

Abstract
Background:
Rosai–Dorfman disease (RDD) is a rare benign histioproliferative disease. It is typically characterized by benign histiocyte proliferation with lymphadenopathy, fever, and leukocytosis and was first described in 1969 by Rosai and Dorfman. Extranodal involvement has been reported in approximately up to 43% of the cases with isolated central nervous system (CNS) manifestations being even rarer.
Case Description:
We report our management of a 41-year-old female with extranodalpurely CNS RDD presenting as a benign scalp lump. Her lump progressed from an asymptomatic benign lesion to one causing localized cerebral edema. Treatment was surgical excision of both the cervical and CNS lesions achieving complete removal of the lesions and resolution of her symptoms.
Conclusion:
RDD is a rare condition and isolated CNS RDD is even less common. Benign scalp lumps have a myriad of differential diagnoses, but RDD should be a consideration in the presence of preexisting RDD lesions at other sites given its potential to progress and result in morbidity. It is imperative to be aware that symptoms may be especially deceiving as the absence of lymphadenopathy may point away from RDD as the diagnosis.
Keywords: Intracranial, Rosai–Dorfman, spinal, surgery
INTRODUCTION
Rosai–Dorfman disease (RDD) or sinus histiocytosis with massive lymphadenopathy (SHML) was first described in 1965 by Destombes and then recognized as a clinically distinct pathology by Dorfman and Rosai in 1969.[15] It is a rare, benign, lymphoproliferative disorder characterized by “emperipolesis”.[6] They coined the term SHML. In its classic description, the patients tend to be young males in their twenties or thirties and present with bilateral painless cervical lymphadenopathy, fever, and weight loss.[22] In one-third cases, extranodal involvement is present and most common sites are the skin, upper respiratory tract, thyroid, or testes.[17]
About 5% of cases involve the central nervous system (CNS) and in such circumstances, patients tend to be from an older age group (mean 39.4 years).[5] Isolated intracranial RDD has been documented in about 50 previous case reports.[19] They are mostly dural-based lesions presenting clinically and radiologically like meningiomas. The natural history of the disease is usually nonprogressive and self-limiting with symptoms often due to local mass effect such as dysphagia, shortness of breath and proptosis depending on the site of the disease. However, the disease can sometimes manifest as autoimmune hemolyticanemia with massive hemoptysis.[7] We report a rare case of intracranial RDD with a concomitant spinal lesion, how it initially presented as a benign scalp lump, and describe our management of this rare disorder.
CASE REPORT
A 41-year-old Indian female first presented to the outpatient clinic complaining of a tender lump over her right parieto-occipital region. A computed tomography (CT) scan of her head was done, revealing a small, well defined lytic lesion of the skull extending from the inner table to the outer table. The initial impression was that the lump was consistent with sinus pericranii [Figure 1].
Figure 1.
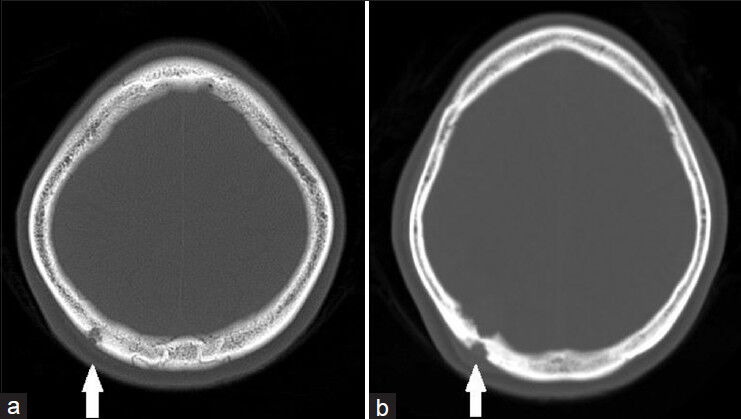
(a) Initial CT brain bone window axial cuts showing lesion extending from the inner table to the outer table of the skull vault with fairly well-defined margin, in keeping with sinus pericranii with no other lytic abnormality seen in the skull vault and (b) progression of the lesion 2 years later with increased extent of bony involvement (white arrow)
She presented to an outside hospital 1 year later with left shoulder pain weakness and numbness of both lower extremities. On examination, power in both lower limbs was 4 out of 5 but there were no motor deficits in her upper limbs. There was mild sensory deficit to light touch and pain over her left cervical segment 8 (C8) and thoracic segment 1 (T1) dermatomes. There were no cerebellar signs or higher mental function deficits. No lymphadenopathy was clinically evident and biochemically there were no abnormalities. A contrast magnetic resonance imaging (MRI) scan of her cervical spine was done, which showed an extradural lesion extending from cervical segment 7 (C7) to thoracic segment 2 (T2) occupying the left posterolateral spinal canal. She underwent a C7 to T2 laminectomy, excision of lesion and instrumentation of the spine. The histological diagnosis was that of RDD.
She subsequently presented to our institute 2 months after surgery with numbness extending from her left mid torso to her left leg. A contrast MRI scan of the brain and spine was done. It showed enhancement in the first and second thoracic segments’ left lateral recesses and neural foramina likely representing postsurgical changes in the absence of residual tumor. It also revealed the previously noted osteolytic lesion involving the right posterior parietal bone, which now had extradural and extracalvarial enhancing soft tissue components [Figure 2]. However, she was asymptomatic from the cranial lesion, erthyrocyte sedimentation rate (ESR) was not elevated, there was no lymphocytosis, and gamma-globulin levels were normal. In addition, the lesion was isolated and small, thus conservative management was adopted and she was placed on outpatient observation with close follow-up.
Figure 2.
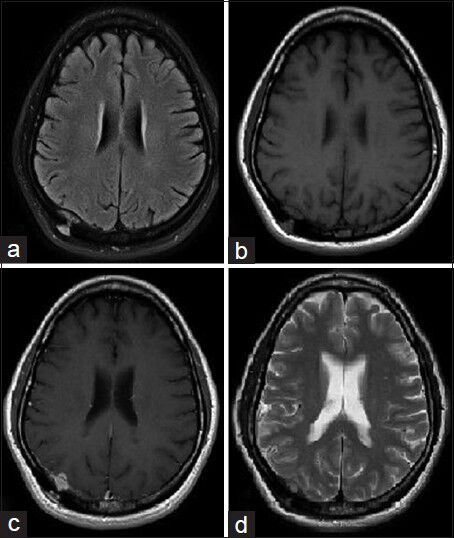
Initial MRI brain axial cuts (a) T2 Flair (b) T1-weighted (c) T1-weighted with gadolinium contrast (d) T2-weighted sequences showing an hour glass shaped 2.1 × 1.6 cm T2 isointense enhancing extradural lesion in the right supraventricular parietal cortex with its waist extending through the calvarium, a subgaleal component beneath the skin and an intracranial component indenting the parietal cortex
A follow-up MRI of her brain and spine was done 4 months later, which showed resolution of the enhancement seen at the region of the spinal lesion. The brain lesion showed marginal increase in size but as the patient continued to remain asymptomatic, we elected to continue observation with close follow-up and interval imaging.
Her subsequent follow-up MRI brain at 6 months now showed a significant interval increase in the size of the right parieto-occipital extraaxial dural-based enhancing soft-tissue lesion. Dimensions were approximately 2 × 3.2 × 1.7 cm in maximum orthogonal dimensions. It also showed an interval increase in protrusion into the underlying right cerebrum with significant interval development of vasogenicedema in the right parieto-occipital lobe, extending to reach the right atrial subependyma. The lesion was transcalvarial with a subgaleal component, which also shows interval increase in size [Figure 3]. Biochemically, she had markedly raised lactate dehydrogenase levels (LDH) (1198, reference: 222-454 U/L) and lymphocytosis.
Figure 3.
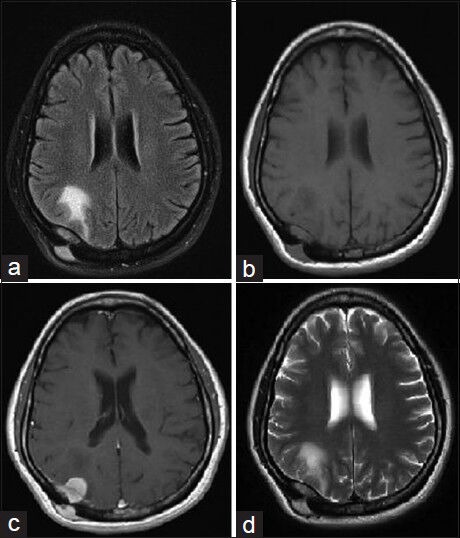
Follow up MRI brain axial cuts (a) T2 Flair (b) T1-weighted (c) T1-weighted with gadolinium contrast (d) T2-weighted sequences showing significant interval progressive increase in size of the rightparieto-occipital extraaxial dural base enhancing soft-tissue lesion, now measuring approximately 2 × 3.2 × 1.7 cm in maximum orthogonal dimensions. Interval increase in protrusion into the underlying right cerebrum with significant interval development of vasogenic edema in the right parieto-occipital lobe, extending to reach the right atrial subependyma. The lesion is transcalvarial with a subgaleal component, which also showed interval increase in size
The patient was due for outpatient review a week after the MRI brain, but in view of the finding, she was quickly contacted to assess her clinical state. She had, however, presented to the emergency department with headaches, neck stiffness, increasing drowsiness, and nausea, and was thus admitted for treatment. She was treated with dexamethasone 4 mg three times a day and analgesics. Her symptoms subsequently improved. We decided to take her for surgery because of the increasing size of the lesion, vasogenic cerebral edema secondary to the lesion as evident on her scan, and evidence of raised intracranial pressure (ICP). She underwent a craniotomy and excision of the lesion. The tumor was found to be an intradiploic tumor eroding into surrounding soft tissue superficially and deep with dural involvement and extension into the brain. The lesion was excised en-bloc and the calvarial defect was closed with an acrylic cranioplasty. The patient made an uneventful recovery. On last follow-up, a month after surgery, a first time positron emission tomography-computed tomography (CT-PET) was done to assess for residual disease or recurrence and also screen for systemic disease. This showed successful removal of the intracranial lesion with no other systemic lesions.
Histopathology showed the lesion to be involving both the inner and outer tables of the skull as well as the dura mater. There was nodular proliferation of foamy histiocytes, with admixture by lymphocytes, plasma cells and neutrophils, with numerous foci of sclerosedstroma. Immunohistochemistry staining of the histiocytes was positive for S-100 and CD68 and negative for CD1a. In the dura, the histiocytic proliferation showed intervening foci of sclerosedstroma [Figure 4]. These findings were highly diagnostic of RDD.
Figure 4.
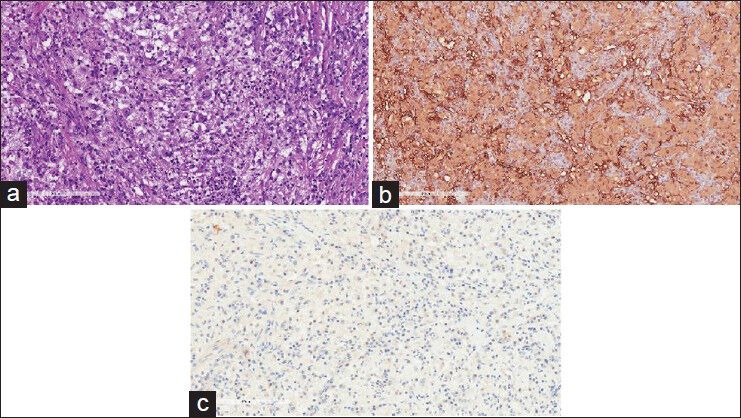
Histological images of (a) Pale staining enlarged histiocytes admixed with other inflammatory cells and with foci of emperipolesis (b) S100 immunostainpositive in larger histiocytes (c) CD1a immunostain negative in larger histiocytes
DISCUSSION
RDD though uncommon is a well described entity. A cytokine-mediated migration of monocytes has been postulated to be involved in histiocyte accumulation and activation. The triggering stimuli seem to be varied. Due to the observed coexistence of RDD and autoimmune diseases, hematological malignancies and postinfectious conditions are hypothesized causes. For example, RDD has been reported following bone marrow transplant for acute lymphoblastic leukemia, and concurrently or after various forms of lymphoma. Many viruses like herpesvirus 6 (HHV-6) and Epstein-Barr virus (EBV) have also been implicated as potential causative agents. However, there is no strong evidence for this at the moment.[14]
Perhaps that explains why biochemically and hematologically, the presence of neutrophilia, a high ESR, elevation of serum LDH, rouleaux formation of red blood cells, polyclonal hypergammaglobulinemia, and hypermetabolism in lymph nodes and spleen on CT-PET are suggestive for the diagnosis.[20]
RDD is a benign disorder with most patients being young males (male to female ratio 3:1)[3] who present with painless lymphadenopathy and systemic symptoms of fever and weight loss. Extranodal disease is rare. Isolated CNS involvement is even less reported in only 5% of cases.[11] To date, the largest review of pure extranodal RDD was by Andriko et al.,[2] who described 11 cases of isolated CNS RDD without nodal disease, and suggested the ability of this entity to mimic meningiomas, lymphomas, and chronic inflammation. Patients who present with intracranial disease often become symptomatic at a later mean age (34.9).[9] A review of the literature showed that to date there are about 50 reported cases of isolated extranodal CNS RDD described in 6 other articles.[1,3,8,10,13,16,18] Most cases have a florid presentation with seizures or headaches and neurological deficits like visual loss and limb weakness or numbness being most common.
Our patient was a middle-aged female with pure CNS disease. Her initial presentation of the painful scalp lump could have been attributed to the intracalvarial lesion but interestingly, the diagnosis was not made then as it had features suggesting sinuspericranii. Her main symptom onset was almost a year and a half later due to the mass effect of the spinal lesion and it took 2 years for progression of the intracranial lesion before it resulted in significant perilesionaledema. She did not have any systemic manifestations and radiologically, the intracranial lesion had typical meningioma features. This highlights how deceivingly benign an RDD lesion may appear initially.
Our patient's histopathology was typical for RDD. Diagnosis is based on histopathology and immunohistochemistry. In extranodal sites, lesions tend to recapitulate the sinus growth in lymph node with light-staining bands alternating with dark-staining bands. The light-staining bands are formed by aggregates of the distinctive Rosai–Dorfman histiocytes, while the dark-staining bands are formed by lympho-plasmacytic cells. The distinctive Rosai–Dorfman histiocytes predominate in the paler foci–these are very large cells with round vesicular nuclei, central nucleoli, and voluminous pale cytoplasm. Some of these cells can have enlarged or hyperchromatic nuclei, mimicking a malignant neoplasm. Emperipolesis can be difficult to appreciate. Regressive changes can be present, with decreased Rosai–Dorfman histiocytes, accumulation of foamy histiocytes, and fibrosis. As a result of fibrosis and spindling of some of the Rosai-Dorfman histiocytes, the histologic picture can mimic fibrohistiocytic tumor or inflammatory pseudo tumor.[21]
Surgical excision is the treatment of choice though there have been reports of regression of intracranial RDD with the use of long-term prednisolone.[4] In most cases, surgery is indicated only when clear disease progression causing significant morbidity is documented. Pulsoni et al. reported in their analysis of the available literature for various treatment methods of RDD (i.e., chemotherapy, immunosuppression, surgery, radiotherapy) that surgery is clearly beneficial and indicated in nodal or extranodal RDD of vital organs causing compression or other life-threatening manifestations.[12] It showed that surgical excision of resectable lesions induced complete remission in eight out of nine patients (with the last one lost to follow up) albeit not all the patients had pure CNS RDD.
As RDD tends to arise extradurally, surgical resection of our patient's spinal lesion was beneficial to alleviate compression of the cord with a low risk of entering dura and causing cord damage. As for the intracranial lesion, in view of the superficial location, relatively easy access, as well as evidence of raised ICP with vasogenic edema and mass effect, surgery was indicated. The considerations were that of low morbidity and a high chance to achieve full clearance. As it turned out, our patient recovered well with complete resolution of symptoms, no neurological deficits and no need for steroids.
CONCLUSION
RDD is a benign, histioproliferative disorder, which usually manifests as painless massive lymphadenopathy. Pure CNS involvement is fairly rare and is less commonly seen in middle-aged females. It may sometimes present as other benign lesions but should be a consideration given its potential to cause significant morbidity. When indicated, surgical excision is the treatment of choice and potentially offers cure if complete removal of the lesion is achieved. This avoids the need for long-term steroid use and its side effects. Our patient is a clear example of the presentation, investigation, and management of isolated CNS RDD.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2014/5/1/99/134912
Contributor Information
Min Wei Chen, Email: minwei.chen@mohh.com.sg.
Nicolas K. K. King, Email: Nicolas_kon@nni.com.sg.
Sathiyamoorthy Selvarajan, Email: sathiyamoorthy.selvarajan@sgh.com;.
David C. Y. Low, Email: David.Low.CY@kkh.com.sg.
REFERENCES
- 1.Ambekar S, Sommana S, Bhat DI, Ranjan M. Isolated Cranio-spinal Involvement of Rosai-Dorfman disease: Case report. Br J Neurosurg. 2011;25:297–9. doi: 10.3109/02688697.2010.508849. [DOI] [PubMed] [Google Scholar]
- 2.Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system. A report of 11 cases. Mod Pathol. 2001;14:172–8. doi: 10.1038/modpathol.3880278. [DOI] [PubMed] [Google Scholar]
- 3.Antuña Ramos A, Alvarez Vega MA, Alles JV, Antuña Garcia MJ, MeilánMartínez A. Multiple involvement of the central nervous system in Rosai-Dorfman disease. Pediatr Neurol. 2012;46:54–6. doi: 10.1016/j.pediatrneurol.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Mcpherson CM, Brown J, Kim AW, DeMonte F. Regression of intracranial Rosai-Dorfman disease following corticosteroid therapy. J Neurosurg. 2006;104:840–4. doi: 10.3171/jns.2006.104.5.840. [DOI] [PubMed] [Google Scholar]
- 5.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai- Dorfman disease): Review of the entity. Semin Diagn Pathol. 1990;7:19–73. [PubMed] [Google Scholar]
- 6.Humble JG, Jaynee WH, Pulvertaft RJ. Biological interaction between lymphocyte and other cells. Br J Hematol. 1956;2:283–94. doi: 10.1111/j.1365-2141.1956.tb06700.x. [DOI] [PubMed] [Google Scholar]
- 7.Varadarajalu L, Thirugnanasambandan S, Thangaraj P. J, Parameswaran A, Mathew M. A Rare Presentation of Rosai Dorfman Disease. Chest. 2012;142:998A–998A. 4_MeetingAbstracts. [Google Scholar]
- 8.Kaminsky J, Koerbel A, Mittelbronn M, Beschorner R, Ernemann U, Tatagiba M. Rosai-Dorfman disease involving the cranial base, paranasal sinuses and spinal cord. Clin Neuropathol. 2005;24:194–200. [PubMed] [Google Scholar]
- 9.Kattner KA, Stroink AR, Roth TC, Lee JM. Rosai-Dorfman disease mimicking parasagittal meningioma: Case presentation and review of literature. Surg Neurol. 2000;53:452–7. doi: 10.1016/s0090-3019(00)00197-x. [DOI] [PubMed] [Google Scholar]
- 10.Kidd DP, Revesz T, Miller NR. Rosai-Dorfman disease presenting with widespread intracranial and spinal cord involvement. Neurology. 2006;67:1551–5. doi: 10.1212/01.wnl.0000242893.55416.8e. [DOI] [PubMed] [Google Scholar]
- 11.Lungren MP, Petrella JR, Cummings TJ, Grant GA. Grant Isolated intracranial Rosai-Dorfman disease in a child. AJNR Am J Neuroradiol. 2009;30:E148–9. doi: 10.3174/ajnr.A1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulsoni A, Anghel G, Falcucci P, Matera R, Pescarmona E, Ribersani M, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Report of a case and literature review. Am J Hematol. 2002;69:67–71. doi: 10.1002/ajh.10008. [DOI] [PubMed] [Google Scholar]
- 13.Raslan O, Ketonen LM, Fuller GN, Schellingerhout D. Intracranial Rosai-Dorfman disease with relapsing spinal lesions. J Clin Oncol. 2008;26:3087–9. doi: 10.1200/JCO.2007.15.7701. [DOI] [PubMed] [Google Scholar]
- 14.Abla O. Rosai-Dorfman Disease. Histiocyte Society. 2011 [Google Scholar]
- 15.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63–70. [PubMed] [Google Scholar]
- 16.Sato A, Sakurada K, Sonoda Y, Saito S, Kayama T, Jokura H, et al. Rosai-Dorfman disease presenting with multiple intracranial and intraspinal masses: A case report. No Shinkei Geka. 2003;31:1199–204. [PubMed] [Google Scholar]
- 17.Sanchez R, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: An analysis of 113 cases with special emphasis on its extranodal manifestation. Lab Invest. 1977;36:21–2. [Google Scholar]
- 18.Seyednejad F, Tubbs RS, Shoja MM, Daghigi MH, Oakes WJ. Presumed recurrence of intracranial Rosai-Dorfman disease as a cervical spine tumor. Acta Neurochir (Wien) 2007;149:425–7. doi: 10.1007/s00701-007-1125-1. [DOI] [PubMed] [Google Scholar]
- 19.Sundaram C, Uppin SG, Prasad BC, Sahu BP, Devi MU, Prasad VS, et al. Isolated RosaiDorfman disease of the central nervous system presenting as dural based and intraparenchymal lesions. Clin Neuropathol. 2005;24:112–7. [PubMed] [Google Scholar]
- 20.Rew SY, Jang HC, Park KH, Ahn JS, Kim GE, Choi YD, et al. A case of Rosai-Dorfman Disease with highly elevated serum ferritin. Ann Lab Med. 2012;32:158–61. doi: 10.3343/alm.2012.32.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnes L. Surgical pathology of the head and neck. Informa healthcare. 2009;2:1023–4. 2008. [Google Scholar]
- 22.Warnke RA, Weiss LM, Chan JK, Cleary ML, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) In: Rosai J, editor. Atlas of Tumor Pathology, 3rd Series: Tumors of the Lymph Nodes and Spleen. Washington: Armed Forces Institute of Pathology; 1995. pp. 349–60. [Google Scholar]