

Abstract
An essential link between the kidney and blood pressure control has long been known. Here, we review evidence supporting the premise that an impaired capacity of the kidney to excrete sodium in response to elevated blood pressure is a major contributor to hypertension, irrespective of the initiating cause. In this regard, recent work suggests that novel pathways controlling key sodium transporters in kidney epithelia have a critical impact on hypertension pathogenesis, supporting a model in which impaired renal sodium excretion is a final common pathway through which vascular, neural, and inflammatory responses raise blood pressure. We also address recent findings calling into question long-standing notions regarding the relationship between sodium intake and changes in body fluid volume. Expanded understanding of the role of the kidney as both a cause and target of hypertension highlights key aspects of pathophysiology and may lead to identification of new strategies for prevention and treatment.
Introduction
Hypertension is one of the most common chronic diseases of humankind, affecting more than 1 billion people worldwide (1). Although elevated blood pressure per se does not typically cause overt symptoms, the consequences of chronic hypertension, including cardiac hypertrophy, heart failure, stroke, and kidney disease, are responsible for substantial morbidity and mortality (2). Treatments that effectively reduce blood pressure can prevent these complications (2–4). However, in a recent analysis of data from the National Health and Nutrition Examination Survey (NHANES) covering the period from 2009 to 2010, blood pressures were reduced to target levels in less than 50% of patients receiving hypertension treatment, and this rate was under 40% in individuals who also had chronic kidney disease (CKD) (5). The reasons for these poor outcomes are complex and include health services issues around processes of care, compliance, and patient education. Moreover, the precise cause of hypertension is not apparent in the vast majority of patients with hypertension. Limitations in our understanding of hypertension pathogenesis in individual patients are an obstacle to applying individualized approaches for prevention and treatment and to identifying new, specific therapies.
A connection between the kidney and blood pressure control
The idea that the kidney plays a role in hypertension dates back almost 200 years. In the 19th century, Richard Bright proposed that abnormalities in urine production by the kidney altered blood in such a way as to increase vascular resistance, leading to high blood pressure and increased cardiac mass (6). A century later, Harry Goldblatt induced malignant hypertension in dogs by obstructing one of the renal arteries (7). In the 1970’s, Arthur Guyton and colleagues advanced a mature hypothesis suggesting that the kidney governs the level of blood pressure by regulating extracellular fluid volume. They argued that balance is normally achieved by matching urinary excretion of salt and water with dietary intake, thereby maintaining a constant extracellular fluid volume and blood pressure (8). In this construct, when blood pressure increases from any cause, renal perfusion pressure increases with a consequent enhancement of sodium and water excretion, which Guyton called pressure-natriuresis. Based on the substantial capacity for the kidney to excrete sodium, this blood pressure-tempering mechanism should have sufficient gain to limit intravascular volume and thereby lower blood pressure in response to a range of stimuli from increased heart rate to elevated peripheral vascular resistance (9, 10). Furthermore, the hypothesis predicts that a permissive modification of the pressure-natriuresis response is required to perpetuate a chronic elevation in intra-arterial pressure, whereby the equilibrium point for salt and water excretion is shifted to a higher level of arterial blood pressure (8). While this hypothesis has been largely embraced by the nephrology community, it remains controversial in some circles based on work suggesting independent control of blood pressure by neural and vascular pathways, for example (11–13). In addition, as discussed below, classic assumptions about direct associations between sodium retention and expansion of extracellular fluid volume have recently been questioned.
Hypertension follows the kidney
Over the years, a series of kidney cross-transplantation studies have supported a key role for intrinsic functions of the kidney in the pathogenesis of hypertension (14–17). Generally, these studies have been performed using genetically compatible donor and recipient strains to circumvent rejection, with both native kidneys removed such that the full extent of excretory function is provided by the transplanted kidney. For example, transplantation of a kidney from a Dahl hypertensive rat into a normotensive, salt-resistant recipient causes hypertension in the recipient regardless of whether the transplant is performed before or after a high salt diet is introduced (14, 15). By contrast, reciprocal transplantation of a kidney from a normotensive, salt-resistant animal into a Dahl salt-sensitive rat abrogates hypertension (14, 15). Likewise, studies in spontaneously hypertensive rats and Milan hypertensive rats recapitulated these findings (16, 17). The same principle seems to also hold true in humans where resistant hypertension can be alleviated after successful kidney transplantation (18). Collectively, these studies indicate that a defect in sodium excretion by the kidney confers susceptibility to elevated blood pressure.
Intrarenal actions of the renin-angiotensin system in blood pressure control
The renin-angiotensin system (RAS) is a powerful modulator of blood pressure, and dysregulation of the RAS causes hypertension. Pharmacological blockade of the RAS with renin inhibitors, angiotensin-converting enzyme (ACE) inhibitors, or angiotensin receptor blockers effectively lowers blood pressure in a substantial proportion of patients with hypertension (19), reflecting the important role for RAS activation as a cause of human hypertension. Similarly, in rodent models, deletion of RAS genes lowers blood pressure whereas overexpression causes hypertension (20).
A role for the actions of angiotensin II in the kidney to influence blood pressure is well established. For example, studies by John Hall and associates in the 1980s showed that chronic infusion of low doses of angiotensin II directly into the kidney caused hypertension with impaired natriuresis due to a shift of the pressure-natriuresis relationship (21). In addition, Navar and associates have posited the existence of local and independent control of RAS activity within the kidney influencing sodium excretion and blood pressure regulation (ref. 22 and Figure 1). In this hypothetical construct, elevated circulating levels of angiotensin II are associated with accumulation of angiotensin peptides in the kidney, upregulated expression of angiotensinogen, the primary RAS substrate, in proximal tubule epithelium, and increased excretion of angiotensinogen and angiotensin peptides in urine (23). In this feed-forward pathway, angiotensin II acting via type 1 angiotensin (AT1) receptors in the kidney induces local activation of the RAS inside the kidney and increases generation of angiotensin II in the lumen of renal tubules, resulting in autocrine and paracrine stimulation of epithelial transporters (24, 25). Consistent with this idea, recent studies have verified the critical requirement of ACE within the kidney to fully manifest stimulation of sodium transporter expression, renal sodium reabsorption, and hypertension in the setting of RAS activation (26, 27).
Figure 1. A model for local control of RAS activity within the kidney.

High levels of angiotensin II (ANGII) in circulation, derived from angiotensinogen (AGT) generated primarily by the liver, are associated with (i) increased ANGII in the kidney, (ii) upregulation of AGT in the proximal tubule epithelium, (iii) increased levels of AGT in the tubular lumen, (iv) generation of ANGII requiring angiotensin-converting enzyme (ACE) expression in the brush border of the proximal tubule (PT), and (v) increased excretion of AGT and ANG peptides in urine. Within the kidney circulation and the tubule lumen, ANGII binds to AT1 receptors (AT1Rs) stimulating AGT transcription in renal epithelial cells and influencing the synthesis and activity of epithelial solute transporters with critical actions to influence body fluid volume and blood pressure. These transporters include NHE3, the major luminal sodium transporter in PT epithelia, along with paracrine stimulation of downstream epithelial transporters such as ENaC and PENDRIN in the collecting duct. In the collecting duct, control of sodium transport involves complex interactions between ANGII, acting via AT1Rs, and aldosterone (ALDO), acting via the mineralocorticoid receptor (MR). In principal cells, ANGII and ALDO both stimulate the abundance and activity of ENaC, but direct effects of ANGII seem to predominate. Rac1 signaling may also stimulate ENaC, independent of ALDO. In intercalated cells, ANGII influences responsiveness of the MR to ALDO by regulating dephosphorylation of the MR at S893, allowing ALDO to bind, leading to activation of PENDRIN and enhanced chloride and sodium reabsorption.
Recent work by Matsusaka and Ichikawa using mice with cell-specific deletion of angiotensinogen from the kidney or liver have called into question some features of this scheme (28). Their study indicated that most of the angiotensinogen and angiotensin II within the kidney arise from angiotensinogen produced by the liver. On the other hand, genetic elimination of angiotensinogen from the kidney significantly reduced excretion of angiotensinogen in urine, consistent with a major tenet of Navar’s theory that angiotensin II acting on the luminal surface of the nephron is the critical effector component of the intrarenal RAS, where it directly modulates sodium transporters along the nephron. Subsequent work by Peti-Peterdi using intravital microscopy is consistent with the idea that the major source of angiotensinogen in the urine is, indeed, the renal epithelium (29).
To assess the capacity of RAS activation in the kidney to affect blood pressure, we turned to the strategy of kidney cross transplantation discussed above. In this case, we carried out transplants between mice with genetic deletion of the AT1A angiotensin receptor and congenic wild-type controls (30). AT1 receptors mediate the classical actions of the RAS including vasoconstriction and stimulation of aldosterone release; the AT1A receptor is the major murine AT1 receptor representing the homologue to the human AT1 receptor. In our cross-transplantation studies, we found that expression of AT1 receptors in the kidney is both necessary and sufficient for induction of angiotensin II-dependent hypertension (31).
To further dissect the specific cellular targets of angiotensin II driving the development of hypertension, we subsequently generated mice with cell-specific deletion of AT1 receptors in key cell lineages in the kidney (32). These studies identified the population of AT1 receptors in the proximal tubular epithelium as critical mediators of angiotensin II-dependent hypertension. Specifically, conditional deletion of AT1A receptors from the proximal tubule provided robust protection from hypertension and facilitated natriuresis by reducing the accumulation of key epithelial sodium transporters (32). In addition, recent studies have suggested that direct actions of angiotensin II on AT1 receptors in the collecting duct can directly stimulate the activity of the epithelial sodium channel (ENaC) (33 and Figure 1). Moreover, relative to aldosterone, these direct effects of angiotensin II in the collecting duct have a predominant effect on ENaC abundance and activity in angiotensin II-dependent hypertension.
In aggregate, these studies indicate that the powerful RAS mediates its effects on blood pressure through direct effects on the kidney. Furthermore, there is compartmentalized control of the RAS along the nephron contributing to hypertension pathogenesis.
Novel control mechanisms and sites of action for aldosterone in hypertension
AT1 receptors in the zona glomerulosa of the adrenal gland stimulate aldosterone release, making aldosterone a downstream effector of the RAS. Classically, activation of the mineralocorticoid receptor (MR) in aldosterone-sensitive nephron segments stimulates assembly and translocation of the subunits of the ENaC. Mutations in ENaC subunits that impair its degradation result in enhanced membrane density and open probability of the channels, resulting in Liddle’s syndrome, characterized by severe, early onset hypertension resembling hyperaldosteronism, but with low levels of aldosterone (34, 35). Similarly, activating mutations in the gene encoding the MR also cause hypertension that is exacerbated by steroid hormone alterations during pregnancy (36). While rare, these syndromes highlight the capacity for dysregulation of the MR/ENaC signaling pathway in the kidney to promote hypertension.
In addition to stimulation of sodium reabsorption as discussed above, aldosterone also has important actions to promote secretion of potassium into urine. Until recently, it has not been clear how the kidney distinguishes between the need for these distinct functions. Recent studies from Shibata et al. indicate that regulated phosphorylation of the MR modulates aldosterone responses in the kidney (Figure 1). They showed that phosphorylation of S843 on the MR prevents ligand binding. In the kidney, this form of the MR is present only in intercalated cells of the collecting duct where its phosphorylation is differentially regulated by volume depletion and hyperkalemia. For example, in volume depletion, the MR in intercalated cells is dephosphorylated, resulting in potentiation of chloride and sodium reabsorption, allowing a distinct response to volume depletion (37). Although the MR is classically activated by aldosterone, recent studies suggest that the small GTPase Rac1 may promote hypertension through an MR-dependent pathway, even in the setting of suppressed aldosterone levels (38 and Figure 1).
The WNKs: novel pathways regulating renal solute transport
Some of the most powerful lines of evidence implicating a predominant role for the kidney in the regulation of blood pressure are the classic studies from the Lifton laboratory, which have defined the genetic basis of virtually all of the known Mendelian disorders associated with abnormal blood pressure phenotypes in humans (36, 39, 40). In each case, these mutations impact sodium and fluid reabsorption along the nephron (39). One of these disorders is pseudo-hypoaldosteronism type II (PHAII), a Mendelian syndrome characterized by the unusual combination of hypertension and hyperkalemia, found to be caused by mutations in the genes encoding WNK1 (with no lysine [K]) kinase and WNK4 (40). This discovery triggered intense study of these unique kinases, identifying roles for WNK1 and WNK4 in the regulation of sodium and potassium flux in the distal nephron (refs. 41, 42, and Figure 2). These actions are primarily mediated through control of the relative levels and activities of the thiazide-sensitive sodium chloride cotransporter (NCC) and/or the renal outer medullary potassium channel (ROMK) (41, 42). The NCC represents a major pathway for sodium reabsorption in the distal nephron and is the target for thiazide diuretics, which are effective and widely used antihypertensive agents (43). Thiazides are a mainstay of treatment for PHAII, consistent with findings that NCC overactivity is a key feature of the disorder (44). It is worth noting that while the actions of WNK4 to suppress ROMK activity have been consistent in these studies, variable effects of WNK4 on NCC activity have been observed, perhaps relating to the relative levels of WNK4 in experimental systems. In this regard, mutations causing accumulation of endogenous WNK4 enhance NCC activity possibly through phosphorylation of STE20/SPS-1–related proline-alanine–rich protein kinase (SPAK), whereas deliberate overexpression of WNK4 appears to target NCC for lysosomal degradation (42, 45, 46).
Figure 2. Mechanisms regulating sodium and potassium flux in the distal nephron.
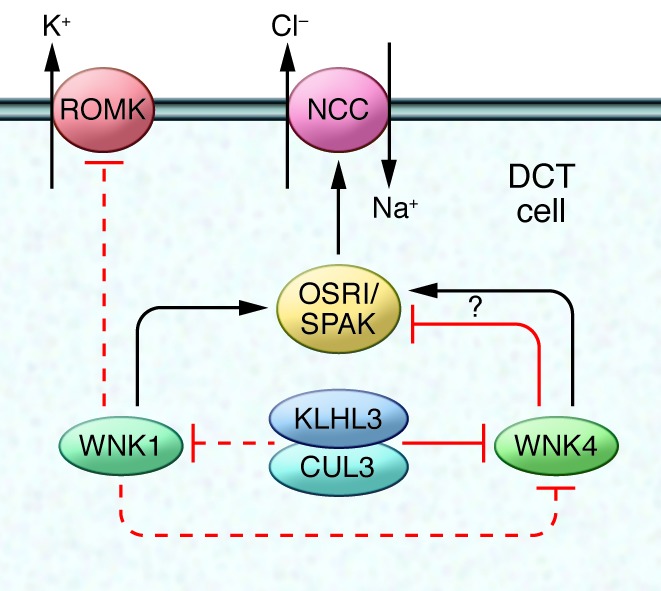
WNK family kinases control the activity of the sodium chloride cotransporter (NCC) and the renal outer medullary potassium channel (ROMK) in distal convoluted tubule (DCT) cells in the kidney. WNK1 phosphorylates and stimulates the SPS1-related proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase 1 (OSR1) protein kinases, which in turn, promotes NCC-dependent sodium transport. WNK1 may also inhibit ROMK. WNK4 inhibits ROMK but has been reported to have both stimulating and inhibitory actions on NCC depending on the experimental system used. Levels of WNK4 are regulated by the activity of the cullin 3-KLHL3 ubiquitin ligase, which has also been suggested to modulate WNK1. Individual mutations in WNK1, WNK4, cullin 3, and KLHL3 generate a similar phenotype: the syndrome of pseudo-hypoaldosteronism type II (PHAII), a Mendelian syndrome characterized by the unusual combination of hypertension and hyperkalemia, highlighting the continuity and importance of this pathway in the normal control of sodium and potassium handling in the distal nephron.
Enhanced activity of NCC through modulation of WNKs seems to be a final common pathway for the development of hypertension in a number of scenarios. For example, β-adrenergic stimulation increases blood pressure by suppressing WNK4 and, in turn, enhancing NCC activity (47). In addition, calcineurin inhibitors, commonly used to treat autoimmune disease and prevent transplant rejection, frequently cause hypertension. Recent studies by Ellison and colleagues indicate that the mechanism of hypertension associated with calcineurin inhibitor use involves stimulation of NCC through upregulation of WNK3 (48).
While the ongoing delineation of WNK functions has provided significant insights into kidney physiology, only a small subset of patients with PHAII have mutations in WNK genes. Using exome sequencing, Lifton’s group uncovered mutations in the kelch-like 3 (KLHL3) and cullin 3 (CUL3) genes in patients with PHAII (49). Moreover, mutations in these two genes accounted for disease in approximately 80% of individuals affected with PHAII (49). KLHL3 is one of a family of more than 50 broad-complex, tramtrack, bric-a-brac complex–containing (BTB-containing) kelch proteins, characterized by six-bladed, β-propeller domains for binding specific target proteins. CUL3 provides the scaffold for the complex, which includes BTB-domain proteins such as KLHL3 and a RING domain protein that serves as an E3 ubiquitin ligase, targeting specific protein substrates for ubiquitination (50).
The observations that KLHL3 and CUL3 mutations result in similar phenotypes suggests that disrupted ubiquitination of KLHL3-specific substrates might be a common mechanistic pathway in PHAII. Subsequent studies showed that KLHL3 binds both WNK1 and WNK4, and these interactions are required for polyubiquitination and degradation of WNK4 (51). Furthermore, abrogation of this pathway for ubiquitination resulted in intracellular accumulation of WNK4. Finally, this alteration in WNK4 ubiquitination reduced levels of cell surface-associated ROMK, which, in kidney tubules, results in diminished excretion of potassium in urine, leading to hyperkalemia characteristic of PHAII (51). These studies raise a number of interesting questions regarding the role of this pathway in regulating NCC, its role in global regulation of electrolyte excretion in the distal nephron and, perhaps most importantly, the relevance of these findings to more common forms of hypertension.
Salt homeostasis gets under your skin
Salt sensitivity, defined as an exaggerated change in blood pressure in response to extremes in dietary salt intake, is relatively common and is associated with an increased risk for the development of hypertension. Classic Guytonian models suggest that a defect in sodium excretion by the kidney is the basis for salt sensitivity, with impaired elimination of sodium during high-salt feeding leading directly to expanded extracellular fluid volume, which promotes increased blood pressure (8). This model presumes that the two major components of extracellular volume within the intravascular and interstitial spaces are in equilibrium. As such, accumulation of sodium would be accompanied by commensurate retention of water to maintain iso-osmolality and would thereby proportionally expand the intravascular volume. On the other hand, recent innovative studies by Titze and associates indicate that sodium handling is more complex than this classical two-compartment model; the interstitium of the skin may act as a sodium reservoir, buffering the impact of sodium accumulation on intravascular volume and blood pressure (52). During high-salt feeding, sodium accumulates in the subdermal interstitium at hypertonic concentrations in complexes with proteoglycans (52, 53). Macrophages infiltrating the interstitial space sense hypertonicity caused by this accumulation of sodium in excess of water, triggering expression of TonEBP, a transcription factor regulating the expression of osmo-protective genes. One of the genes induced downstream of TonEBP is vascular endothelial growth factor-C (VEGF-C) (52), a potent inducer of lymphangiogenesis.
Indeed, in response to high-salt feeding, Titze’s group found robust lymphatic vessel hyperplasia in the dermal interstitium (52). Depletion of macrophages, cell-specific deletion of TonEBP from macrophages, or specific blockade of VEGF-C prevented hyperplasia of lymphatic vessels and enhanced the level of sodium-dependent hypertension (52–54), demonstrating that this pathway has a key role in the extrarenal control of sodium and fluid volumes.
The nature and regulation of proteoglycans mediating hypertonic storage of sodium in the skin, as well as mechanisms for control of macrophage gene expression by hypertonicity, remain unclear. The authors have found elevated plasma levels of VEGF-C in patients with refractory hypertension, indicating that this system might be perturbed in the human disorder. However, preclinical models predict that reduced levels of VEGF-C would promote hypertension. Consistent with this notion, a recent report showed that overexpression of VEGF-C lowered blood pressure in a model of salt-sensitive hypertension, whereas blocking VEGF-C activity exaggerated the severity of hypertension (55). Nonetheless, chronic hypertension in humans is a complex disorder; it is possible that the observed elevation in VEGF-C levels may reflect tissue resistance to VEGF-C or even a compensatory response. The specific relevance of this pathway in human hypertension and its potential value as a therapeutic target are interesting questions for future studies.
A subsequent study by Titze et al. described a unique, long-term evaluation of physiological responses within the highly controlled environment of a space travel-simulation training program called Mars500 (56). In this enclosed, restricted environment, subjects were provided diets with scrupulously defined sodium content while undergoing continuous monitoring of a range of parameters including urinary sodium excretion, blood pressure, body weight, and steroid hormone levels. Aspects of these findings are consistent with Guytonian principles whereby abrupt increases of salt intake caused expansion of total-body sodium and extracellular water, brisk suppression of aldosterone, and consequent increases in urinary sodium excretion. However, the authors unexpectedly found considerable day-to-day variability in 24 hour urinary sodium excretion, accompanied by fluctuations in excretion of aldosterone, cortisol, and cortisone, despite the fixed sodium content in the diet. Power spectral analysis demonstrated a regular pattern, peaking with a period interval of about 6 days. Regular fluctuations of total body sodium (TBNa), estimated using bioimpedance spectroscopy, were also observed but with much longer periodicity. This uncoupling of changes in TBNa from body weight and blood pressure may reflect this process of sodium storage in the subdermal interstitium (52). Dissociation of blood pressure from fluid volumes likely reflects complexities of sodium handling not accounted for by Guyton’s hypothesis. Along with the endogenous rhythms of sodium balance, the authors also observed increases in blood pressure and urinary aldosterone excretion following night shift duty, reminiscent of mouse studies wherein disruption of the clock gene Cry is associated with hypertension caused by chronically enhanced aldosterone levels (57).
Immune mechanisms in hypertension
A prominent emerging area of hypertension research over the past decade suggests that the immune system may provide an independent cardiovascular control mechanism, whereby its cellular constituents and inflammatory mediators regulate blood pressure. Moreover, we suggest below that immunological effector mechanisms impacting blood pressure can be mapped to the kidney.
Although not rigorously controlled, small clinical studies indicate that broad immunosuppression lowers blood pressure in hypertensive patients with rheumatologic disorders (58), pointing to a role for immune activation in human hypertension. Within the innate immune system, adoptive transfer of monocytes, which are precursors to dendritic cells and macrophages, promotes the hypertensive response to angiotensin II (59). Studies by Guzik and associates convincingly established a role for the adaptive immune response in hypertension pathogenesis, showing that mice with targeted disruption of the recombinase-activating gene-1 (Rag1), and thus lacking T and B cells, were resistant to the development of hypertension induced by chronic infusion of angiotensin II. Adoptive transfer of T but not B cells restored the hypertensive response, clearly illustrating the capacity for T lymphocytes to drive blood pressure elevation (60). Although specific antigens triggering hypertension have not been identified, dual involvement of antigen presenting cells (APCs) and T cells has been well documented. Furthermore, blocking the interaction of B7 ligands on APCs with costimulatory molecules on T cells can abrogate hypertension, indicating that both T cell receptor engagement and costimulation are required (61).
While susceptibility to hypertension primarily depends on CD8+ rather than CD4+ T cells (62), the CD4+ population also may modulate hypertension and hypertensive end-organ damage. In this regard, Th1 cells, a proinflammatory CD4+ T cell subset characterized by expression of the transcription factor Tbet, promote renal damage during hypertension without altering blood pressure (63). Moreover, Th17 cells, another CD4+ subset secreting IL-17, potentiate chronic hypertension (64). In contrast, CD4+ Foxp3+ T regulatory cells, which suppress immune responses, have been reported to protect against hypertension in one study (65), a finding that was not confirmed in work from another group (66). In the first study, three separate doses of T regulatory cells were administered, whereas only one adoptive transfer was performed in the second study, suggesting that the beneficial response may depend on the absolute number of regulatory cells present.
In immune responses, the NF-κB signaling cascade is a prototypical inflammatory pathway mobilizing cytokine transcription and triggering reactive oxygen species (ROS) generation. This pathway seems to play a central role in blood pressure control since its blockade protects against hypertension (67). NF-κB activation within the vascular endothelium drives hypertensive renal damage without measurable effects on blood pressure suggesting a direct role in end-organ damage (68). Within cardiovascular control centers in the brain, NF-κB activation potently enhances sympathetic outflow (69–71), which could potentially promote sodium retention in the kidney via stimulation of renal sympathetic nerves. Within kidney parenchymal cells, induction of oxidative stress leads to NF-κB nuclear translocation, impaired sodium excretion, and blood pressure elevation by disrupting D1 dopamine receptor function (72). Thus, NF-κB signaling may directly contribute to the hypertensive response in kidney cells.
Once activated, macrophages and T cells release cytokines. These soluble mediators may drive blood pressure elevation or mediate end-organ injury in hypertension as illustrated in several experiments using gene knockout models. For example, genetic deletion of the receptor for interferon-γ, the prototypical inflammatory cytokine produced by immune cells, affords protection against end-organ damage in hypertension without altering blood pressure (73). On the other hand, interleukin-6 (IL-6) promotes blood pressure elevation, as mice genetically deficient in IL-6 have a blunted hypertensive response to chronic angiotensin II infusion (74). Finally, tumor necrosis factor-α (TNF-α) seems to have complex effects on blood pressure regulation. In this regard, some studies have shown that genetic deficiency of TNF-α protects against hypertension (60, 75), whereas others indicate TNF-α may have negative or neutral effects on blood pressure (76, 77). Perhaps the influence of TNF-α on blood pressure control may depend on its local levels or the relative expression of its two receptor isoforms (78). In sum, inflammatory cytokines have pleiotropic effects on blood pressure elevation and/or target organ damage in hypertension. As discussed below, several lines of evidence suggest that these cytokines may influence blood pressure by modulating sodium handling by the kidney.
Regardless of the initial trigger for immune activation, immune cells and the inflammatory mediators they secrete appear to alter blood pressure through effects on kidney function. During hypertension, clusters of T lymphocytes infiltrate the adventitia surrounding blood vessels in the kidney, while macrophages disperse throughout the renal interstitium (60, 79, 80). These cells release ROS that can interfere with vascular relaxation to reduce renal blood flow, and thereby diminish sodium excretion following a hypertensive stimulus. Accordingly, scid mice lacking functional T cells have attenuated renal oxidative stress, exaggerated natriuresis, and blunted hypertension during chronic angiotensin II infusion (81). Similarly, preventing infiltration of immune cells into the kidney reduces oxidative stress and lowers blood pressure in rat models of salt-sensitive hypertension (82). Various cytokines prominent in inflammatory responses have direct effects on kidney function. For example, IL-6 stimulates ENaC activity in the collecting duct, promoting sodium retention (83). Similarly, although its role in hypertension is not clearly established, IL-1 stimulates vasoconstriction and modulates sodium excretion (84, 85). Finally, in the thick ascending limb of the loop of Henle, TNF-α suppresses eNOS expression (86), which would enhance sodium retention and increase blood pressure. Thus, in hypertension, T lymphocytes infiltrate the kidney, propagating oxidative stress and the secretion of prohypertensive cytokines, which promotes sodium reabsorption and blood pressure elevation.
Conclusions
Sodium handling by the kidney is a major determinant of the level of arterial pressure and is under complex physiological control by hormones, inflammatory mediators, and the sympathetic nervous system. It is self-evident that a basic mechanism of efficacy for diuretics and dietary sodium restriction in hypertension is to favorably influence sodium balance and homeostasis. We submit that other antihypertensive agents such as RAS inhibitors, vasodilators, and β-blockers work through a similar mechanism by facilitating pressure-natriuresis. Recent studies reviewed above suggest that WNK signaling pathways, soluble inflammatory mediators, and pathways regulating extra-renal sodium disposition might also be useful targets for enhancing elimination of sodium and reducing blood pressure in hypertension.
Acknowledgments
The authors’ work in this area has been supported by funding from the Medical Research Service of the Veterans Administration, the NIH (DK087893 to S.D. Crowley and DK069896 to T.M. Coffman), the Mandel Center for Hypertension and Atherosclerosis at Duke, and the American Heart Association.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(6):2341–2347. doi:10.1172/JCI72274.
References
- 1.Lawes CM, Vander Hoorn S, Rodgers A, International Society of Hypertension Global burden of blood-pressure-related disease, 2001. Lancet. 2008;371(9623):1513–1518. doi: 10.1016/S0140-6736(08)60655-8. [DOI] [PubMed] [Google Scholar]
- 2.[No authors listed]. Effects of treatment on morbidity in hypertension. Results in patients with diastolic blood pressures averaging 115 through 129 mm Hg. JAMA. 1967202111028–1034. [PubMed] [Google Scholar]
- 3.[No authors listed]. Five-year findings of the hypertension detection follow-up program. I. Reduction in mortality of persons with high blood pressure, including mild hypertension. Hypertension Detection and Follow-up Program Cooperative Group. JAMA. 1979242232562–2571. [PubMed] [Google Scholar]
- 4.[No authors listed]. MRC trial of treatment of mild hypertension: principal results. Medical Research Council Working Party. Br Med J (Clin Res Ed). 1985291648897–104. 10.1136/bmj.291.6488.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu Q, Burt VL, Dillon CF, Yoon S. Trends in antihypertensive medication use and blood pressure control among United States adults with hypertension: the National Health And Nutrition Examination Survey, 2001 to 2010. Circulation. 2012;126(17):2105–2114. doi: 10.1161/CIRCULATIONAHA.112.096156. [DOI] [PubMed] [Google Scholar]
- 6. Bright, R. Observations on the treatment of fever. Case of simple fever, protracted by irritation of the bowels, and attended by relapse. In: Guy’s Hospital Reports. Vol 1. London, United Kingdom: Churchill Livingstone; 1836:1–8. [Google Scholar]
- 7.Goldblatt H, Lynch J, Hanzal RF, Summerville WW. Studies on experimental hypertension: I. The production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med. 1934;59(3):347–379. doi: 10.1084/jem.59.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guyton AC, Coleman TG, Cowley AV, Jr, Scheel KW, Manning RD, Jr, Norman RA., Jr Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. 1972;52(5):584–594. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 9.Aperia AC, Broberger CG, Soderlund S. Relationship between renal artery perfusion pressure and tubular sodium reabsorption. Am J Physiol. 1971;220(5):1205–1212. doi: 10.1152/ajplegacy.1971.220.5.1205. [DOI] [PubMed] [Google Scholar]
- 10.Guyton AC. Blood pressure control — special role of the kidneys and body fluids. Science. 1991;252(5014):1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 11.Osborn JW, Fink GD, Kuroki MT. Neural mechanisms of angiotensin II-salt hypertension: implications for therapies targeting neural control of the splanchnic circulation. Curr Hypertens Rep. 2011;13(3):221–228. doi: 10.1007/s11906-011-0188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCurley A, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18(9):1429–1433. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michael SK, et al. High blood pressure arising from a defect in vascular function. Proc Natl Acad Sci U S A. 2008;105(18):6702–6707. doi: 10.1073/pnas.0802128105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahl LK, Heine M, Thompson K. Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ Res. 1974;40(4):94–101. doi: 10.1161/01.res.40.4.94. [DOI] [PubMed] [Google Scholar]
- 15.Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res. 1975;36(6):692–696. doi: 10.1161/01.RES.36.6.692. [DOI] [PubMed] [Google Scholar]
- 16.Bianchi G, Fox U, Di Francesco GF, Giovanetti AM, Pagetti D. Blood pressure changes produced by kidney cross-transplantation between spontaneously hypertensive rats and normotensive rats. Clin Sci Mol Med. 1974;47(5):435–448. doi: 10.1042/cs0470435. [DOI] [PubMed] [Google Scholar]
- 17.Kawabe K, Watanabe TX, Shiono K, Sokabe H. Influence on blood pressure of renal isografts between spontaneously hypertensive and normotensive rats, utilizing the F1 hybrids. Jpn Heart J. 1978;19(6):886–894. doi: 10.1536/ihj.19.886. [DOI] [PubMed] [Google Scholar]
- 18.Curtis JJ, et al. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309(17):1009–1015. doi: 10.1056/NEJM198310273091702. [DOI] [PubMed] [Google Scholar]
- 19.Matchar DB, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148(1):16–29. doi: 10.7326/0003-4819-148-1-200801010-00189. [DOI] [PubMed] [Google Scholar]
- 20.Le TH, Coffman TM. Targeting genes in the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2008;17(1):57–63. doi: 10.1097/MNH.0b013e3282f2fd39. [DOI] [PubMed] [Google Scholar]
- 21.Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol. 1986;250(6 pt 2):R960–R972. doi: 10.1152/ajpregu.1986.250.6.R960. [DOI] [PubMed] [Google Scholar]
- 22.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57(3):355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 24.Navar LG, Lewis L, Hymel A, Braam B, Mitchell KD. Tubular fluid concentrations and kidney contents of angiotensins I and II in anesthetized rats. J Am Soc Nephrol. 1994;5(4):1153–1158. doi: 10.1681/ASN.V541153. [DOI] [PubMed] [Google Scholar]
- 25.Navar LG, Harrison-Bernard LM, Wang CT, Cervenka L, Mitchell KD. Concentrations and actions of intraluminal angiotensin II. J Am Soc Nephrol. 1999;10(suppl 11):S189–S195. [PubMed] [Google Scholar]
- 26.Gonzalez-Villalobos RA, et al. Intrarenal angiotensin-converting enzyme induces hypertension in response to angiotensin I infusion. J Am Soc Nephrol. 2011;22(3):449–459. doi: 10.1681/ASN.2010060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Villalobos RA, et al. The absence of intrarenal ACE protects against hypertension. J Clin Invest. 2013;123(5):2011–2023. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsusaka T, et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol. 2012;23(7):1181–1189. doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano D, et al. Multiphoton imaging of the glomerular permeability of angiotensinogen. J Am Soc Nephrol. 2012;23(11):1847–1856. doi: 10.1681/ASN.2012010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowley SD, et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest. 2005;115(4):1092–1099. doi: 10.1172/JCI23378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowley SD, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103(47):17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gurley SB, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13(4):469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamenko M, et al. Chronic angiotensin II infusion drives extensive aldosterone-independent epithelial Na+ channel activation. Hypertension. 2013;62(6):1111–1122. doi: 10.1161/HYPERTENSIONAHA.113.01797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura H, et al. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. 1996;97(7):1780–1784. doi: 10.1172/JCI118606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Firsov D, Schild L, Gautschi I, Merillat A-M, Schneeberger E, Rossier BC. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci U S A. 1996;93(26):15370–15375. doi: 10.1073/pnas.93.26.15370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geller DS, et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. 2000;289(5476):119–123. doi: 10.1126/science.289.5476.119. [DOI] [PubMed] [Google Scholar]
- 37.Shibata S, et al. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013;18(5):660–671. doi: 10.1016/j.cmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shibata S, et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest. 2011;121(8):3233–3243. doi: 10.1172/JCI43124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104(4):545–556. doi: 10.1016/S0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 40.Wilson FH, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293(5532):1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 41.Kahle KT, Ring AM, Lifton RP. Molecular physiology of the WNK kinases. Annu Rev Physiol. 2008;70:329–355. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- 42.McCormick JA, Ellison DH. The WNKs: atypical protein kinases with pleiotropic actions. Physiol Rev. 2011;91(1):177–219. doi: 10.1152/physrev.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ALLHAT Officers Coordinators for the ALLHAT Collaborative Research Group The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288(23):2981–2997. doi: 10.1001/jama.288.23.2981. [DOI] [PubMed] [Google Scholar]
- 44.Lalioti MD, et al. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38(10):1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 45.Ahlstrom R, Yu AS. Characterization of the kinase activity of a WNK4 protein complex. Am J Physiol Renal Physiol. 2009;297(3):F685–F692. doi: 10.1152/ajprenal.00358.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai H, et al. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 2006;69(12):2162–2170. doi: 10.1038/sj.ki.5000333. [DOI] [PubMed] [Google Scholar]
- 47.Mu S, et al. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17(5):573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- 48.Hoorn EJ, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. 2011;17(10):1304–1309. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prag S, Adams JC. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4:42. doi: 10.1186/1471-2105-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A. 2013;110(19):7838–7843. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Machnik A, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 53.Machnik A, et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension. 2010;55(3):755–761. doi: 10.1161/HYPERTENSIONAHA.109.143339. [DOI] [PubMed] [Google Scholar]
- 54.Wiig H, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. 2013;123(7):2803–2815. doi: 10.1172/JCI60113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang GH, et al. Overexpression of VEGF-C attenuates chronic high salt intake-induced left ventricular maladaptive remodeling in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;306(4):H598–H609. doi: 10.1152/ajpheart.00585.2013. [DOI] [PubMed] [Google Scholar]
- 56.Rakova N, et al. Long-term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell Metab. 2013;17(1):125–131. doi: 10.1016/j.cmet.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 57.Doi M, et al. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16(1):67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 58.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17(12 suppl 3):S218–S225. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 59.Wenzel P, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124(12):1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 60.Guzik TJ, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vinh A, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122(24):2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thabet SR, et al. The role of CD8+ T cells, IP-10 and MMP12 in hypertension. FASEB J. 2011;25:640.37. [Google Scholar]
- 63.Zhang JD, et al. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110(12):1604–1617. doi: 10.1161/CIRCRESAHA.111.261768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Madhur MS, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barhoumi T, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57(3):469–476. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 66.Kvakan H, et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119(22):2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 67.Muller DN, et al. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35(1 pt 2):193–201. doi: 10.1161/01.hyp.35.1.193. [DOI] [PubMed] [Google Scholar]
- 68.Henke N, et al. Vascular endothelial cell–specific NF-κB suppression attenuates hypertension-induced renal damage. Circ Res. 2007;101(3):268–276. doi: 10.1161/CIRCRESAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 69.Kang YM, et al. Brain nuclear factor-κB activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res. 2009;82(3):503–512. doi: 10.1093/cvr/cvp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guggilam A, et al. TNF-α blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am J Physiol Heart Circ Physiol. 2007;293(1):H599–H609. doi: 10.1152/ajpheart.00286.2007. [DOI] [PubMed] [Google Scholar]
- 71.Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-β and NF-κB. Nat Med. 2011;17(7):883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banday AA, Fazili FR, Lokhandwala MF. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor-κB and protein kinase C. J Am Soc Nephrol. 2007;18(5):1446–1457. doi: 10.1681/ASN.2006121373. [DOI] [PubMed] [Google Scholar]
- 73.Markó L, et al. Interferon-γ signaling inhibition ameliorates angiotensin II–induced cardiac damage. Hypertension. 2012;60(6):1430–1436. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 74.Lee DL, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290(3):H935–H940. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 75.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-α in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51(5):1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen CC, Pedraza PL, Hao S, Stier CT, Ferreri NR. TNFR1-deficient mice display altered blood pressure and renal responses to ANG II infusion. Am J Physiol Renal Physiol. 2010;299(5):F1141–F1150. doi: 10.1152/ajprenal.00344.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muller DN, et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161(5):1679–1693. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramseyer VD, Garvin JL. Tumor necrosis factor-α: regulation of renal function and blood pressure. Am J Physiol Renal Physiol. 2013;304(10):F1231–F1242. doi: 10.1152/ajprenal.00557.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Crowley SD, et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295(2):F515–F524. doi: 10.1152/ajprenal.00527.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007;292(1):F330–F339. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1089–R1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300(3):F734–F742. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li K, et al. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299(2):R590–R595. doi: 10.1152/ajpregu.00207.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cooper AL, Beasley D. Hypoxia stimulates proliferation and interleukin-1alpha production in human vascular smooth muscle cells. Am J Physiol. 1999;277(4 pt 2):H1326–H1337. doi: 10.1152/ajpheart.1999.277.4.H1326. [DOI] [PubMed] [Google Scholar]
- 85.Sakairi Y, Ando Y, Tabei K, Kusano E, Asano Y.Interleukin-1 inhibits sodium and water transport in rabbit cortical collecting duct. Am J Physiol. 19942664 Pt 2)(4) [DOI] [PubMed] [Google Scholar]
- 86.Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension. 2012;59(6):1145–1150. doi: 10.1161/HYPERTENSIONAHA.111.189761. [DOI] [PMC free article] [PubMed] [Google Scholar]