

Abstract
This study reports the metagenomic detection and complete genome characterization of a novel turkey picornavirus from faecal samples of healthy (1/3) and affected (6/8) commercial turkeys with enteric and/or stunting syndrome in Hungary. The virus was detected at seven of the eight farms examined. The turkey/M176-TuASV/2011/HUN genome (KC465954) was genetically different from the currently known picornaviruses of turkey origin (megriviruses and galliviruses), and showed distant phylogenetic relationship and common genomic features (e.g. uncleaved VP0 and three predicted and unrelated 2A polypeptides) to duck hepatitis A virus (DHAV) of the genus Avihepatovirus. The complete genome analysis revealed multiple distinct genome features like the presence of two in-tandem aphthovirus 2A-like sequence repeats with DxExNPG/P ‘ribosome-skipping’ sites (76 %, 23/30 amino acids identical), with the first aphthovirus 2A-like sequence being located at the end of the VP1 capsid protein (VP1/2A1 ‘ribosome-skipping’ site). The phylogenetic analyses, low sequence identity (33, 32 and 36 % amino acid identity in P1, P2 and P3 regions) to DHAV, and the type II-like internal ribosome entry site suggests that this turkey picornavirus is related to, but distinct from the genus Avihepatovirus and it could be the founding member of a novel Avihepatovirus sister-clade genus. This is the third, taxonomically highly distinct picornavirus clade identified from turkeys exhibiting varied symptoms.
Introduction
The family Picornaviridae is currently divided into 12 officially recognized genera (Aphthovirus, Avihepatovirus, Cardiovirus, Enterovirus, Erbovirus, Hepatovirus, Kobuvirus, Parechovirus, Sapelovirus, Senecavirus, Teschovirus and Tremovirus) and five formally proposed genera (‘Aquamavirus’, ‘Cosavirus’, ‘Dicipivirus’, ‘Megrivirus’ and ‘Salivirus’) (Knowles et al., 2012).
Picornaviruses are small, non-enveloped viruses with single-stranded, positive-sense genomic RNA. In general, the 7.2–9.1 kb polyadenylated picornaviral genomes have a common organization pattern: viral polyprotein coding a single ORF [except for the proposed genus ‘Dicipivirus’, which has two ORFs separated by a UTR containing a second internal ribosomal entry site (IRES)] is flanked by the highly structured 5′ and 3′ UTRs (Racaniello, 2007; Woo et al., 2012). Generally, the viral polyprotein is processed to form several proteins: three or four capsid monomer proteins (VP4-VP2-VP3-VP1 or VP0-VP3-VP1) and at least seven non-structural proteins (2A-2B-2C-3A-3B-3C-3D); however, slight differences occur in picornaviruses of different genera. The numbers of the cleaved individual proteins can be influenced by the presence or absence of several genomic features e.g. presence of a leader (L) protein upstream of the capsid proteins, the cleavage of VP0 into VP4 and VP2 or the cleavage of 2A (Racaniello, 2007; Semler and Ertel, 2008). Most of the picornaviruses have only one mature 2A, although some picornaviruses, e.g. ‘swine pasivirus’ (SPaV), ‘bluegill picornavirus’ (BGPV) and Ljungan virus (LV) of the genus Parechovirus, are suggested to have two or, in case of duck hepatitis A viruses (DHAV) of the genus Avihepatovirus, up to three individual 2A proteins (Sauvage et al., 2012; Barbknecht, 2009, Johansson et al., 2002; Tseng and Tsai, 2007, Tseng et al., 2007). However, there is some uncertainty about the actual release of all of these mature 2A peptides during the viral infection cycle (Johansson et al., 2003; Tseng et al., 2007). The presence of an aphthovirus 2A-like ‘ribosome-skipping’ motif DxExNPG/P (where x is a non-conserved amino acid) is common among picornaviruses e.g. members of the genera Aphthovirus, Cardiovirus, Avihepatovirus, Erbovirus, Senecavirus and Teschovirus and a number of recently identified picornaviruses e.g. ‘Hungarovirus’, ‘Aquamavirus’ and ‘Pasivirus’ (Knowles et al., 2012; Reuter et al., 2012, Kapoor et al., 2008b; Sauvage et al., 2012).
Birds are well-known reservoirs of numerous viral pathogens such as avian influenza virus, West Nile virus and Japanese encephalitis virus in humans (Woo et al., 2006, 2010). However, compared with the thousands of known bird species (~10 000), rather few picornavirus species (n = 15) have been described from avian sources and ten of these have been identified in the present decade. The ‘avian’ picornaviruses are currently classified into three official genera: Tremovirus [avian encephalomyelitis virus 1 (AEV-1)], Avihepatovirus (DHAV) and Sapelovirus (avian sapelovirus 1) (Knowles et al., 2012), and five candidate and formally proposed genera, which were described in the last 3 years: ‘Orthoturdivirus’ and ‘Paraturdivirus’ (Woo et al., 2010), ‘Megrivirus’ (Honkavuori et al., 2011), ‘Gallivirus’ (Farkas et al., 2012; Boros et al., 2012a), and two unclassified picornaviruses (pigeon picornavirus A and B) from feral pigeons (Kofstad & Jonassen, 2011); furthermore, a quail picornavirus has been suggested as a potential novel picornavirus genus (Pankovics et al., 2012). Members of the genus ‘Megrivirus’ (turkey hepatitis virus), with the longest picornavirus genome (>9040 nt) so far described, and ‘Gallivirus’ (turkey gallivirus) were originally described from turkeys (later, these viruses were also identified from chickens) and both are phylogenically distantly related to the recently identified turdiviruses. The role of these picornaviruses as causative agents of ‘turkey viral hepatitis syndrome’ or poult enteritis complex in young turkeys is plausible (Honkavuori et al., 2011, Farkas et al., 2012; Boros et al., 2012a).
One of the well-studied avian picornaviruses is the internationally widespread DHAV, which can cause a highly lethal, acute, contagious infection in ducklings under 6 weeks of age (OIE, 2010). The rapid course of clinical disease is characterized by lethargy and ataxia followed by opisthotonos and death. Characteristic pathological changes appear primarily in the liver, which is enlarged and displays distinct petechial and ecchymotic haemorrhages (OIE, 2010). Recent studies have identified three serotypes (DHAV 1–3) (Tseng and Tsai, 2007; Fu et al., 2008; Kim et al., 2007). To our current knowledge, the DHAV types have only been identified from birds of the family Anatidae (geese, ducks); so far there have been no avian picornaviruses related to DHAVs identified from other avian sources.
This study reports the detection and complete genome characterization of a novel turkey picornavirus from faecal samples of healthy and affected commercial turkey (Meleagris gallopavo), from different turkey flocks in Hungary. Besides the phylogenetic relationship to DHAVs, presence of DHAV-like genomic features e.g. uncleaved VP0, three unrelated 2A, the low sequence identity to DHAVs and the distinct IRES types suggests that turkey picornavirus is related to, but distinct from, the genus Avihepatovirus (DHAV 1–3) and could be the founding member of a novel Avihepatovirus sister-clade genus.
Results
Genome characterization of turkey picornavirus
In faecal sample M176, a viral metagenomic analysis of viral-particle associated nucleic acids using 454 pyrosequencing (Kapoor et al., 2008a) revealed three sequencing reads (singletons) (Fig. 1) covering only 6.5 % of the picornaviral genome related to DHAV-3 (HQ654774) as the closest match using GenBank blastx. Based on the singletons, eight different primers (Table S1, available in JGV Online) were designed to verify the reads and amplify the complete genome of the picornavirus. The PCR products generated were sequenced using the primer-walking technique. The complete RNA genome of this turkey picornavirus (turkey/M176-TuASV/2011/HUN, KC465954) – which follows similar genome organization to DHAV: 5′ UTR-P1(VP0-3-1)-P2(2A-B-C)-P3(3A-B-C-D)-3′ UTR – consists of 7532 nt, excluding the poly(A) tail (Fig. 1). The base composition of the genome was found to be 28.53 % A, 23.78 % G, 21.19 % C and 26.50 % U. The G+C content (44.97 %) of the entire genome is similar to the DHAVs (43–44 %). The single 6720 nt ORF encodes the viral polyprotein precursor of 2239 aa, preceded by a 548 nt 5′ UTR and followed by a 264 nt 3′ UTR and a poly(A) tail. The complete P1 (2181 nt; 727 aa), P2 (2343 nt; 781 aa) and P3 (2196 nt; 731 aa) regions show 33, 32 and 36 % amino acid identity to the closest relatives, DHAV-1 (EU106097 and EF151312) and DHAV-3 (EU877916). The P2 is the longest compared with P1 and P3, because of the relatively long 2A (1010 nt/339 aa), which was presumed to cleave into three parts (2A1-2A2-2A3), similar to that predicted for DHAVs. Interestingly, the highest sequence identities were found not in 3D (38 % amino acid identity), but in VP3 (44 %) and 2C (42 %) compared with the representatives of the 12 officially recognized and one candidate picornavirus genera (Table 1).
Fig. 1.

(a) Genome organization with conserved picornaviral motifs and predicted cleavage sites with the enlarged VP1-2A3 genome region of turkey picornavirus turkey/M176-TuASV/2011/HUN. Nucleotide (upper numbers) and amino acid (lower numbers) lengths are indicated in each gene box. The positions of the conserved picornaviral amino acid motifs are indicated with the first amino acid positions of the motif. The sequence singletons acquired from pyrosequencing are indicated with grey bars. (b) Sequence alignment of the C-terminal VP1 and complete 2A1 of the study sequence. Letters with black backgrounds represent identical amino acids. Possible cleavage sites are indicated with white arrows and question marks. (c) 2A1-3 sequence alignment of turkey/M176-TuASV/2011/HUN with the prototype strains of DHAV 1–3 (DQ219396, EF067924, EU747874). Predicted cleavage sites of the study strain (white arrows) and the DHAV types (black arrows) together with the identified picornaviral motifs are indicated. Letters with black backgrounds represent similar amino acids. *, Conservative amino acids identical in the study sequence and all three prototype DHAV strains; #, conservative amino acids identical in the study sequence and at least one of the DHAV types.
Table 1. Amino acid sequence identity (%) comparisons of the turkey/M176-TuASV/2011/HUN sequence to the representative members of the 12 official picornavirus genera and the proposed genus ‘Pasivirus’.
Genera: 1, Avihepatovirus (DHAV-1; DQ249299); 2, Parechovirus (HPeV-1; L02971); 3, Parechovirus (LV-1; AF327920); 4, Sapelovirus (PSV-1; AF406813); 5, Enterovirus (PV-1; V01149); 6, Aphthovirus (FMDV-C; AF274010); 7, Cardiovirus (EMCV-1; M81861); 8, Hepatovirus (HAV-1; M14707); 9, Kobuvirus (AiV-1; AB010145); 10, Erbovirus (ERBV-2; AF361253); 11, Teschovirus (PTV-1; AJ011380); 12, Tremovirus (AEV-1; AJ225173); 13, Senecavirus (SVV-1; DQ641257); 14, ‘Pasivirus’ (SPaV-1; Q316470). Bold numbers indicate the highest levels of amino acid identity. ns, No significant alignment (sequence identity lower than 10 %).
| Genera | ||||||||||||||
| Region | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| VP0 | 32 | 23 | 24 | 15 | 17 | 16 | 16 | 16 | 13 | 16 | 16 | 18 | 14 | 23 |
| VP3 | 44 | 25 | 25 | 17 | 14 | 13 | 16 | 17 | 14 | 17 | 17 | 16 | 15 | 27 |
| VP1 | 26 | 27 | 18 | 11 | 11 | 13 | 14 | 12 | 15 | 14 | 12 | 11 | 11 | 23 |
| 2A1 | 10 | ns | 11 | ns | ns | 11 | 15 | ns | ns | 11 | 11 | ns | ns | 11 |
| 2A2 | 25 | ns | ns | ns | 11 | ns | ns | ns | ns | ns | ns | ns | ns | 12 |
| 2A3 | 32 | 21 | 22 | ns | 16 | ns | ns | 12 | 12 | ns | ns | 15 | ns | ns |
| 2B | 28 | 26 | 28 | 10 | 11 | 14 | 10 | 10 | ns | ns | ns | ns | 15 | 18 |
| 2C | 42 | 31 | 31 | 21 | 24 | 22 | 23 | 22 | 23 | 22 | 22 | 23 | 22 | 31 |
| 3A | 21 | 17 | 17 | 10 | 11 | ns | 17 | ns | 15 | ns | ns | 15 | 14 | 21 |
| 3B | 11 | 21 | 13 | 27 | 27 | 17 | 23 | 13 | 22 | 20 | 20 | 23 | 26 | 20 |
| 3C | 36 | 26 | 24 | 18 | 20 | 16 | 16 | 17 | 19 | 17 | 17 | 18 | 17 | 23 |
| 3D | 38 | 28 | 30 | 24 | 24 | 22 | 20 | 20 | 19 | 22 | 22 | 19 | 19 | 28 |
Analysis of the 5′ UTR and 3′ UTR
The predicted 548 nt 5′ UTR of turkey picornavirus was significantly shorter than those of the DHAV 1–3 reference sequences (DQ249299; EF067924; DQ256132) (623, 653 and 652 nt) and showed only 43, 42 and 44 % nucleotide identity, respectively. The predicted initiation codon was mapped at position 549, which was found in an optimal Kozak context (AxxA549UGG) (Fig. 2). There was an additional in-frame AUG triplet 18 nt upstream from the predicted initiation codon but not in a Kozak context. The 5′ UTR of turkey picornavirus contained at least two significantly long (24 and 23 nt) pyrimidine-rich tracts starting at positions 1 and 508 (Fig. 2). The predicted secondary structure of the study 5′ UTR sequence showed various stem–loops and two predicted domains (I and II) contained branches with hairpins of different sizes and multiple GNRA motifs (Fig. 2). The results of GenBank blastn search and clustal w alignment showed that the 5′ UTR of turkey/M176-TuASV/2011/HUN shared only 45 % sequence identity with the closest match ‘turkey gallivirus’ (TuGV, candidate genus ‘Gallivirus’) (JQ691613), although higher sequence identity was found in certain regions of the study sequence (apical part of domain I and hairpins of domain II) which could correspond to the apical part of domains I, J and K of the IRES of TuGV and encephalomyocarditis virus (EMCV, genus Cardiovirus) (Fig. 2). Based on the predicted secondary structure of the 5′ UTR, turkey/M176-TuASV/2011/HUN has a potential type II-like IRES. In contrast to EMCV and TuGV, a specific binding site of pyrimidine tract binding protein (PTB) in domain II (analogous to domain J–K of EMCV and TuGV) was not found in the study sequence (Fig. 2).
Fig. 2.

Predicted secondary RNA structure of the complete 5′ UTR including the IRES of the turkey picornavirus (turkey/M176-TuASV/2011/HUN; KC465954). The predicted structure of the EMCV 5′ UTR with type II IRES is shown in-frame for comparison. Light-grey circles represent identical amino acids compared with the IRES of ‘turkey gallivirus’ (JQ691613). Black arrow shows the site of a 12 nt (possible pyrimidine tract binding protein-binding site) deletion compared with TuGV.
The 264 nt 3′ UTR sequence of the turkey/M176-TuASV/2011/HUN was considerably shorter than those of the DHAV 1–3 reference sequences (DQ249299; EF067924; DQ256132) (314, 366 and 367 nt) and showed only 44, 44 and 45 % nucleotide identity, respectively. The predicted secondary RNA structure of the 3′ UTR showed a complex structure containing multiple stem–loops similar to the DHAVs (Fig. 3). The presence of a 48 nt ‘barbell-like’ structure identified in the 3′ UTR of TuGV, DHAV-1 and other viruses of different picornavirus genera (Boros et al., 2012a) was not detectable in the study sequence.
Fig. 3.
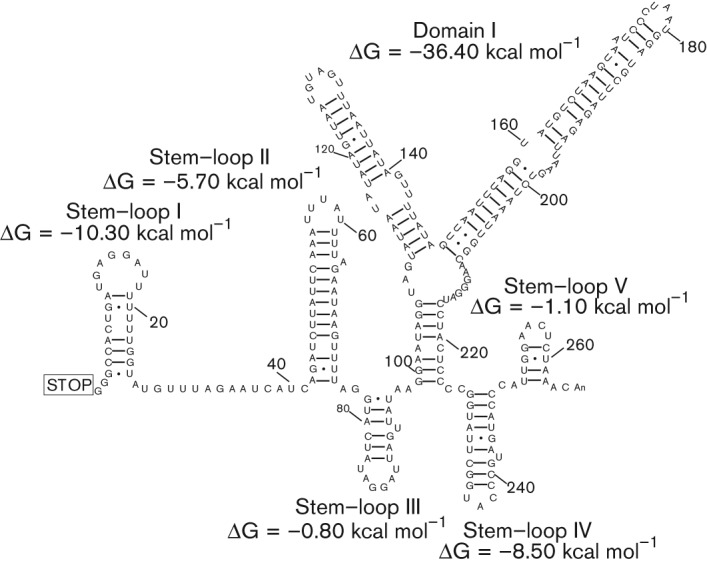
Predicted secondary RNA structure of the complete 3′ UTR of the turkey/M176-TuASV/2011/HUN.
Analysis of the polyprotein processing
The possible cleavage sites of the polyprotein of turkey/M176-TuASV/2011/HUN were mapped based on (i) the amino acid alignment with the selected strains of the closest relatives DHAV 1–3, human parechovirus (HPeV) and LV, and (ii) NetPicoRNA predictions (Blom et al., 1996). The Q/G (Gln/Gly), E/G (Glu/Gly) and Q/S (Gln/Ser) cleavage sites of 3Cpro were strongly supported by the polyprotein alignments and the NetPicoRNA predictions (Fig. 1). The E/G cleavage sites were not common among avihepato- and parechoviruses, but are found in other picornaviruses e.g. foot-and-mouth disease virus (Aphthovirus), equine rhinitis B virus (Erbovirus) and hepatitis A virus (Hepatovirus). The 3A/3B cleavage site prediction proved to be difficult because of the relatively low 3B sequence identity (Table 1) and the absence of known 3Cpro processing sites. The cleavage site was mapped to two presumed positions: A1578/R (Ala/Arg, based on the alignment of HPeVs and LVs) or S1583/N (Ser/Asn, based on the alignment of DHAVs). In both cases the tyrosine (Y) at position 3 of the study sequence, as well as in all of the known 3BVPg of picornaviruses was present (Fig. S1). The 20 aa 3B was used in the sequence comparison analyses based on the overall closer sequence relationship of the study sequence to DHAVs rather than to HPeVs or LVs. The prediction of the cleavage site of VP1/2A presented a special challenge. The polyprotein analysis showed that two identical autocleavage motifs DxExNPG727 and 803/P were located at the presumed VP1/2A site. Surprisingly, the analysis of this region revealed significantly high amino acid sequence identity between sequences upstream from the first and second NPG/P (Fig. 1). The alignments of this region did not reveal any of the known VP1/2A cleavage sites identified among the closest relatives (e.g. E/S; E/M or Q/S presented in DHAVs, LVs and HPeVs) before the first autocatalytic DxExNPG727/P motif (Fig. 1). Therefore the VP1/2A cleavage site of turkey/M176-TuASV/2011/HUN was hypothetically mapped to the first DxExNPG727/P motif site similar to that presumed for LVs (Johansson et al., 2003). The presence of another cleavage site (like the S669/N, S741/N, which is also a presumed cleavage site of 3A/3B or E685/R) upstream from the first NPG/P motif could not be ruled out (Fig. 1); in this case the number of presumed 2As would increase to four (or in case of active S/N sites, up to five) (Fig. 1).
Analysis of capsid proteins
The analysis of the P1 region alignments did not support the presence of L protein or as in kobu-, avihepato-, parecho-, ‘turdi-’, ‘pasi-’ and ‘galliviruses’ the cleavage of VP0 into VP4 and VP2, therefore the turkey/M176-TuASV/2011/HUN virion is probably built up from only three capsid monomers (VP0-VP3-VP1). The integrin-binding RGD motif typical in some HPeV types (Williams et al., 2009) was not found in the VP1. The VP0 (253 aa), VP3 (231 aa) and VP1 (243 aa) of turkey/M176-TuASV/2011/HUN were similar in length to those of DHAV1s (DQ249299–DQ249301) (256 aa/231 aa/244 aa). The amino acid identity of different capsid proteins of the study sequence and other picornaviruses is presented in Table 1.
Analysis of non-structural proteins
The analysis of the P2 region of the turkey/M176-TuASV/2011/HUN polyprotein revealed the presence of three unrelated 2A proteins (2A1–3). The N-terminal 2A1 contains the second DxExNPG803/P motif, which is a typical aphthovirus-like 2A cleavage site (Doherty et al., 1999; Johansson et al., 2002) similar to those found in 2A1 of DHAV types. The 76 aa 2A1 was considerably longer and possessed minor sequence identity (11 %) compared with the similar aphthovirus-like 2A1 (20 aa) of different DHAV types (Table 1; Fig. 1). The highest level of 2A1 sequence identity was found with the 143 aa aphthovirus-like 2A of EMCV (Table 1). Besides the viral NPGP motif, the 2A1 shows a distant relationship to S-adenosylmethionine-dependent methyltransferases (E-value 0.14) as the closest match using the conserved domain database (CDD) search of NCBI (Marchler-Bauer et al., 2011).
The 140 aa 2A2 was shorter than the 2A2 of DHAVs (161 aa) and possessed only 25 % sequence identity, despite the fact that DHAV was the closest match found (Table 1). Similar to DHAV 2A2, the presence of GxxGXGKS (X = uncharged, x = variable) potentially responsible for a 2C-like NTPase function (Ghazi et al., 1998) was also identifiable. This protein is related to the proteins of P-loop-containing nucleoside triphosphate hydrolases (E-value 0.01) based on the results of a CDD search and shows no sequence relationship to the AIG1 proteins related to the DHAV 2A2 (Fig. 1).
The 123 aa 2A3 of turkey/M176-TuASV/2011/HUN was similar in length (124 aa) and possessed the highest sequence identity compared with the 2A3 of DHAV types, 2A of HPeV and 2A2 of LV (Table 1), and also contains H-box/NC regions (Hughes and Stanway, 2000). Therefore, it appears to belong to the parechovirus-like 2A proteins (Fig. 1).
The 2C protein of turkey/M176-TuASV/2011/HUN possessed the highly conserved GxxGXGKS (X = uncharged, x = variable) motif for NTP binding sites – a similar but not identical motif was found in the 2A2 – and the QxxxxxDDLxQ motif (x = variable) for putative helicase activity (Gorbalenya et al., 1989, 1990) (Fig. 1). The 20 aa 3B (small genome-linked or VPg) protein of the turkey/M176-TuASV/2011/HUN was considerably shorter than VPg of DHAVs (34 aa), possessed low sequence identity (Table 1) and had no sequence repeats. The conservative catalytic triad (H, E/D, C) of the 3C viral cysteine-active-centre protease was seen in the turkey/M176-TuASV/2011/HUN (Bazan & Fletterick, 1988) and the active site cysteine in the motif GxCG (x = variable) was also present (Gorbalenya et al., 1989) (Fig. 1). The highly conserved motifs (e.g. GxxPSG, YGDD and FLKR) (x = variable) of the RNA-dependent-RNA-polymerase (3Dpol) were also present in the study sequence although the common KDELR contained a unique L (Leu)/V (Val) substitution (KDEVR) (Fig. 1).
Phylogenetic analysis
The phylogenetic relationships inferred by using the complete P1, and the most conservative 2C and 3CD deduced amino acid sequences of turkey/M176-TuASV/2011/HUN and the representative members of the 12 official, five proposed and nine candidate genera of the family Picornaviridae are shown in Fig. 4. The study sequence and the DHAVs constituted a monophyletic clade in all three phylogenetic trees; however, the turkey picornavirus was clearly distinct from the members of the genus Avihepatovirus, supported by high (≥95 %) bootstrap values (Fig. 4).
Fig. 4.



Phylogenetic relationship between turkey picornavirus turkey/M176-TuASV/2011/HUN (underlined), representative members of the 12 picornavirus genera and unassigned picornaviruses based on amino acid sequences of the different picornavirus coding regions: P1 (a), 2C (b) and 3CD (c). Bars, amino acid substitutions per site.
Prevalence and phylogenetic analysis of additional turkey picornavirus sequences
A specific primer pair was used to amplify the complete VP1 and VP1-2A1 genome regions and examine the existence of two NPG/P of turkey picornavirus and to screen for the presence of this turkey picornavirus in the faecal samples from healthy and affected commercial meat-type turkeys collected from an additional seven different turkey farms in Hungary (Table 2). Seven of the 11 tested faecal samples were reverse transcription-PCR (RT-PCR)-positive for turkey picornavirus with two specific bands corresponding to the amplified VP1 and VP1-2A1 genome regions (the antisense primer was designed to recognize both NPG/P sites). Four additional VP1-positive samples were randomly selected (three affected and one healthy) for sequence and phylogenetic analysis (Fig. 5). The selected VP1 sequences formed two distinct but related clusters which were most closely related to the members of the genus Parechovirus (Fig. 5). The closer relationship of turkey picornavirus VP1 to parechoviruses rather than DHAVs was also noticeable in the sequence identity table (Table 1). Cluster I contained the sequences of turkey/M176-TuASV/2011/HUN (KC465954), turkey/B232-TuASV/2011/HUN (KC465955) and turkey/PP3-TuASV/2011/HUN (KC465956) while cluster II contained the turkey/B415-TuASV/2011/HUN (KC465957) and turkey/B308-TuASV/2011/HUN (KC465958) sequences (Fig. 5). The pairwise sequence analysis shows 98 % nucleotide and amino acid identity between the sequences of the same cluster, and 74 % nucleotide and 86 % amino acid identity compared with sequences of different clusters.
Table 2. Epidemiological and clinical background of the 11 turkeys tested from eight turkey farms at different sites in Hungary, and the diagnostic results of the collected faecal samples for turkey picornavirus (KC465954).
| Location of farm | Sample ID | Age (days) | Symptoms | Routine diagnostic tests* | VP1 RT-PCR† | GenBank accession number |
| Bogyiszló | B-177 | 8 | Catarrhal enteritis, | Par. neg. | Positive | |
| foamy–watery caecum content | Bact. neg. | |||||
| B-415 | 14 | Rickets | Par. neg. | Positive | KC465957 | |
| Bact. neg. | ||||||
| Szalánta | B-195 | 7 | Catharral enteritis, | Par. neg. | Negative | |
| foamy–watery caecum content | Bact. neg. | |||||
| Karakószörcsök | B-232 | 21 | Growth depression | Par. neg. | Positive | KC465955 |
| Bact. neg. | ||||||
| Nagyalásony | B-308 | 47 | Growth depression, | Par. neg. | Positive | KC465958 |
| skeletal disorders | Bact. neg. | |||||
| Bakonypölöske | B-356 | 14 | Uneven growth, | Par. neg. | Negative | |
| catarrhal enteritis, | Bact. pos. | |||||
| foamy–watery caecum content | (Escherichia. coli bacteraemia) | |||||
| Bikal | B-407 | 42 | Skeletal disorders | Par. neg. | Positive | |
| Bact. neg. | ||||||
| Nemesbőd | PP-1 | 7 | Healthy | None | Negative | |
| PP-2 | 14 | Healthy | None | Negative | ||
| PP-3 | 21 | Healthy | None | Positive | KC465956 | |
| Bögöte | M176 | 20 | Growth depression | Par. neg. | Positive | KC465954‡ |
| Bact. neg. |
Par., Pathogen parasite diagnostics based on microscopic observation using native faecal samples; Bact., routine bacterial culture of heart blood and liver or bone marrow.
For the primer sequences, see Table S1.
Complete genome.
Fig. 5.

Unrooted phylogenetic tree based on the deduced VP1 capsid protein sequences of the study sequences (in shaded boxes), and the representative members of the family Picornaviridae. CL-I and CL-II, Clusters I and II, respectively. Bar, 0.2 amino acid substitutions per site.
Discussion
Taxonomic classification of turkey picornavirus
In this study, using metagenomic and RT-PCR approaches, we report the complete genome characterization and an initial prevalence analysis of a novel turkey picornavirus in Hungary phylogenetically related to the members of the genus Avihepatovirus. According to the current guidelines as suggested by the International Committee on Taxonomy of Viruses (ICTV) Picornaviridae Study Group (http://www.picornastudygroup.com/definitions/genus_definition.htm), novel picornavirus genera are defined by amino acid identities in the P1, P2 and P3 regions being less than 40, 40 and 50 %, respectively, from all other genera. Based upon this theoretical agreement – and the supporting results of the phylogenetic analysis – the turkey picornavirus could be the founding member of a novel picornavirus genus. Based on the phylogenic position relative to DHAV types in most genome regions (P1, 2C, 3CD) and the presence of DHAV-like genome features (e.g. uncleaved VP0, three unrelated 2A motifs), we propose the name ‘Avisivirus’ for Avihepatovirus sister-clade virus, with ‘turkey avisivirus 1’ (TuASV-1) as the type species.
Analysis of the UTR structures of turkey picornavirus
To our current knowledge the picornaviral 5′ UTR possess two distinct functional domains, the 5′ part contains structural elements necessary for the replication of the RNA genome, while the domains adjacent to the initiation codon make up the IRES, the key genome element of viral polyprotein synthesis (Racaniello, 2007). To date, five types of IRES (IRES I–V) have been identified: (i) the enteroviral type I IRES; (ii) the type II of parecho-, cardio-, erbo- and aphthoviruses; (iii) the type III of hepatitis A virus; (iv) the Hepacivirus/Pestivirus-like type IV IRES of tescho-, tremo-, sapelo- and avihepatoviruses; and (v) the proposed type V IRES of Kobuvirus, ‘Salivirus’ and ‘Paraturdivirus’ genera (Belsham, 2009; Ding and Zhang, 2007, Sweeney et al., 2012). Analysis of the 5′ UTR revealed that turkey picornavirus turkey/M176-TuASV/2011/HUN has a type II-like IRES based upon the sequence and secondary structural similarities to TuGV and EMCV (Boros et al., 2012a; Yu et al., 2011), although due to some characteristic differences (e.g. the absence of a region corresponding to Domain L of EMCV; only the apical part of Domain I corresponds to the Domain J of EMCV; and the missing sequence of PTB-binding site from Domain II) the exact classification of the IRES of the turkey picornavirus requires further experimental investigation. The presence of conservative (both in structure and sequence) IRES elements in the study sequence, turkey/M176-TuASV/2011/HUN, and TuGV (turkey/M176/2011/HUN) suggests the pivotal and uniform role of these elements during viral polyprotein synthesis in turkey despite the notable phylogenetic separation of these two turkey picornavirus species (TuGV being related to turdiviruses) (Boros et al., 2012a). The constant relationship of the study sequence to avihepatoviruses throughout the coding region and the heterogeneity of IRES types (type II vs type IV of DHAVs) raise the possibility of a recombinational event during the evolution of turkey/M176-TuASV/2011/HUN, although the results of recombination analysis did not reveal any evidence to support this (data not shown) due to the absence of potential parental sequences.
The 3′ UTRs of the known picornaviruses range from 40 nt (e.g. rhinoviruses) up to 654 nt (JF424832; turkey hepatitis virus 1; proposed genus ‘Megrivirus’) and contain stem–loops of variable size and number, which may be specific binding sites for viral and/or cellular proteins (Rohll et al., 1995). The same complexity of the 3′ UTRs of the study sequence and DHAVs (Tseng et al., 2007) suggests the same role of this genome region in the process of virus replication. The identification of the exact roles of the stem–loops and the suspected structural analogy between the 3′ UTR structures of these two virus species requires further experiments.
Analysis of the viral polyprotein
During the genome analysis, characteristic picornaviral motifs (e.g. Rhv-like capsid domains, 2C, 3C and 3D functional domains) were recognized in the viral polyprotein with a unique variance in motif KDELR → KDEVR. The role and function of this amino acid substitution remains to be elucidated. The most interesting feature of the viral polyprotein is the presence of the aphthovirus 2A-like ‘ribosome-skipping’ motif NPG/P (Sharma et al., 2012) as the presumed site of VP1/2A cleavage and the presence of the same NPG/P motif 76 aa downstream, as the predicted cleavage site of 2A1/2A2. The NPG/P repeat was present in all of the VP1 RT-PCR positive samples. The presence of two nearly identical NPG/P motifs is rare among picornaviruses. Similar features have only been found in BGPV (Barbknecht, 2009) and two related members of the family Iflaviridae, Perina nuda virus (PnPV) and Evtropis obliqua virus (Wu et al., 2002; Wang et al., 2004). Moreover, the analysis of this region revealed significant sequence identity between the C-terminal end of VP1 and the 2A1, which suggests a NPG/P motif-containing gene duplication event during the evolution of turkey/M176-TuASV/2011/HUN. The role of this presumed gene duplication is currently unknown; although the presence of nucleotide sequence duplications has been identified in other picornavirus genomes e.g. in the L-protein in quail picornavirus and 2B in porcine kobuvirus (Pankovics et al., 2012; Reuter et al., 2009). The presence of an NPG motif at the C-terminal end of a capsid protein VP1 was demonstrated among LVs using SDS-PAGE, where both the presence of NPG at the C-terminal end of VP1 of the mature virions and therefore the absence of 2A1 was suggested (Johansson et al., 2003), and also among the insect PnPVs, where the first active 2A-like NPG/P was experimentally demonstrated to be the ‘cleavage site’ of two capsid proteins (Wu et al., 2002). Without experimental results we cannot rule out the possibility of the presence of an unusual VP1/2A cleavage site upstream from the first NPG/P motif and therefore the release of four (or more) different 2A polypeptides. In this case the members of the candidate genus ‘Avisivirus’ would represent the most ancient group of known picornaviruses showing multi-2A genome features (Tseng et al., 2007; Johansson et al., 2002). Nevertheless, the release of one or more aphthovirus-like 2A peptides in TuASV and other picornaviruses containing NPG/P-motifs (e.g. DHAVs, seal picornaviruses) is worthy of investigation as previously suggested (Tseng et al., 2007). The role of the three different 2A of TuASV is currently unknown, although similar 2A features to DHAVs suggest similar replication properties of avihepatoviruses and ‘avisiviruses’.
Turkey picornavirus in healthy and affected turkeys
The high occurrence of TuASVs in faecal samples of healthy and affected turkeys collected from different sites in Hungary strongly suggests the endemic circulation of this virus at least in (but probably not restricted to) this country. Furthermore, the detection of related TuASV strains, which were distributed in at least two clusters (probably distinct geno/serotypes) in samples from different farms, suggested that turkeys are the natural hosts of this proposed novel genus of the family Picornaviridae. As discussed earlier, the presence a picornavirus in healthy animals does not rule out its pathogenic potential (e.g. turkey astro-, rota-, reo- and galliviruses) since these viruses are also detectable in apparently healthy turkey flocks (Pantin-Jackwood et al., 2007, 2008; Day et al., 2010; Boros et al., 2012a).
This is the third, and probably not the last, enteric picornavirus, along with the ‘turkey megrivirus’ and the ‘turkey gallivirus’, identified from turkeys with different manifested symptoms (Honkavuori et al., 2011; Boros et al., 2012a). It should be noted that the ‘turkey gallivirus’ (Boros et al., 2012a) and the novel turkey picornavirus presented in this study were identified in the same turkey faecal sample. In addition, co-infection with these two picornaviruses was detected in seven (63.6 %) specimens and six (75 %) of the investigated turkey farms (Boros et al., 2012a). In this context, any contribution of these viruses to the development of stunting syndrome and/or poultry enteritis complex as a single infection or part of co-infections or with other pathogens, as well as the potential worldwide distribution, remains unknown and should be investigated. A clear connection between this novel turkey picornavirus infection and the possible clinical outcomes will require experimental animal infection studies and periodic surveys. The increasing number of avian picornaviruses indicates that many unidentified and highly divergent avian picornaviruses still circulate among domestic and wild birds.
Methods
454 Pyrosequencing and metagenomic analysis.
Faecal sample (M176) collected in April 2011 from a commercial meat-type turkey (Meleagris gallopavo domesticus) showing stunting syndrome in a turkey farm located in north-west Hungary was selected for viral metagenomic analysis (Boros et al., 2012a). PBS diluted specimens were passed through a 0.45 µm sterile filter and centrifuged at 6000 g for 5 min. Pellet was mixed with a mixture of nucleases to enrich for particle-protected nucleic acids (Victoria et al., 2009). Nucleic acids were extracted using a QIAamp Viral RNA Mini kit (Qiagen) according to the manufacturer’s instructions. Viral nucleic acid libraries containing both RNA and DNA molecules were constructed by sequence independent random RT-PCR amplification as previously described (Victoria et al., 2009). Pyrosequencing using 454 GS FLX technology was then performed as previously described (Kapoor et al., 2008a; Victoria et al., 2009). The pyrosequencing reads (singletons) and assembled sequence contigs were compared with the GenBank nucleotide and protein databases using blastn and blastx, respectively.
Complete genome acquisition of turkey picornavirus.
Specific primer pairs (Table S1) were designed based on the available sequence reads from the pyrosequencing analysis to determine the complete nucleotide sequence of the turkey picornavirus using RT-PCR and long-range RT-PCR. RNA was extracted from 150 µl faecal suspension (35–40 %, v/v, in 0.1 M PBS) using TRIzol Reagent (Invitrogen) according to the manufacturer’s instructions. The RT-PCR and the long-range RT-PCR was the same as described previously (Reuter et al., 2002; Boros et al., 2012a, b). The 5′ and the 3′ end of the genome were determined using a 5′/3′ RACE PCR kit (Roche) as previously described (Boros et al., 2011). PCR-products were sequenced directly with the BigDye Terminator Cycle Sequencing Ready Reaction kit (PE Applied Biosystems) using the primer-walking method and run on an automated sequencer (ABI PRISM 310 Genetic Analyzer; Applied Biosystems).
Detection of turkey picornavirus in different turkey flocks.
Additional faecal samples from seven different turkey flocks located in western Hungary were collected in 2011 from turkeys of different age, showing different symptoms, and three samples (PP1–3) were taken from a healthy turkey at three consecutive weeks after hatching (Table 2). Native faecal samples were initially tested by microscopic observation for pathogen parasites and routine bacterial culture of heart blood and liver or bone marrow. RNA samples were tested for turkey picornavirus by RT-PCR using specific primer pairs targeting the VP1/2A1 region. The antisense primer was designed to recognize the two NPG/P motifs. (Table S1).
Sequence and phylogenetic analysis.
Sequences from representative members of different picornaviruses were obtained from the GenBank database and the study sequences were aligned using clustal x software (version 2.0.3) (Thompson et al., 1997) and similarity calculations were performed by using GeneDoc software (version 2.7) (Nicholas and Nicholas, 1997). Phylogenetic trees of the amino acid alignments were created using the neighbour-joining method based on the Jones-Taylor-Thornton matrix-based model of mega software (version 5) (Tamura et al., 2011). Bootstrap values (based on 1000 replicates) for each node are given if >50 %. The secondary structure of the 5′ and 3′ UTR was predicted using the Mfold program (Zuker, 2003) and the 2D model was drawn by using the Corel Draw Graphics Suite (version 12). Recombination analysis was performed with RDP software ver. 4.14 using the Jukes–Cantor model using a window size of 400 nt, a step size of 20 nt and 1000 bootstrap replicates (Martin et al., 2010). The complete genome sequence of turkey/M176-TuASV/2011/HUN and partial VP1 sequences were submitted to GenBank and assigned the accession numbers KC465954 to KC465958.
Acknowledgements
This work was supported by grants from the Hungarian Scientific Research Fund (OTKA, K83013 and PD108482). Á.B. was supported by TÁMOP 4.2.4.A/2-11-1-2012-0001 National Excellence Program of the European Union and European Social Fund. G.R. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and E.D. by NHLBI grant R01HL083254. We thank Dr Nick Knowles (Head, ICTV Picornaviridae Study Group) for careful reading of the manuscript and helpful comments.
Footnotes
One supplementary figure and table are available with the online version of this paper.
References
- Barbknecht M. (2009). Characterization of a unclassified virus and survey for its presence in Wisconsin bluegill populations. MSc thesis, University of Wisconsin–La Crosse [Google Scholar]
- Bazan J. F., Fletterick R. J. (1988). Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc Natl Acad Sci U S A 85, 7872–7876 10.1073/pnas.85.21.7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham G. J. (2009). Divergent picornavirus IRES elements. Virus Res 139, 183–192 10.1016/j.virusres.2008.07.001 [DOI] [PubMed] [Google Scholar]
- Blom N., Hansen J., Brunak S., Blaas D. (1996). Cleavage site analysis in picornaviral polyproteins: discovering cellular targets by neural networks. Protein Sci 5, 2203–2216 10.1002/pro.5560051107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros A., Pankovics P., Simmonds P., Reuter G. (2011). Novel positive-sense, single-stranded RNA (+ssRNA) virus with di-cistronic genome from intestinal content of freshwater carp (Cyprinus carpio). PLoS ONE 6, e29145 10.1371/journal.pone.0029145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros A., Nemes C., Pankovics P., Kapusinszky B., Delwart E., Reuter G. (2012a). Identification and complete genome characterization of a novel picornavirus in turkey (Meleagris gallopavo). J Gen Virol 93, 2171–2182 10.1099/vir.0.043224-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros A., Pankovics P., Knowles N. J., Reuter G. (2012b). Natural interspecies recombinant bovine/porcine enterovirus in sheep. J Gen Virol 93, 1941–1951 10.1099/vir.0.041335-0 [DOI] [PubMed] [Google Scholar]
- Day J. M., Ballard L. L., Duke M. V., Scheffler B. E., Zsak L. (2010). Metagenomic analysis of the turkey gut RNA virus community. Virol J 7, 313 10.1186/1743-422X-7-313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C., Zhang D. (2007). Molecular analysis of duck hepatitis virus type 1. Virology 361, 9–17 10.1016/j.virol.2007.01.007 [DOI] [PubMed] [Google Scholar]
- Doherty M., Todd D., McFerran N., Hoey E. M. (1999). Sequence analysis of a porcine enterovirus serotype 1 isolate: relationships with other picornaviruses. J Gen Virol 80, 1929–1941 [DOI] [PubMed] [Google Scholar]
- Farkas T., Fey B., Hargitt E., III, Parcells M., Ladman B., Murgia M., Saif Y. (2012). Molecular detection of novel picornaviruses in chickens and turkeys. Virus Genes 44, 262–272 10.1007/s11262-011-0695-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Pan M., Wang X., Xu Y., Yang H., Zhang D. (2008). Molecular detection and typing of duck hepatitis A virus directly from clinical specimens. Vet Microbiol 131, 247–257 10.1016/j.vetmic.2008.03.011 [DOI] [PubMed] [Google Scholar]
- Ghazi F., Hughes P. J., Hyypiä T., Stanway G. (1998). Molecular analysis of human parechovirus type 2 (formerly echovirus 23). J Gen Virol 79, 2641–2650 [DOI] [PubMed] [Google Scholar]
- Gorbalenya A. E., Donchenko A. P., Blinov V. M., Koonin E. V. (1989). Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases: a distinct protein superfamily with a common structural fold. FEBS Lett 243, 103–114 10.1016/0014-5793(89)80109-7 [DOI] [PubMed] [Google Scholar]
- Gorbalenya A. E., Koonin E. V., Wolf Y. I. (1990). A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett 262, 145–148 10.1016/0014-5793(90)80175-I [DOI] [PubMed] [Google Scholar]
- Honkavuori K. S., Shivaprasad H. L., Briese T., Street C., Hirschberg D. L., Hutchison S. K., Lipkin W. I. (2011). Novel picornavirus in Turkey poults with hepatitis, California, USA. Emerg Infect Dis 17, 480–487 10.3201/eid1703.101410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P. J., Stanway G. (2000). The 2A proteins of three diverse picornaviruses are related to each other and to the H-rev107 family of proteins involved in the control of cell proliferation. J Gen Virol 81, 201–207 [DOI] [PubMed] [Google Scholar]
- Johansson S., Niklasson B., Maizel J., Gorbalenya A. E., Lindberg A. M. (2002). Molecular analysis of three Ljungan virus isolates reveals a new, close-to-root lineage of the Picornaviridae with a cluster of two unrelated 2A proteins. J Virol 76, 8920–8930 10.1128/JVI.76.17.8920-8930.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson E. S., Niklasson B., Tesh R. B., Shafren D. R., Travassos da Rosa A. P., Lindberg A. M. (2003). Molecular characterization of M1146, an American isolate of Ljungan virus (LV) reveals the presence of a new LV genotype. J Gen Virol 84, 837–844 10.1099/vir.0.18792-0 [DOI] [PubMed] [Google Scholar]
- Kapoor A., Victoria J., Simmonds P., Slikas E., Chieochansin T., Naeem A., Shaukat S., Sharif S., Alam M. M. & other authors (2008a). A highly prevalent and genetically diversified Picornaviridae genus in South Asian children. Proc Natl Acad Sci U S A 105, 20482–20487 10.1073/pnas.0807979105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A., Victoria J., Simmonds P., Wang C., Shafer R. W., Nims R., Nielsen O., Delwart E. (2008b). A highly divergent picornavirus in a marine mammal. J Virol 82, 311–320 10.1128/JVI.01240-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. C., Kwon Y. K., Joh S. J., Kim S. J., Tolf C. J., Kim J. H., Sung H. W., Lindberg A. M., Kwon J. H. (2007). Recent Korean isolates of duck hepatitis virus reveal the presence of a new geno- and serotype when compared to duck hepatitis virus type 1 type strains. Arch Virol 152, 2059–2072 10.1007/s00705-007-1023-0 [DOI] [PubMed] [Google Scholar]
- Knowles N. J., Hovi T., Hyypiä T., King A. M. Q., Lindberg A. M., Pallansch M. A., Palmenberg A. C., Simmonds P., Skern T. & other authors (2012). Picornaviridae. In Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses, pp. 855–880 Edited by King A. M. Q., Adams M. J., Carstens E. B., Lefkowitz E. J. San Diego: Elsevier [Google Scholar]
- Kofstad T., Jonassen C. M. (2011). Screening of feral and wood pigeons for viruses harbouring a conserved mobile viral element: characterization of novel astroviruses and picornaviruses. PLoS ONE 6, e25964 10.1371/journal.pone.0025964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A., Lu S., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C. & other authors (2011). CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39 (Database issue), D225–D229 10.1093/nar/gkq1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P., Lemey P., Lott M., Moulton V., Posada D., Lefeuvre P. (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas K. B., Nicholas H. B. (1997). GeneDoc: a tool for editing and annotating multiple sequence alignments. National Resource for Biomedical Supercomputing http://www.nrbsc.org Accessed 02 Dec 2011. [Google Scholar]
- OIE (2010). Duck virus hepatitis. In Manual of Diagnostic Tests & Vaccines for Terrestrial Animals, 7th edn, Chapter 2.3.8 Paris: OIE (Office International des Epizooties) http://www.oie.int/manual-of-diagnostic-tests-and-vaccines-for-terrestrial-animals/ Accessed 07 Jan. 2013. [Google Scholar]
- Pankovics P., Boros A., Reuter G. (2012). Novel picornavirus in domesticated common quail (Coturnix coturnix) in Hungary. Arch Virol 157, 525–530 10.1007/s00705-011-1192-8 [DOI] [PubMed] [Google Scholar]
- Pantin-Jackwood M. J., Spackman E., Day J. M., Rives D. (2007). Periodic monitoring of commercial turkeys for enteric viruses indicates continuous presence of astrovirus and rotavirus on the farms. Avian Dis 51, 674–680 10.1637/0005-2086(2007)51[674:PMOCTF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Pantin-Jackwood M. J., Day J. M., Jackwood M. W., Spackman E. (2008). Enteric viruses detected by molecular methods in commercial chicken and turkey flocks in the United States between 2005 and 2006. Avian Dis 52, 235–244 10.1637/8174-111507-Reg.1 [DOI] [PubMed] [Google Scholar]
- Racaniello V. (2007). Picornaviridae: the viruses and their replication. In Fields Virology, 5th edn, pp. 795–838 Edited by Knipe D. M., Howley P. M., Griffin D. E., Lamb R. A., Martin M. A., Riozman B., Straus S. E. Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- Reuter G., Farkas T., Berke T., Jiang X., Matson D. O., Szücs G. (2002). Molecular epidemiology of human calicivirus gastroenteritis outbreaks in Hungary, 1998 to 2000. J Med Virol 68, 390–398 10.1002/jmv.10216 [DOI] [PubMed] [Google Scholar]
- Reuter G., Boldizsár A., Pankovics P. (2009). Complete nucleotide and amino acid sequences and genetic organization of porcine kobuvirus, a member of a new species in the genus Kobuvirus, family Picornaviridae. Arch Virol 154, 101–108 10.1007/s00705-008-0288-2 [DOI] [PubMed] [Google Scholar]
- Reuter G., Pankovics P., Knowles N. J., Boros A. (2012). Two closely related novel picornaviruses in cattle and sheep in Hungary from 2008 to 2009, proposed as members of a new genus in the family Picornaviridae. J Virol 86, 13295–13302 10.1128/JVI.01142-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohll J. B., Moon D. H., Evans D. J., Almond J. W. (1995). The 3′ untranslated region of picornavirus RNA: features required for efficient genome replication. J Virol 69, 7835–7844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage V., Ar Gouilh M., Cheval J., Muth E., Pariente K., Burguiere A., Caro V., Manuguerra J. C., Eloit M. (2012). A member of a new Picornaviridae genus is shed in pig feces. J Virol 86, 10036–10046 10.1128/JVI.00046-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semler B. L., Ertel K. J. (2008). Picornaviruses: molecular biology. In Encyclopedia of virology, 3rd edn, pp. 129–140 Edited by Mahy B. W. J., Van Regenmortel M. H. V. Oxford: Academic Press; 10.1016/B978-012374410-4.00471-4 [DOI] [Google Scholar]
- Sharma P., Yan F., Doronina V. A., Escuin-Ordinas H., Ryan M. D., Brown J. D. (2012). 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res 40, 3143–3151 10.1093/nar/gkr1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney T. R., Dhote V., Yu Y., Hellen C. U. (2012). A distinct class of internal ribosomal entry site in members of the Kobuvirus and proposed Salivirus and Paraturdivirus genera of the Picornaviridae. J Virol 86, 1468–1486 10.1128/JVI.05862-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28, 2731–2739 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997). The clustal_x windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25, 4876–4882 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C. H., Tsai H. J. (2007). Molecular characterization of a new serotype of duck hepatitis virus. Virus Res 126, 19–31 10.1016/j.virusres.2007.01.012 [DOI] [PubMed] [Google Scholar]
- Tseng C. H., Knowles N. J., Tsai H. J. (2007). Molecular analysis of duck hepatitis virus type 1 indicates that it should be assigned to a new genus. Virus Res 123, 190–203 10.1016/j.virusres.2006.09.007 [DOI] [PubMed] [Google Scholar]
- Victoria J. G., Kapoor A., Li L., Blinkova O., Slikas B., Wang C., Naeem A., Zaidi S., Delwart E. (2009). Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol 83, 4642–4651 10.1128/JVI.02301-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhang J., Lu J., Yi F., Liu C., Hu Y. (2004). Sequence analysis and genomic organization of a new insect picorna-like virus, Ectropis obliqua picorna-like virus, isolated from Ectropis obliqua. J Gen Virol 85, 1145–1151 10.1099/vir.0.19638-0 [DOI] [PubMed] [Google Scholar]
- Williams C. H., Panayiotou M., Girling G. D., Peard C. I., Oikarinen S., Hyöty H., Stanway G. (2009). Evolution and conservation in human parechovirus genomes. J Gen Virol 90, 1702–1712 10.1099/vir.0.008813-0 [DOI] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Yuen K. Y. (2006). Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Curr Opin Infect Dis 19, 401–407 10.1097/01.qco.0000244043.08264.fc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Huang Y., Lam C. S., Poon R. W., Tsoi H. W., Lee P., Tse H., Chan A. S. & other authors (2010). Comparative analysis of six genome sequences of three novel picornaviruses, turdiviruses 1, 2 and 3, in dead wild birds, and proposal of two novel genera, Orthoturdivirus and Paraturdivirus, in the family Picornaviridae. J Gen Virol 91, 2433–2448 10.1099/vir.0.021717-0 [DOI] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Choi G. K., Huang Y., Teng J. L., Tsoi H. W., Tse H., Yeung M. L., Chan K. H. & other authors (2012). Natural occurrence and characterization of two internal ribosome entry site elements in a novel virus, canine picodicistrovirus, in the picornavirus-like superfamily. J Virol 86, 2797–2808 10.1128/JVI.05481-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. Y., Lo C. F., Huang C. J., Yu H. T., Wang C. H. (2002). The complete genome sequence of Perina nuda picorna-like virus, an insect-infecting RNA virus with a genome organization similar to that of the mammalian picornaviruses. Virology 294, 312–323 10.1006/viro.2001.1344 [DOI] [PubMed] [Google Scholar]
- Yu Y., Abaeva I. S., Marintchev A., Pestova T. V., Hellen C. U. T. (2011). Common conformational changes induced in type 2 picornavirus IRESs by cognate trans-acting factors. Nucleic Acids Res 39, 4851–4865 10.1093/nar/gkr045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]