

Abstract
The phylogenetic diversity of the bacteria in hot composting samples collected from three spatial locations was investigated by molecular tools in order to determine the influence of gradient effect on bacterial communities during the thermophilic phase of composting swine manure with rice straw. Total microbial DNA was extracted and bacterial near full-length 16S rRNA genes were subsequently amplified, cloned, restriction fragment length polymorphism-screened and sequenced. The superstratum sample had the highest microbial diversity among the three samples which was possibly related to the surrounding conditions of the sample resulting from the location. The results showed that the sequences related to Bacillus sp. were most common in the composts. In superstratum sample, 45 clones (33%) and 36 clones (27%) were affiliated with the Bacillus sp. and Clostridium sp., respectively; 74 clones (58%) were affiliated with the Clostridium sp. in the middle-level sample; 52 clones (40%) and 29 clones (23%) were affiliated with the Clostridium sp. and Bacillus sp. in substrate sample, respectively. It indicated that the microbial diversity and community in the samples were different for each sampling site, and different locations of the same pile often contained distinct and different microbial communities.
Keywords: Spatial Heterogeneity, 16S rRNA Gene, Swine Manure, Composting)
INTRODUCTION
Composting is an aerobic, biological process that uses naturally occurring microorganisms to convert biodegradable organic matter into a humus-like product. The process destroys pathogens, converts nitrogen from unstable ammonia to stable organic forms, and reduces the volume of waste (Imbeah, 1998). It has become an important method to dispose of organic solid wastes (Stentiford, 1987; Wei, 2000; Zhu et al., 2004; Zhu, 2006).
It is known that composting is essentially a microbiological process that depends highly on composting temperature. Generally, a composting process goes through three classic phases: mesophilic-heating phase, thermophilic phase, and cooling phase. In different phases, composting temperature reflects different functions: 35 to 40°C to maximize the microbial diversity, 45 to 55°C to maximize the biodegradation rate, and >55°C to maximize sanitation (Stentiford, 1996; Hassen et al., 2001). Apparently, thermophilic phase is a key phase because sanitation mostly depends on it and it plays a selective role in the evolution and succession of the microbial communities (Peters et al., 2000; Tiquia et al., 2005).
In fact, the microbial community in the composting process might be quite different because of variation of the environmental factors (Strom, 1985a, b; Peters et al., 2000; Tiquia et al., 2005). Coincidently, gradient effect can cause variation between the different height locations in environmental factors, such as temperature, oxygen, moisture content and other physico-chemical conditions (Ekinci, 2001; Zhu et al., 2004). Therefore, the microbial communities in the different height locations should be theoretically quite different due to this gradient effect.
The microbial community composition in natural systems can be determined by analyzing the 16S rDNA sequences obtained directly from samples by PCR amplification, cloning, and sequencing (Muyzer et al., 1993; Dees and Ghiorse, 2001; Leser et al., 2002). Dees and Ghiorse (2001) analyzed the microbial diversity in hot synthetic compost, and the results showed that the microbial community was complex and that many microbes were present that could not be obtained by classic culture-depended methods. From the point view of molecular microbial ecology analysis the general protocols for microbial total genomic DNA extraction and purification, Primers and PCR amplification, cloning library construction and screening have been sufficiently detailed to guarantee the outcome (Lane, 1991; Barns et al., 1994; Holben, 1994; Heuer et al., 1997; Wintzingerode et al., 1997; LaMontagne et al., 2002; von Arbeli and Fuentes, 2007; Yang et al., 2007; Wu et al., 2009). Recently, using a culture-independent method, we found that composting temperature significantly affected the microbial community in the initial stage of the composting and that the microbial community in different locations was quite different due to the gradient effect (Guo et al., 2007).
In order to improve the understanding of the gradient effect during the composting process, this paper presents the analysis of three libraries of bacteria 16S rDNA sequences cloned from three hot composting samples. This study aimed to provide molecular evidence for the spatial heterogeneity and to clarify the reason for the gradient effect.
MATERIALS AND METHODS
Composting facility, operation and sampling
The composting experiment was conducted on the Huazhong Agricultural University Swine Farm, which is subordinate to the National Engineering Research Center-Animal Science (NERC-AS). A 1.5 m×1.2 m×1.2 m (length ×width×height) composting bin was designed and built, which was covered with a shed to shield against sunshine and rain. The swine manure was from the swine farm, and rice straw was obtained from a local farm. The rice straw was passed through a cutting mill adjusted to yield a 4 cm particle size. For the reactor, ~500 kg of swine manure and 25 kg of rice straw were mixed according to the previous report (Zhu, 2007). The initial moisture content of the mixture was adjusted to about 60%.
Temperature at different locations in the pile, including bottom (20 cm from bottom), core and surface (20 cm to surface) temperature, and environmental temperature were monitored by Ttime system (Zhu, 2006; 2007). The temperature data were refurbished per 10 s, and saved automatically per hour.
When the core temperature reached 60°C in the sustained high temperatures phase, about 1 kg of fresh composting sample was collected from different locations, bottom (20 cm from bottom), core and surface (20 cm to surface), respectively. The exact temperatures of superstratum and substrate were 63°C and 47.1°C, respectively. The collected samples were immediately placed on ice for transportation to the laboratory and then stored at −20°C.
Cell extraction, lysis, DNA purification, and PCR amplification and cloning
The cell extraction, lysis and DNA purification followed the method described by (Guo et al., 2007). Bacterial 16S rRNA genes were amplified by PCR using the universal primer pair 1492r (5′-TAC CTT GTT ACGACT T-3′) and 27f (5′-AGA GTT TGA TCC TGG CTCAG-3′) (Lane, 1991; Dojka et al., 1998). The PCR reaction system was: 10 pmol of both primers, 3 μl genome DNA, 1.5 μl of 2 mM mixed dNTPs, 2.5 μl of 10×Taq DNA polymerase buffer, 2.5 μl of 25 mM MgCl2, 2.0 units of Taq DNA polymerase (1 U/μl) (Jingmei Biotech, Shenzhen) and 15 μl of sterile water. The PCR raction cycle was as follows: 94°C for 40 s, 52°C for 45 s, 72°C for 10 min, 25 cycles, then 72°C extension for 10 min, finally 4°C to terminate the reaction. The amplified products were electrophoretically separated and visualized in 1.5% agarose gels stained with ethidium bromide. Bands of interest were cut from the gel and purified with UNIQ-10 DNA Purification Kit (Bio Basic Inc).
Finally, the purified product was ligated into the pMD18-T Vector (Takara) and the ligation product was transformed into Escherichia coli DH5α competent cells. White transformant colonies were selected randomly and inserted size was detected by using M13⁄pUC universal primers P47 and P48.
Screening of rRNA gene clones
Plasmids were extracted from the positive transformant colonies, and then were subjected to restriction fragment length polymorphism (RFLP) by separate endonuclease digestions with HhaI (Mentas) and HaeIII (Mentas) following the manufacturer’s instructions, and the digested DNA fragments were electrophoresed in 2.5% agarose gels. After staining with ethidium bromide, the gels were photographed and scanning image analyses were performed manually.
DNA sequencing and phylogenetic analysis
One to three representative clones from each unique RFLP type were selected for sequencing. The rDNA inserts were sequenced using M13⁄pUC universal sequencing primers P47 and P48. All near-full-length sequences were tested for possible chimeric structures. Chimeras were detected by the RDP analysis service Check-Chimera (Larsen et al., 1993) by comparing the phylogenetic positions of the near-full-length sequences from the clones and during the manual inspection of the alignment.
The spelled nearly entire sequences (~1,500 bp) except for the Chimeras were aligned by using the Clustalw program (http://www.ebi.ac.uk/clustalw/). Sequences differing only slightly (≤3%) were considered as a single relatedness group, and each group was represented by one sequence type. Every sequence type was compared with those available in GenBank by performing BLASTn program (http://www.ncbi.nlm.nih.gov) to determine their approximate phylogenetic affiliation. Phylogenetic trees were constructed based on the Kimura two-parameter model and the neighbour-joining algorithm using the PAUP software (version 4.0 b8) (Swofford, 1999).
Nucleotide sequence accession numbers
The retrieved 138 16S rDNA sequences have been deposited in the EMBL (The European Molecular Biology Laboratory) nucleotide sequence database under accession numbers AM500715-AM500852.
RESULTS
PCR amplification and clone library analysis
Total community genomic DNA extracted from the three layers (supertratum, middle-level and substrate) of composts was subjected to PCR amplification using the primer pair (27f⁄1492r). The ~1,500-bp PCR product was used to create a 16S rDNA clone library, and three bacterial 16S rRNA gene libraries were finally established to represent the superstratum, middle-level and substrate composts. To exclude false positives, a total of 150 recombinant clones (called SMD, SME and SMF) from each sample were randomly selected and screened by restriction endonuclease analysis. From the 150 clones, 98, 68 and 74 unique RFLP types were identified and 15, 22 and 21 false-positive clones were excluded from the superstratum, middle-level and substrate samples, respectively. One representative of each of the RFLP types was selected for sequencing, and the phylogenetic analysis was done on these near-full-length 16S rDNA sequences.
The clone library analysis results showed that 94 phylotypes were detected only once, and the percentage of coverage was 76% in all samples. It was indicated that 24% of the clones produced from the three samples would represent previously undetected phylotypes.
Bacterial diversity determination
Under the 97% sequence similarity criterion, 48, 46 and 44 sequence types were found from the superstratum sample, middle-level sample and substrate sample, respectively. Each of these represents a phylotype and may be a representative of a bacterial species. The number and distribution of sequence types and hence the bacterial diversity varies among the three sampling locations. The Shannon-Wiener index for the superstratum was 3.27, decreasing to 3.13 and 3.08 in the middle-level and substrate, respectively.
Isolates and cloned sequences of the superstratum compost
A total of 48 sequence types were observed in the 136 positive clones from the superstratum compost. Within these bacterial clones, 103 were represented by 16 sequence types with at least two clones. The other 32 sequence types had only one detected clone. The number of clone(s) of each sequence type is marked in the bracket after SMD in the phylogenetic tree (Figure 1).
Figure 1.
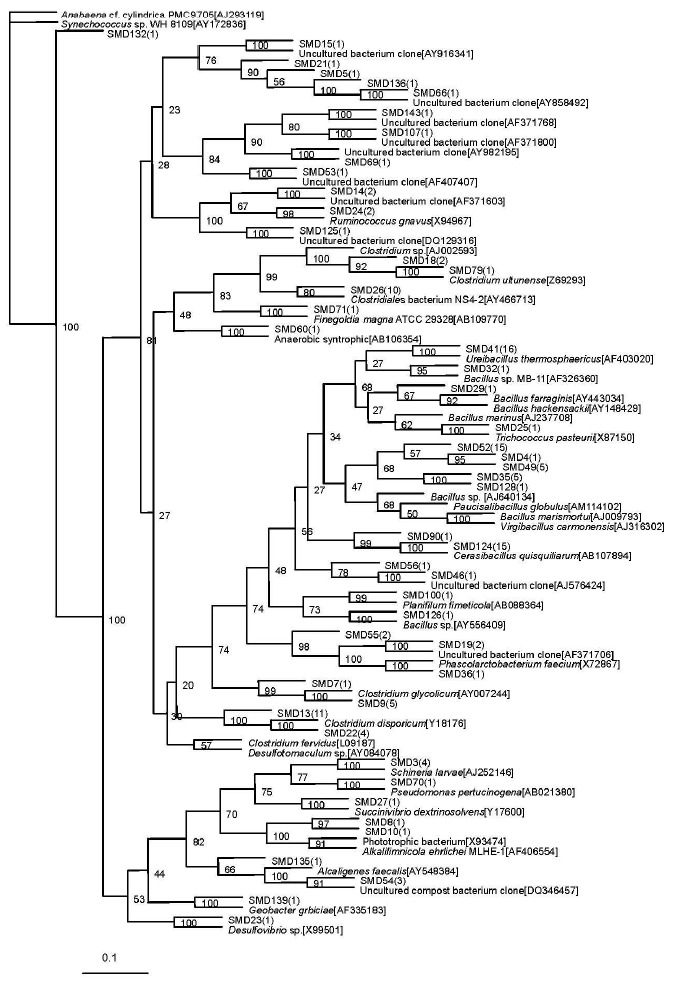
Phylogenetic tree showing the relationship of 16S rDNA sequences cloned from the superstratum sample compared with previously described bacterium species rDNA sequences deposited in public databases. Synechococcus sp. WH 8109[AY172836] as outgroup.
The most abundant clones (45 clones total) represented by nine phylotypes (i.e. SMD52) belonged to the Bacillus sp. (92 to 94% similarities). The sequence type SMD5 included 36 bacterial clones which were related to Clostridium sp. (90 to 98%). The sequence type SMD41 representing 10 clones, was affiliated with Ureibacillus thermosphaericus (97%) which was isolated from an aerobic digestor of swine waste (Gagné et al., 2001).
Sequence type SMD19 (2 clones) was related to AF371706 (98%) which was isolated from pig gut bacteria (Leser et al., 2002). Sequence type SMD14 (2 clones) was nearly related to AF371603 (95%), also obtained from pig gut bacteria (Leser et al., 2002). Sequence type SMD15 (one clone) was nearly related to AY916341 (99%), obtained from human intestinal microbial flora (Eckburg et al., 2005). The phylotype represented by SMD66 and SMD136 were related to the same known sequence AY858492, isolated from microbial populations in the wild herbivore gastrointestinal tract (Nelson et al., 2003).
The other four sequence types represented by SMD 46, SMD 143, SMD 53, and SMD 107, respectively, were all with only one bacterial clone and had no relationship with known cultivated bacterial members, but they were related to the bacterium clone sequence. SMD 46 was nearly related to a bacterium clone designated AJ576424 (96%), which was obtained from midgut and hindgut of the humus-feeding larva of Pachnoda ephippiata (Egert et al., 2003). SMD 143 and SMD 107 were nearly related to AF371768 (93%) and AF371800 (98%), respectively, both obtained from pig gut bacteria (Leser et al., 2002). SMD 53 was nearly related to AF407407 (95%), isolated from the monochlorobenzene contaminated groundwater (Alfreider et al., 2002).
Isolates and cloned sequences of the middle-level compost
A total of 46 sequence types were observed in the 128 positive clones from the middle-level compost. Within these bacterial clones, 97 were represented by 15 sequence types including at least two clones, while the other 31 sequence types were detected in only one clone. The number of clone(s) of each sequence type is indicated in the bracket after SME in the phylogenetic tree (Figure 2).
Figure 2.
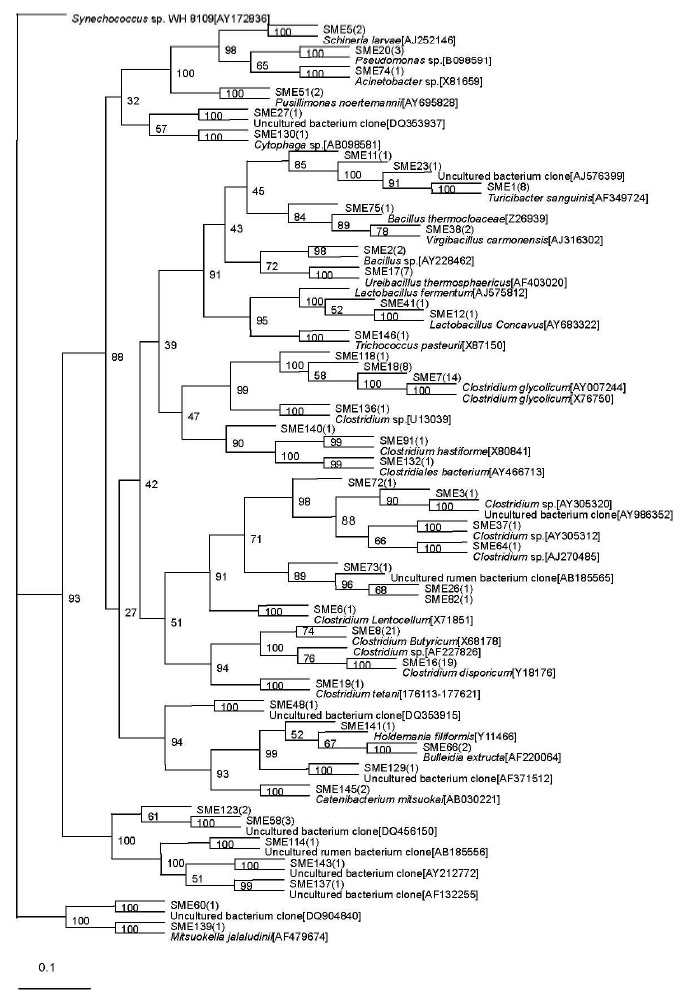
Phylogenetic tree showing the relationship of 16S rDNA sequences cloned from the middle-level sample compared with previously described bacterium species rDNA sequences deposited in public databases. Synechococcus sp. WH 8109[AY172836] as outgroup.
The most abundant sequence type in the middle-level sample, SME3, included 74 clones that were related to the genus Clostridium and displayed relatively high levels of similarity with their closest cultivated Clostridium sp. Sequence types represented by SME1 (9 clones) were related to Turicibacter sanguinis (99%), isolated from a blood culture of a febrile patient with acute appendicitis (Bosshard et al., 2002). The sequence type SME17 included 7 bacterial clones that were related to Ureibacillus thermosphaericus (96 to 97%), isolated from an aerobic digestor of swine waste (Gagné et al., 2001). Exceptionally, the sequence type represented by SME 130 with only one bacterial clone was related to Cytophaga sp. (90%), isolated from composting household biowaste composting (Hiraishi et al., 2003).
Sequence type SME 58 (3 clones) had no relationship to known cultivated bacteria members, but it was related to the known sequence DQ815725 (97%), isolated from the germ-free recipients transplanted by the gut microbiota of zebrafish and mice (Rawls et al., 2006). The other 10 phylotypes represented by SME 26, SME 82, SME 11, SME 48, SME 72, SME 60, SME 114, SME 137, SME 129, and SME 143 were all with only one bacterial clone and had no relationship with known cultivated bacterial members, but they were related to the bacterium clone sequence. Among them, the phylotype represented by SME 26 and SME 82 were related to the same known sequence AB185565, isolated from rumen bacteria in cattle (Ozutsumi et al., 2005). SME 11 was nearly related to AJ576399 (94%), obtained from the midgut and hindgut of Pachnoda ephippiat (Egert et al., 2003). SME 48 was nearly related to DQ353915 (99%), obtained from fecal bacteria of a wild gorilla (Frey et al., 2006). SME 72 was related to a bacterium clone designated AY986352 (93%), which was obtained from human intestinal microbial (Eckburg et al., 2005). SME 60 was nearly related to DQ904840 (99%), obtained from human intestinal microbial (Mai et al., 2006). SME114 was nearly related to AB185556 (96%), isolated from rumen bacteria in cattle (Ozutsumi et al., 2005). SME 137 was related to a bacterium clone designated AF132255 (94%), which was obtained from human gut microbial (Suau et al., 1999). SME129 was nearly related to AF371512 (99%), obtained from pig gut bacterium (Leser et al., 2002). SME 143 was related to a bacterium clone designated AY212772 (95%), which was obtained from equine fecal bacterium (Simpson et al., 2004).
Isolates and cloned sequences of the substrate compost
A total of 44 sequence types were observed in the 128 positive clones from the substrate compost. Within these bacterial clones, 98 were represented by 13 sequence types including at least two clones, while the remaining 31 sequence types were detected in only one clone. The number of clone(s) of each sequence type is indicated in the bracket after SMF in phylogenetic tree (Figure 3).
Figure 3.
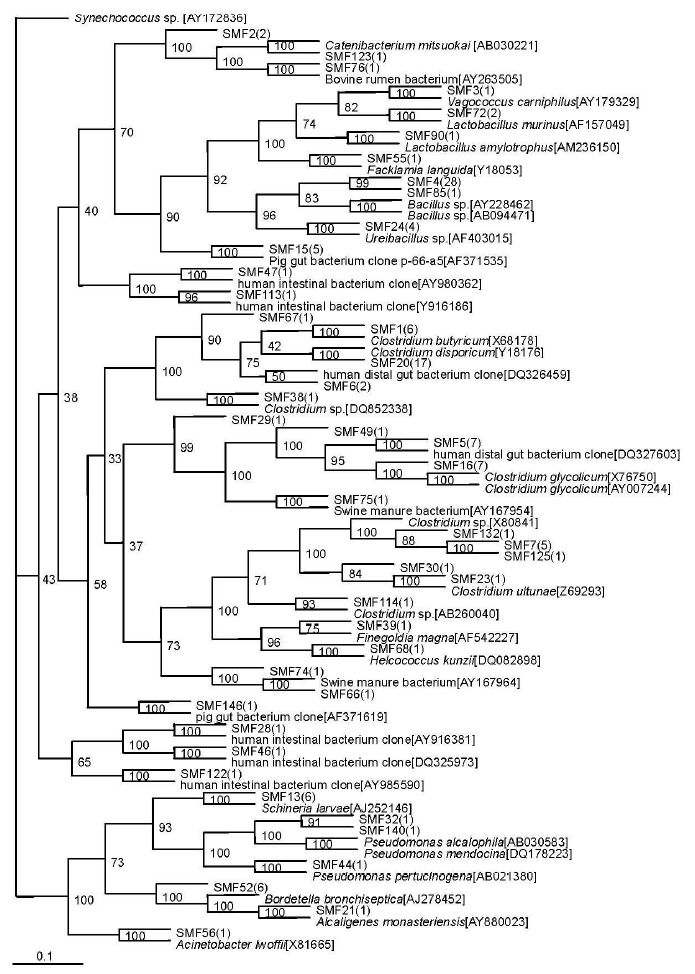
Phylogenetic tree showing the relationship of 16S rDNA sequences cloned from the substrate sample compared with previously described bacterium species rDNA sequences deposited in public databases. Synechococcus sp. WH 8109[AY172836] as outgroup.
Isolate SMF 1, the most abundant sequence type in the substrate sample, represented 52 clones that were closely related to Clostridium sp. (90 to 98%). The second most abundant sequence type SMF 4 included 29 bacterial clones that were nearly related to Bacillus sp. (91 to 94%). The six bacterial clones represented by SMF13, the third most abundant sequence type, were related to Schineria larvae (98%) isolated from the 1st and 2nd larval stages of Wohlfahrtia magnifica (Diptera:Sarcophagidae) (Toth et al., 2001). The sequence type SMF24 included 4 bacterial clones that were related to Ureibacillus thermosphaericus (97%), isolated from an aerobic digestor of swine waste (Gagné et al., 2001). Sequence types represented by SMF90 (3 clones) were related to Lactobacillus amylotrophus strain NRRL B-4435 (94 to 98%) (Naser et al., 2006).
Sequence type SMF15 (5 clones) had no relationship to known cultivated bacteria members, but it was nearly related to the known sequence AF371535 (99%), isolated from pig gut bacterium (Leser et al., 2002). The phylotypes represented by SMF74 (1 clone) and SMF66 (1 clone) were related to the same known sequence AY167964, isolated from Swine manure bacterium (Whitehead and Cotta, 2004). Sequence type SMF75 (1 clone) was nearly related to AY167954 (99%), obtained from Swine manure bacterium (Whitehead and Cotta, 2004). Sequence type SMF76 (1 clone) was nearly related to AY263505 (98%), obtained from Bovine rumen bacterium (Wang et al., 2005). The sequence types SMF29 (1 clone), SMF46 (1 clone) and SMF67 (1 clone) were nearly related to a bacterium clone designated DQ327603 (97%), DQ325973 (99%) and DQ326459 (99%), respectively, which were all obtained from human distal gut microbiome (Gill et al., 2006). The sequence types SMF28 (1 clone), SMF47 (1 clone), SMF122 (1 clone) and SMF113 (1 clone) were nearly related to a bacterium clone designated AY916381 (99%), AY980362 (97%), AY985590 (91%) and AY916186 (91%), respectively, which were all obtained from human intestinal microbial flora (Eckburg et al., 2005).
DISCUSSION
Clostridium sp. was detected in the three all samples. It always predominated, being both the most abundant (41% of the total clones examined), and the most diverse (27% of the total sequence types detected). The genera Clostridum has been detected in manure (Peters et al., 2000; Guo et al., 2007) and chicken feather composts (Tiquia et al., 2005). The bacteria related to genera Clostridum were assumed to play an important role in the degradation of cellulose in the initial and thermophilic phases of composting (van Dyke and McCarthy 2002; Tiquia et al., 2005; Guo et al., 2007).
In the present study, we observed that members of the genus Bacillus and those belonging to the phylum Bacteroidetes were the second most abundant group (20% of the total clones examined) and the second most diverse group (10% of the total sequence types detected) in the three samples. Most of them displayed relatively high levels of similarity with cultivated Bacillus sp., which were generally detected in hot composts (Blanc et al., 1999). The number of clones related to the genera Bacillus is dominant in superstratum and substrate samples. The genus Bacillus has been known to be widely distributed in various compost raw materials (Sasaki et al., 2009). This was expected as Bacillus bacteria has the ability to decompose protein and complex polysaccharides, plays an important role in the organic matter degradation process, and are ubiquitous in nature.
Clones similar to Ureibacillus thermosphaericus and Pseudomonas sp. were also found in the three all samples. Thermophilic strains, identified as Ureibacillus thermosphaericus using 16S rRNA gene sequence analysis were isolated from an aerobic digestor of swine waste (Gagné et al., 2001). In the Gagné study, the Ureibacillus thermosphaericus strains were detected with esterase activity and were found to grow between 35°C and 65°C with strictly aerobic metabolism. Pseudomonas sp. bacteria can promote the swine manure composting digest and increase the quality of the composting products (Liu et al., 2003).
In this study, the representation of the domain bacteria was extremely diverse with 107 sequence types distributing among 36 well-established bacterial divisions, and 31 sequences were grouped within 29 novel, proposed bacterial divisions AF371706, AY916341, AF371603, AY858492, AJ576424, AF371768, AF407407, AF371800, AJ576399, AB185565, DQ353915, AY986352, DQ904840, DQ815725, AB185556, AF132255, AF371512, AY212772, AY916381, AF371535, DQ327603, DQ325973, AY980362, AY985590, AY916186, DQ326459, AY167964, AY167954 and AY263505. Sequence types grouping into genera or taxa that were identified by using classical culture-dependent methods and expected with known functions were retrieved, including Bacillus sp., Clostridium sp., Ureibacillus thermosphaericus, Turicibacter sanguinis, Cytophaga sp., Lactobacillus sp. and Schineria larvae. However, thirty-nine clones were similar to sequences in the database not assigned to any genera, indicating that a portion of bacterial species associated with the composting remains to be identified.
The difference of the bacterial community composition confirmed the presence of the phenomenon of gradient effect, and on the other hand the difference can reveal the nature of the gradient effect from the point of view of microbiology. There is a recent report that different layers of the same heterogeneous mat often contained distinct and different communities of cyanobacteria (Roeselers et al., 2007). Futhermore, the difference of the microbial distribution will lead to a difference of temperature, oxygen consumption and moisture content of the composting mixture that appears in different levels (Ekinci, 2001; Zhu et al., 2004; Zhu, 2005).
Only 33 of the sequence types had sequence similarity of 97 per cent or more to any previously cultured species for which the 16S rRNA sequence is available, i.e., only 24% of the phylotypes likely represented known bacterial species. Large fractions of uncharacterized phylotypes in 16S rDNA libraries have been found since the introduction of the rRNA approach to microbial ecology (Stahl et al., 1985; Giovannoni et al., 1990; Ward et al., 1990; DeLong, 1992). This indicates that bacterial diversity in natural systems extends far beyond what can currently be perceived from traditional culturing methods (Stahl, 1995; Pace, 1996; Hugenholtz et al., 1998; Olsson et al., 2003; Schwarza et al., 2007).
This paper investigated the spatial distribution of bacterial community in the hot swine manure composting by using molecular tools, and tried to annotate the reason for the phenomenon of gradient effect. However, the function and spatial distribution of microbial community composition should be subjected to the future investigations. To monitor the continuous and dynamic succession progress, more samples at different times will be collected and analysised by using PCR-DGGE technology, which will enhance the understanding of the role and the succession order of the microbial community.
ACKNOWLEDGEMENTS
This work was financially supported by the National Natural Science Foundation of China (30471271), International Foundation for Science (W/4370-1), “11th five year” National Science and Technology Program (2006BAD14B05-2), The Emphasis Science and Technology Program of Hubei Province (2006AA201B104), and Natural Science Foundation of Guangdong Province (04300060).
REFERENCES
- Alfreider A, Vogt C, Babel W. Microbial diversity in an in situ reactor system treating monochlorobenzene contaminated groundwater as revealed by 16S ribosomal DNA analysis. Syst Appl Microbiol. 2002;25:232–240. doi: 10.1078/0723-2020-00111. [DOI] [PubMed] [Google Scholar]
- Arbeli Z, Fuentes CL. Improved purification and PCR amplification of DNA from environmental samples. FEMS Microbiol Lett. 2007;272:269–275. doi: 10.1111/j.1574-6968.2007.00764.x. [DOI] [PubMed] [Google Scholar]
- Barns SM, Fundyga RE, Jeffries MW, Pace NR. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc Natl Acad Sci USA. 1994;91:1609–1613. doi: 10.1073/pnas.91.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M, Marilley L, Beffa T, Aragno M. Thermophilic bacterial communities in hot composts as revealed by most probable number counts and molecular (16S rDNA) methods. FEMS Microbiol Ecol. 1999;28:141–149. [Google Scholar]
- Bosshard PP, Zbinden R, Altwegg M. Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, Gram-positive bacterium. Int J Syst Evol Microbiol. 2002;52:1263–1266. doi: 10.1099/00207713-52-4-1263. [DOI] [PubMed] [Google Scholar]
- Dees PM, Ghiorse WC. Microbial diversity in hot synthetic compost as revealed by PCR-amplified rRNA sequences from cultivated isolates and extracted DNA. FEMS Microbiol Ecol. 2001;35:207–216. doi: 10.1111/j.1574-6941.2001.tb00805.x. [DOI] [PubMed] [Google Scholar]
- DeLong EF. Archaea in coastal marine environments. Proc Natl Acad Sci USA. 1992;89:5685–5689. doi: 10.1073/pnas.89.12.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dojka MA, Hugenholtz P, Haack SK, Pace NR. Microbial diversity in a hydrocarbon- and chlorinated-solvent contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol. 1998;64:3869–3877. doi: 10.1128/aem.64.10.3869-3877.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egert M, Wagner B, Lemke T, Brune A, Friedrich MW. Microbial community structure in midgut and hindgut of the humus-feeding larva of Pachnoda ephippiata(Coleoptera: Scarabaeidae) Appl Environ Microbiol. 2003;69:6659–6668. doi: 10.1128/AEM.69.11.6659-6668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekinci K. PhD Thesis. The Ohio State University; Columbus, Ohio, USA: 2001. Theoretical and experimental studies on the effects of aeration strategies on the composting process. [Google Scholar]
- Frey JC, Rothman JM, Pell AN, Nizeyi JB, Cranfield MR, Angert ER. Fecal bacterial diversity in a wild gorilla. Appl Environ Microbiol. 2006;72:3788–3792. doi: 10.1128/AEM.72.5.3788-3792.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagné A, Chicoine M, Morin A, Houde A. Phenotypic and genotypic characterization of esterase-producing Ureibacillus thermosphaericus isolated from an aerobic digestor of swine waste. Can J Microbiol. 2001;47:908–1015. doi: 10.1139/w01-096. [DOI] [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Britschgi TB, Moyer CL, Field KG. Genetic diversity in Sargasso Sea bacterioplankton. Nature. 1990;345:60–63. doi: 10.1038/345060a0. [DOI] [PubMed] [Google Scholar]
- Guo Y, Zhu N, Zhu S, Deng C. Molecular phylogenetic diversity of bacteria and its spatial distribution in composts. J Appl Microbiol. 2007;103:1344–1354. doi: 10.1111/j.1365-2672.2007.03367.x. [DOI] [PubMed] [Google Scholar]
- Hassen A, Belguith K, Jedidi N, Cherif A. Microbial characterization during composting of municipal solid waste. Bioresour Technol. 2001;80:217–225. doi: 10.1016/s0960-8524(01)00065-7. [DOI] [PubMed] [Google Scholar]
- Heuer H, Krsek M, Baker P, Smalla K, Wellington EMH. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol. 1997;63:3233–3241. doi: 10.1128/aem.63.8.3233-3241.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraishi A, Narihiro T, Yamanaka Y. Microbial community dynamics during start-up operation of flowerpot-using fed-batch reactors for composting of household biowaste. Environ Microbiol. 2003;5:765–776. doi: 10.1046/j.1462-2920.2003.00473.x. [DOI] [PubMed] [Google Scholar]
- Holben WE. Isolation and purification of bacterial DNA from soil. In: Weaver RW, Angle S, Bottomley P, editors. Methods of soil analysis. Part 2. Microbiogical and Biochemical Properties. Soil Science Society of America; Madison, WI, USA: 1994. pp. 727–751. [Google Scholar]
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbeah M. Composting piggery waste: a review. Bioresour Technol. 1998;63:197–203. [Google Scholar]
- LaMontagne MG, Michel FC, Holden PA, Reddy CA. Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J Microbiol Methods. 2002;49:255–264. doi: 10.1016/s0167-7012(01)00377-3. [DOI] [PubMed] [Google Scholar]
- Lane DJ. 16S⁄23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic Acid Techniques in Bacterial Systematics. New York: Wiley; 1991. pp. 115–175. [Google Scholar]
- Larsen N, Olsen GJ, Maidak BL, McCaughey MJ, Overbeek R, Macke TJ, Marsh TL, Woese CR. The ribosomal database project. Nucleic Acids Res. 1993;21:3021–3023. doi: 10.1093/nar/21.13.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leser TD, Amenuvor JZ, Jensen TK, Lindecrona RH, Boye M, Moller K. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl Environ Microbiol. 2002;68:673–690. doi: 10.1128/AEM.68.2.673-690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Liu Y, Lei Z, Li C, Shi A. Effects of different microorganism consortiums on quality of composting pig-dung. J Agro-Environ Sci. 2003;22:311–314. [Google Scholar]
- Muyzer G, de Waal EC, Uitterlinden AG. Profiling of microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction amplified genes coding for 16S rRNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naser SM, Vancanneyt M, Snauwaert C, Vrancken G, Hoste B, De Vuyst L, Swings J. Reclassification of Lactobacillus amylophilus LMG 11400 and NRRL B-4435 as Lactobacillus amylotrophicus sp. nov. Int J Syst Evol Microbiol. 2006;56:2523–2527. doi: 10.1099/ijs.0.64463-0. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Zinder SH, Hance I, Burr P, Odongo D, Wasawo D, Odenyo A, Bishop R. Phylogenetic analysis of the microbial populations in the wild herbivore gastrointestinal tract: insights into an unexplored niche. Environ Microbiol. 2003;5:1212–1220. doi: 10.1046/j.1462-2920.2003.00526.x. [DOI] [PubMed] [Google Scholar]
- Olsson C, Ahrné S, Pettersson B, Molin G. The bacterial flora of fresh and chill-stored pork: analysis by cloning and sequencing of 16S rRNA genes. Int J Food Microiol. 2003;83:245–252. doi: 10.1016/s0168-1605(02)00372-0. [DOI] [PubMed] [Google Scholar]
- Ozutsumi Y, Tajima K, Takenaka A, Itabashi H. The effect of protozoa on the composition of rumen bacteria in cattle using 16S rRNA gene clone libraries. Biosci Biotechnol Biochem. 2005;69:499–506. doi: 10.1271/bbb.69.499. [DOI] [PubMed] [Google Scholar]
- Pace NR. New perspective on the natural microbial world: molecular microbial ecology. ASM News. 1996;62:463–470. [Google Scholar]
- Peters S, Koschinsky S, Schwieger F, Tebbe CC. Succession of microbial communities during hot composting as detected by PCR-single-strand-conformation polymorphism -based genetic profiles of small-subunit rRNA genes. Appl Environ Microbiol. 2000;66:930–936. doi: 10.1128/aem.66.3.930-936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–433. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeselers G, Norris TB, Castenholz RW, Rysgaard S, Glud RN, Kühl M, Muyzer G. Diversity of phototrophic bacteria in microbial mats from Arctic hot springs (Greenland) Environ Microbiol. 2007;9:26–38. doi: 10.1111/j.1462-2920.2006.01103.x. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nonaka J, Otawa K, Kitazume O, Asano R, Sasaki T, Nakai Y. Analysis of the structure of the bacterial community in the livestock manure-based composting process. Asian-Aust J Anim Sci. 2009;22:113–118. [Google Scholar]
- Schwarza JIK, Eckertb W, Conrad R. Community structure of Archaea and Bacteria in a profundal lake sediment Lake Kinneret (Israel). Syst. Appl Microbiol. 2007;30:239–254. doi: 10.1016/j.syapm.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Simpson JM, Santo JW, Domingo, Reasoner DJ. Assessment of equine fecal contamination: the search for alternative bacterial source-tracking targets. FEMS Microbiol Ecol. 2004;47:65–75. doi: 10.1016/S0168-6496(03)00250-2. [DOI] [PubMed] [Google Scholar]
- Stahl DA. Application of phylogenetically based hybridization probes to microbial ecology. Mol Ecol. 1995;4:535–542. [Google Scholar]
- Stahl DA, Lane DJ, Olsen GJ, Pace NR. Characterization of a Yellowstone hot spring microbial community by 5S rRNA sequences. Appl Environ Microbiol. 1985;49:1379–1384. doi: 10.1128/aem.49.6.1379-1384.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stentiford EI. Recent developments in composting. In: de Bertoldi M, Ferranti M, L’Hermite P, Zuicconi F, editors. Compost, Production, Quality and Use. Elsevier; London: 1987. pp. 52–60. [Google Scholar]
- Stentiford EI. Composting control: principles and practice. Chapman and Hall; London, UK: 1996. [Google Scholar]
- Strom PF. Effect of temperature on bacterial species diversity in thermophilic solid waste composting. Appl Environ Microbiol. 1985a;50:899–905. doi: 10.1128/aem.50.4.899-905.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom PF. Identification of thermophilic bacteria in solid waste composting. Appl Environ Microbiol. 1985b;50:906–913. doi: 10.1128/aem.50.4.906-913.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP: Phylogenetic Analysis Using Parsimony. Version 4.0. Champaign, IL: Illinois Natural History Survey; 1999. [Google Scholar]
- Tiquia SM, Ichida JM, Keener HM, Elwell DL, Jr, Burtt EH, Michel FC., Jr Bacterial community profiles on feathers during composting as determined by terminal restriction fragment length polymorphism analysis of 16S rDNA genes. Appl Microbiol Biotechnol. 2005;67:412–419. doi: 10.1007/s00253-004-1788-y. [DOI] [PubMed] [Google Scholar]
- Van Dyke MI, McCarthy AJ. Molecular biological detection and characterization of Clostridium populations in municipal landfill sites. Appl Environ Microbiol. 2002;68:2049–2053. doi: 10.1128/AEM.68.4.2049-2053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wintzingerode F, Gbel UB, Stackebrandt E. Determination of microbial diversity in environmental samples: pit-falls of PCR-based rRNA analysis. FEMS Microbiol Rev. 1997;21:213–229. doi: 10.1111/j.1574-6976.1997.tb00351.x. [DOI] [PubMed] [Google Scholar]
- Wang XL, Shin KH, Hur HG, Kim SI. Enhanced biosynthesis of dihydrodaidzein and dihydrogenistein by a newly isolated bovine rumen anaerobic bacterium. J Biotechnol. 2005;115:261–269. doi: 10.1016/j.jbiotec.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Ward DM, Weller R, Bateson MM. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature. 1990;345:63–65. doi: 10.1038/345063a0. [DOI] [PubMed] [Google Scholar]
- Wei Y. PhD thesis. Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences; 2000. Efficient and economical composting of sewage sludge for small and mid-scale municipal wastewater treatment plants. [Google Scholar]
- Whitehead TR, Cotta MA. Isolation and identification of hyper-ammonia producing bacteria from swine manure storage pits. Curr Microbiol. 2004;48:20–26. doi: 10.1007/s00284-003-4084-7. [DOI] [PubMed] [Google Scholar]
- Wu L, Li FG, Deng CY, Xu DQ, Jiang SW, Xiong YZ. A method for obtaining DNA from compost. Appl Microbiol Biotechnol. 2009;84:389–395. doi: 10.1007/s00253-009-2103-8. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Xiao Y, Zeng GM, Xu ZY, Liu YS. Comparision of methods for total commmunity DNA extraction and purification from compost. Appl Microbiol Biotechnol. 2007;74:918–925. doi: 10.1007/s00253-006-0704-z. [DOI] [PubMed] [Google Scholar]
- Zhu N. Temporal-spatial distribution of pile temperature of aerated static bin composting system. J South China Univ Technol. 2005;33:19–23. [Google Scholar]
- Zhu N. Composting of high moisture content swine manure with corncob in a pilot scale system. Bioresour Technol. 2006;97:1870–1875. doi: 10.1016/j.biortech.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Zhu N. Effect of low initial C/N ratio on the composting of swine manure with rice straw. Bioresour Technol. 2007;98:9–13. doi: 10.1016/j.biortech.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Zhu N, Deng C, Xiong Y, Qian H. Performance characteristics of three aeration systems in the swine manure composting. Bioresour Technol. 2004;95:319–326. doi: 10.1016/j.biortech.2004.02.021. [DOI] [PubMed] [Google Scholar]