Yazdi F.S., Moghaddam N.A., Chehrei A., Kabiri M., Naeimi A., Taheri D. Prognostic value of insulin- like growth factor-I receptor expression in renal cell carcinoma. Saudi. J. Kidney Dis. Transpl. 2010;21:69–74. [PubMed] [Google Scholar]
- 175.Resnicoff M., Sell C., Rubini M., Coppola D., Ambrose D., Baserga R., Rubin R. Rat glioblastoma cells expressing an antisense RNA to the insulin-like growth factor-1 (IGF-1) receptor are nontumorigenic and induce regression of wild-type tumors. Cancer Res. 1994;54:2218–2222. [PubMed] [Google Scholar]
- 176.Resnicoff M., Li W., Basak S., Herlyn D., Baserga R., Rubin R. Inhibition of rat C6 glioblastoma tumor growth by expression of insulin-like growth factor I receptor antisense mRNA. Cancer Immunol. Immunother. 1996;42:64–68. doi: 10.1007/s002620050252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rininsland F., Johnson T.R., Chernicky C.L., Schulze E., Burfeind P., Ilan J. Suppression of insulin-like growth factor type I receptor by a triple-helix strategy inhibits IGF-I transcription and tumorigenic potential of rat C6 glioblastoma cells. Proc. Natl. Acad. Sci. USA. 1997;94:5854–5859. doi: 10.1073/pnas.94.11.5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Resnicoff M., Tjuvajev J., Rotman H.L., Abraham D., Curtis M., Aiken R., Baserga R. Regression of C6 rat brain tumors by cells expressing an antisense insulin-like growth factor I receptor RNA. J. Exp. Ther. Oncol. 1996;1:385–389. [PubMed] [Google Scholar]
- 179.Berns E.M., Klijn J.G., van Staveren I.L., Portengen H., Foekens J.A. Sporadic amplification of the insulin-like growth factor 1 receptor gene in human breast tumors. Cancer Res. 1992;52:1036–1039. [PubMed] [Google Scholar]
- 180.Sekyi-Otu A., Bell R.S., Ohashi C., Pollak M., Andrulis I.L. Insulin-like growth factor 1 (IGF-1) receptors, IGF-1, and IGF-2 are expressed in primary human sarcomas. Cancer Res. 1995;55:129–134. [PubMed] [Google Scholar]
- 181.DiGiovanni J., Bol D.K., Wilker E., Beltran L., Carbajal S., Moats S., Ramirez A., Jorcano J., Kiguchi K. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000;60:1561–1570. [PubMed] [Google Scholar]
- 182.DiGiovanni J., Kiguchi K., Frijhoff A., Wilker E., Bol D.K., Beltran L., Moats S., Ramirez A., Jorcano J., Conti C. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc. Natl. Acad. Sci. USA. 2000;97:3455–3460. doi: 10.1073/pnas.97.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kiess W., Lee L., Graham D.E., Greenstein L., Tseng L.Y., Rechler M.M., Nissley S.P. Rat C6 glial cells synthesize insulin-like growth factor I (IGF-I) and express IGF-I receptors and IGF-II/mannose 6-phosphate receptors. Endocrinology. 1989;124:1727–1736. doi: 10.1210/endo-124-4-1727. [DOI] [PubMed] [Google Scholar]
- 184.Trojan J., Blossey B.K., Johnson T.R., Rudin S.D., Tykocinski M., Ilan J., Ilan J. Loss of tumorigenicity of rat glioblastoma directed by episome-based antisense cDNA transcription of insulin-like growth factor I. Proc. Natl. Acad. Sci. USA. 1992;89:4874–4878. doi: 10.1073/pnas.89.11.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Schlenska-Lange A., Knupfer H., Lange T.J., Kiess W., Knupfer M. Cell proliferation and migration in glioblastoma multiforme cell lines are influenced by insulin-like growth factor I in vitro. Anticancer Res. 2008;28:1055–1060. [PubMed] [Google Scholar]
- 186.Yu H., Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 187.Rosenzweig S.A., Atreya H.S. Defining the pathway to insulin-like growth factor system targeting in cancer. Biochem. Pharmacol. 2010;80:1115–1124. doi: 10.1016/j.bcp.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Goya M., Miyamoto S., Nagai K., Ohki Y., Nakamura K., Shitara K., Maeda H., Sangai T., Kodama K., Endoh Y., et al. Growth inhibition of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice by a ligand-specific antibody to human insulin-like growth factors. Cancer Res. 2004;64:6252–6258. doi: 10.1158/0008-5472.CAN-04-0919. [DOI] [PubMed] [Google Scholar]
- 189.Kimura T., Kuwata T., Ashimine S., Yamazaki M., Yamauchi C., Nagai K., Ikehara A., Feng Y., Dimitrov D.S., Saito S., et al. Targeting of bone-derived insulin-like growth factor-II by a human neutralizing antibody suppresses the growth of prostate cancer cells in a human bone environment. Clin. Cancer Res. 2010;16:121–129. doi: 10.1158/1078-0432.CCR-09-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gucev Z.S., Oh Y., Kelley K.M., Rosenfeld R.G. Insulin-like growth factor binding protein 3 mediates retinoic acid- and transforming growth factor beta2-induced growth inhibition in human breast cancer cells. Cancer Res. 1996;56:1545–1550. [PubMed] [Google Scholar]
- 191.Higo H., Duan C., Clemmons D.R., Herman B. Retinoic acid inhibits cell growth in HPV negative cervical carcinoma cells by induction of insulin-like growth factor binding protein-5 (IGFBP-5) secretion. Biochem. Biophys. Res. Commun. 1997;239:706–709. doi: 10.1006/bbrc.1997.7499. [DOI] [PubMed] [Google Scholar]
- 192.Aleksic T., Chitnis M.M., Perestenko O.V., Gao S., Thomas P.H., Turner G.D., Protheroe A.S., Howarth M., Macaulay V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010;70:6412–6419. doi: 10.1158/0008-5472.CAN-10-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sehat B., Tofigh A., Lin Y., Trocme E., Liljedahl U., Lagergren J., Larsson O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010;3:ra10. doi: 10.1126/scisignal.2000628. [DOI] [PubMed] [Google Scholar]
- 194.Schechter Y., Hernaez L., Schlessinger J., Cuatrecasas P. Local aggregation of hormone-receptor complexes is required for activation by epidermal growth factor. Nature. 1979;278:835–838. doi: 10.1038/278835a0. [DOI] [PubMed] [Google Scholar]
- 195.Carraway K.L., III, Koland J.G., Cerione R.A. Visualization of epidermal growth factor (EGF) receptor aggregation in plasma membranes by fluorescence resonance energy transfer. Correlation of receptor activation with aggregation. J. Biol. Chem. 1989;264:8699–8707. [PubMed] [Google Scholar]
- 196.Gadella T.W., Jr., Jovin T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995;129:1543–1558. doi: 10.1083/jcb.129.6.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mendelsohn J., Baselga J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006;33:369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 198.Van den Eynde M., Baurain J.F., Mazzeo F., Machiels J.P. Epidermal growth factor receptor targeted therapies for solid tumours. Acta Clin. Belg. 2011;66:10–17. doi: 10.1179/ACB.66.1.2062508. [DOI] [PubMed] [Google Scholar]
- 199.Krex D., Klink B., Hartmann C., von D.A., Pietsch T., Simon M., Sabel M., Steinbach J.P., Heese O., Reifenberger G., et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130:2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- 200.Shinojima N., Tada K., Shiraishi S., Kamiryo T., Kochi M., Nakamura H., Makino K., Saya H., Hirano H., Kuratsu J., et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- 201.Quan A.L., Barnett G.H., Lee S.Y., Vogelbaum M.A., Toms S.A., Staugaitis S.M., Prayson R.A., Peereboom D.M., Stevens G.H., Cohen B.H., et al. Epidermal growth factor receptor amplification does not have prognostic significance in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2005;63:695–703. doi: 10.1016/j.ijrobp.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 202.Hegi M.E., Diserens A.C., Godard S., Dietrich P.Y., Regli L., Ostermann S., Otten P., Van M.G., de T.N., Stupp R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.CCR-03-0384. [DOI] [PubMed] [Google Scholar]
- 203.Halatsch M.E., Schmidt U., Behnke-Mursch J., Unterberg A., Wirtz C.R. Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat. Rev. 2006;32:74–89. doi: 10.1016/j.ctrv.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 204.Haas-Kogan D.A., Prados M.D., Tihan T., Eberhard D.A., Jelluma N., Arvold N.D., Baumber R., Lamborn K.R., Kapadia A., Malec M., et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J. Natl. Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 205.Carrasco-Garcia E., Saceda M., Grasso S., Rocamora-Reverte L., Conde M., Gomez-Martinez A., Garcia-Morales P., Ferragut J.A., Martinez-Lacaci I. Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp. Cell Res. 2011;317:1476–1489. doi: 10.1016/j.yexcr.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 206.ClinicalTrials.gov. A service of the U.S. National Institutes of Health. [(accessed on 12 March 2014)]; Available online: http://www.clinicaltrials.gov/ct2/results?term=EGFR%2C+glioblastoma&Search=Search.
- 207.Thiessen B., Stewart C., Tsao M., Kamel-Reid S., Schaiquevich P., Mason W., Easaw J., Belanger K., Forsyth P., McIntosh L., et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010;65:353–361. doi: 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- 208.Guo D., Prins R.M., Dang J., Kuga D., Iwanami A., Soto H., Lin K.Y., Huang T.T., Akhavan D., Hock M.B., et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Learn C.A., Hartzell T.L., Wikstrand C.J., Archer G.E., Rich J.N., Friedman A.H., Friedman H.S., Bigner D.D., Sampson J.H. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin. Cancer Res. 2004;10:3216–3224. doi: 10.1158/1078-0432.CCR-03-0521. [DOI] [PubMed] [Google Scholar]
- 210.Voelzke W.R., Petty W.J., Lesser G.J. Targeting the epidermal growth factor receptor in high-grade astrocytomas. Curr. Treat. Options. Oncol. 2008;9:23–31. doi: 10.1007/s11864-008-0053-5. [DOI] [PubMed] [Google Scholar]
- 211.Maher E.A., Furnari F.B., Bachoo R.M., Rowitch D.H., Louis D.N., Cavenee W.K., DePinho R.A. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 212.Hermanson M., Funa K., Hartman M., Claesson-Welsh L., Heldin C.H., Westermark B., Nister M. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–3219. [PubMed] [Google Scholar]
- 213.Gross D., Bernhardt G., Buschauer A. Platelet-derived growth factor receptor independent proliferation of human glioblastoma cells: Selective tyrosine kinase inhibitors lack antiproliferative activity. J. Cancer Res. Clin. Oncol. 2006;132:589–599. doi: 10.1007/s00432-006-0109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Vassbotn F.S., Ostman A., Langeland N., Holmsen H., Westermark B., Heldin C.H., Nister M. Activated platelet-derived growth factor autocrine pathway drives the transformed phenotype of a human glioblastoma cell line. J. Cell Physiol. 1994;158:381–389. doi: 10.1002/jcp.1041580221. [DOI] [PubMed] [Google Scholar]
- 215.Ranza E., Mazzini G., Facoetti A., Nano R. In-vitro effects of the tyrosine kinase inhibitor imatinib on glioblastoma cell proliferation. J. Neurooncol. 2010;96:349–357. doi: 10.1007/s11060-009-9975-4. [DOI] [PubMed] [Google Scholar]
- 216.Ren H., Tan X., Dong Y., Giese A., Chou T.C., Rainov N., Yang B. Differential effect of imatinib and synergism of combination treatment with chemotherapeutic agents in malignant glioma cells. Basic Clin. Pharmacol. Toxicol. 2009;104:241–252. doi: 10.1111/j.1742-7843.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 217.Neyns B., Sadones J., Chaskis C., Dujardin M., Everaert H., Lv S., Duerinck J., Tynninen O., Nupponen N., Michotte A., et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J. Neurooncol. 2011;103:491–501. doi: 10.1007/s11060-010-0402-7. [DOI] [PubMed] [Google Scholar]
- 218.ClinicalTrials.gov. A service of the U.S. National Institutes of Health. [(accessed on 12 March 2014)]; Available online: http://www.clinicaltrials.gov/ct2/results?term=PDGFR%2C+glioma&Search=Search.
- 219.Hewish M., Chau I., Cunningham D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent Pat. Anticancer Drug Discov. 2009;4:54–72. doi: 10.2174/157489209787002515. [DOI] [PubMed] [Google Scholar]
- 220.Casa A.J., Dearth R.K., Litzenburger B.C., Lee A.V., Cui X. The type I insulin-like growth factor receptor pathway: a key player in cancer therapeutic resistance. Front Biosci. 2008;13:3273–3287. doi: 10.2741/2925. [DOI] [PubMed] [Google Scholar]
- 221.Chitnis M.M., Yuen J.S., Protheroe A.S., Pollak M., Macaulay V.M. The type 1 insulin-like growth factor receptor pathway. Clin. Cancer Res. 2008;14:6364–6370. doi: 10.1158/1078-0432.CCR-07-4879. [DOI] [PubMed] [Google Scholar]
- 222.Chakravarti A., Loeffler J.S., Dyson N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002;62:200–207. [PubMed] [Google Scholar]
- 223.Giovannone B., Scaldaferri M.L., Federici M., Porzio O., Lauro D., Fusco A., Sbraccia P., Borboni P., Lauro R., Sesti G. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res. Rev. 2000;16:434–441. doi: 10.1002/1520-7560(2000)9999:9999<::AID-DMRR159>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 224.Mukohara T., Shimada H., Ogasawara N., Wanikawa R., Shimomura M., Nakatsura T., Ishii G., Park J.O., Janne P.A., Saijo N., et al. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Lett. 2009;282:14–24. doi: 10.1016/j.canlet.2009.02.056. [DOI] [PubMed] [Google Scholar]
- 225.Werner H., Bruchim I. The insulin-like growth factor-I receptor as an oncogene. Arch. Physiol. Biochem. 2009;115:58–71. doi: 10.1080/13813450902783106. [DOI] [PubMed] [Google Scholar]
- 226.Oh Y. IGF-independent regulation of breast cancer growth by IGF binding proteins. Breast Cancer Res. Treat. 1998;47:283–293. doi: 10.1023/A:1005911319432. [DOI] [PubMed] [Google Scholar]
- 227.Girnita A., Girnita L., del Prete F., Bartolazzi A., Larsson O., Axelson M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer Res. 2004;64:236–242. doi: 10.1158/0008-5472.CAN-03-2522. [DOI] [PubMed] [Google Scholar]
- 228.Stromberg T., Ekman S., Girnita L., Dimberg L.Y., Larsson O., Axelson M., Lennartsson J., Hellman U., Carlson K., Osterborg A., et al. IGF-1 receptor tyrosine kinase inhibition by the cyclolignan PPP induces G2/M-phase accumulation and apoptosis in multiple myeloma cells. Blood. 2006;107:669–678. doi: 10.1182/blood-2005-01-0306. [DOI] [PubMed] [Google Scholar]
- 229.Yin S., Girnita A., Stromberg T., Khan Z., Andersson S., Zheng H., Ericsson C., Axelson M., Nister M., Larsson O., et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro. Oncol. 2010;12:19–27. doi: 10.1093/neuonc/nop008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.ClinicalTrials.gov. A service of the U.S. National Institutes of Health. [(accessed on 12 March 2014)]; Available online: http://www.clinicaltrials.gov/ct2/results?term=IGF1R%2C+glioma&Search=Search.
- 231.Moller H.G., Rasmussen A.P., Andersen H.H., Johnsen K.B., Henriksen M., Duroux M. A systematic review of microRNA in glioblastoma multiforme: Micro-modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013;47:131–144. doi: 10.1007/s12035-012-8349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hernando E. microRNAs and cancer: Role in tumorigenesis, patient classification and therapy. Clin. Transl. Oncol. 2007;9:155–160. doi: 10.1007/s12094-007-0029-0. [DOI] [PubMed] [Google Scholar]
- 233.Karsy M., Arslan E., Moy F. Current Progress on Understanding MicroRNAs in Glioblastoma Multiforme. Genes Cancer. 2012;3:3–15. doi: 10.1177/1947601912448068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kefas B., Godlewski J., Comeau L., Li Y., Abounader R., Hawkinson M., Lee J., Fine H., Chiocca E.A., Lawler S., et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68:3566–3572. doi: 10.1158/0008-5472.CAN-07-6639. [DOI] [PubMed] [Google Scholar]
- 235.Papagiannakopoulos T., Friedmann-Morvinski D., Neveu P., Dugas J.C., Gill R.M., Huillard E., Liu C., Zong H., Rowitch D.H., Barres B.A., et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene. 2012;31:1884–1895. doi: 10.1038/onc.2011.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang Y., Chen X., Lian H., Liu J., Zhou B., Han S., Peng B., Yin J., Liu W., He X. MicroRNA-503 acts as a tumor suppressor in glioblastoma for multiple antitumor effects by targeting IGF-1R. Oncol. Rep. 2014;31:1445–1452. doi: 10.3892/or.2013.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Shao M., Rossi S., Chelladurai B., Shimizu M., Ntukogu O., Ivan M., Calin G.A., Matei D. PDGF induced microRNA alterations in cancer cells. Nucleic Acids Res. 2011;39:4035–4047. doi: 10.1093/nar/gkq1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Katakowski M., Zheng X., Jiang F., Rogers T., Szalad A., Chopp M. MiR-146b-5p suppresses EGFR expression and reduces in vitro migration and invasion of glioma. Cancer Invest. 2010;28:1024–1030. doi: 10.3109/07357907.2010.512596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhou X., Ren Y., Moore L., Mei M., You Y., Xu P., Wang B., Wang G., Jia Z., Pu P., et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab Invest. 2010;90:144–155. doi: 10.1038/labinvest.2009.126. [DOI] [PubMed] [Google Scholar]
- 240.Chan J.A., Krichevsky A.M., Kosik K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 241.Stupp R., Mason W.P., Van den Bent M.J., Weller M., Fisher B., Taphoorn M.J., Belanger K., Brandes A.A., Marosi C., Bogdahn U., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 242.Furnari F.B., Fenton T., Bachoo R.M., Mukasa A., Stommel J.M., Stegh A., Hahn W.C., Ligon K.L., Louis D.N., Brennan C., et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 243.Singh S.K., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 244.Jun H.J., Bronson R.T., Charest A. Inhibition of EGFR induces a c-MET-driven stem cell population in glioblastoma. Stem Cells. 2014;32:338–348. doi: 10.1002/stem.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Martinez-Lacaci I., Garcia M.P., Soto J.L., Saceda M. Tumour cells resistance in cancer therapy. Clin. Transl. Oncol. 2007;9:13–20. doi: 10.1007/s12094-007-0004-9. [DOI] [PubMed] [Google Scholar]
- 246.Gottesman M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 247.Gottesman M.M., Fojo T., Bates S.E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 248.Ambudkar S.V., Dey S., Hrycyna C.A., Ramachandra M., Pastan I., Gottesman M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 249.Tan B., Piwnica-Worms D., Ratner L. Multidrug resistance transporters and modulation. Curr. Opin. Oncol. 2000;12:450–458. doi: 10.1097/00001622-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 250.Johnstone R.W., Ruefli A.A., Smyth M.J. Multiple physiological functions for multidrug transporter P-glycoprotein? Trends Biochem. Sci. 2000;25:1–6. doi: 10.1016/S0968-0004(99)01493-0. [DOI] [PubMed] [Google Scholar]
- 251.Decleves X., Fajac A., Lehmann-Che J., Tardy M., Mercier C., Hurbain I., Laplanche J.L., Bernaudin J.F., Scherrmann J.M. Molecular and functional MDR1-Pgp and MRPs expression in human glioblastoma multiforme cell lines. Int. J. Cancer. 2002;98:173–180. doi: 10.1002/ijc.10135. [DOI] [PubMed] [Google Scholar]
- 252.Beaulieu E., Demeule M., Ghitescu L., Beliveau R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997;326:539–544. doi: 10.1042/bj3260539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Toth K., Vaughan M.M., Peress N.S., Slocum H.K., Rustum Y.M. MDR1 P-glycoprotein is expressed by endothelial cells of newly formed capillaries in human gliomas but is not expressed in the neovasculature of other primary tumors. Am. J. Pathol. 1996;149:853–858. [PMC free article] [PubMed] [Google Scholar]
- 254.Nakagawa T., Ido K., Sakuma T., Takeuchi H., Sato K., Kubota T. Prognostic significance of the immunohistochemical expression of O6-methylguanine-DNA methyltransferase, P-glycoprotein, and multidrug resistance protein-1 in glioblastomas. Neuropathology. 2009;29:379–388. doi: 10.1111/j.1440-1789.2008.00983.x. [DOI] [PubMed] [Google Scholar]
- 255.Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J.O., Lindeman N., Gale C.M., Zhao X., Christensen J., et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 256.Karamouzis M.V., Konstantinopoulos P.A., Papavassiliou A.G. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009;10:709–717. doi: 10.1016/S1470-2045(09)70137-8. [DOI] [PubMed] [Google Scholar]
- 257.Wheeler D.L., Huang S., Kruser T.J., Nechrebecki M.M., Armstrong E.A., Benavente S., Gondi V., Hsu K.T., Harari P.M. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Guix M., Faber A.C., Wang S.E., Olivares M.G., Song Y., Qu S., Rinehart C., Seidel B., Yee D., Arteaga C.L., et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest. 2008;118:2609–2619. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lu Y., Zi X., Zhao Y., Mascarenhas D., Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin) J. Natl. Cancer Inst. 2001;93:1852–1857. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 260.Rexer B.N., Engelman J.A., Arteaga C.L. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle. 2009;8:18–22. doi: 10.4161/cc.8.1.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Gorre M.E., Mohammed M., Ellwood K., Hsu N., Paquette R., Rao P.N., Sawyers C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 262.Antonescu C.R., Besmer P., Guo T., Arkun K., Hom G., Koryotowski B., Leversha M.A., Jeffrey P.D., Desantis D., Singer S., et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 2005;11:4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 263.Ogino A., Kitao H., Hirano S., Uchida A., Ishiai M., Kozuki T., Takigawa N., Takata M., Kiura K., Tanimoto M. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res. 2007;67:7807–7814. doi: 10.1158/0008-5472.CAN-07-0681. [DOI] [PubMed] [Google Scholar]
- 264.Daub H., Specht K., Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat. Rev. Drug Discov. 2004;3:1001–1010. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- 265.Morgillo F., Lee H.Y. Resistance to epidermal growth factor receptor-targeted therapy. Drug Resist. Updat. 2005;8:298–310. doi: 10.1016/j.drup.2005.08.004. [DOI] [PubMed] [Google Scholar]