

Summary
The credo that epileptic seizures can be initiated only by “epileptic” neurons has been recently challenged. The recognition of key astrocytic-neuronal communication, and the close interaction and crosstalk between astrocytes and brain endothelial cells, has shifted attention to the blood–brain barrier (BBB) and the “neurovascular unit.” Therefore, the pursuit of mechanisms of seizure generation and epileptogenesis now includes investigations of cerebral blood flow and permeability of cerebral microvessels. For example, leukocyte adhesion molecules at the BBB have been proposed to play a role as an initiating factor for pilocarpine-induced status epilepticus, and a viral infectionmodel with a strong BBB etiology has been used to study epileptogenesis. Finally, the fact that in nonepileptic subjects seizures can be triggered by BBB disruption, together with the antiseizure effects obtained by administration of potent antiinflammatory “BBB repair” drugs, has increased the interest in neuroinflammation; both circulating leukocytes and resident microglia have been studied in this context. The dual scope of this review is the following: (1) outline the proposed role of BBB damage and immune cell activation in seizure disorders; and (2) explain how increased cerebrovascular permeability causes neuronal misfiring. The temporal sequence linking seizures to peripheral inflammation and BBB dysfunction remains to be clarified. For example, it is still debated whether seizures cause systemic inflammation or vice versa. The topographic localization of fundamental triggers of epileptic seizures also remains controversial: Are immunologic mechanisms required for seizure generation brain-specific or is systemic activation of immunity sufficient to alter neuronal excitability? Finally, the causative role of “BBB leakage” remains a largely unresolved issue.
Keywords: Inflammation, Multiple sclerosis, TNF-α, Neuroimmunology, Cell trafficking, Patch clamp, Ion-sensitive electrodes
This review is focused on epilepsy and mechanisms leading to seizures. In the following paragraphs the terms “epilepsy” and “seizures” are sometimes used as synonyms, even though it is clear that a significant distinction exists, seizures being a symptom of epilepsy and not a disease per se. Occurrence of seizures does not always lead to a diagnosis of epilepsy and seizures do not exclusively occur in the context of epilepsy. Within a life span, 5–10% of the population will have at least one seizure without an epilepsy diagnosis. While the distinction between seizures and epilepsies is clinically well understood, translation to the basic scientists’ laboratory has been more problematic. Epilepsy research has sometimes failed to consider the multifaceted clinical reality of seizure disorders; this has sometimes led to a simplistic modeling of the disease and its mechanisms. The gap between clinical reality and research models, if filled, will open a venue for the investigation of new therapeutic approaches. Given the many new and exciting cell types and mechanisms of this disease, one of the goals of this manuscript is to incite a more “holistic” approach to the epileptic disease and its symptoms.
A similar disclaimer can be made on the use of the terms “leakage” and “opening” when referring to increased permeability across the blood–brain barrier (BBB). These terms are useful jargon to describe increased permeability across the endothelial cells constituting the BBB. These terms of course give no specific information of the site of the “leakage”; for example, is increased passage of molecules of ions pericellular (tight junctions), transcellular (pinocytosis and/or vesicular release), or due to a generic failure of the many mechanisms ensuing selective trans-BBB permeability. The accepted term according to current orthodoxy is “BBB disruption” or “failure.” These are more elegant but not much more informative terms. The reality seems to be more complex and difficult to reduce to a few semantic terms. Tight junctions alone are complex “doors” that open and close stochastically in a manner not dissimilar to ion channels. When an agent, for example histamine, is applied to increase permeability, the probability of a single tight junction to be in an open (permissive) state is increased. Conversely, when agents that “tighten” the BBB are applied, the probability of a permissive state is reduced. In the case of “osmotic blood–brain barrier disruption” often mentioned in this review, the data are close to nonexistent, but the consensus is that shrinkage of endothelial cells causes a mechanical disarray of the junctional complex. This is unlikely to be controlled by any specific rules, but is rather due to drastic yet reversible changes in cell morphology leading to a spatial reorganization of the junctions.
Blood–Brain Barrier and Neurologic Diseases
The BBB is the most important vascular barrier of the central nervous system (CNS). The BBB protects the brain from harmful substances circulating in the bloodstream, while also supplying the brain with the nutrients required for proper brain function. Examples of molecules that would be toxic to a brain lacking the BBB are cardioactive drugs and penicillin. The BBB likewise strictly regulates the trafficking of cells of the immune system; it also prevents free movement of chemokines and cytokines from the blood into the brain and vice versa. Recent findings indicate that neurovascular dysfunction is an integral part of many neurologic disorders (Krizanac-Bengez et al., 2004; Zlokovic, 2008; Neuwelt et al., 2011), including epilepsy (Marchi et al., 2010b). In diseases with a compromised BBB, the microenvironment of neurons is altered and brain infiltration of cells, ions, or molecules may initiate, amplify, procrastinate, repair, or disrupt a CNS response.
Failure of the BBB is observed in association with a variety of pathologic events, such as stroke, multiple sclerosis (MS), and epilepsy. Increasing evidence has shown that BBB damage often accompanies abnormal neuronal activity. Therefore, in addition to epilepsy, seizures are observed in patients with MS (Hauser, 2008), as a consequence of stroke (Asconape & Penry, 1991), or during systemic or local inflammation (Hauser, 2008), all conditions associated with a leaky BBB. In addition, clinical and animal model findings show that acute failure of the BBB induced by “mechanical” means (e.g., osmotic shock to disrupt tight junctions between endothelial cells) is a mechanism of acute seizures (Marchi et al., 2007a). More recent experimental and clinical evidence has shown that BBB failure can cause seizures regardless of the means used to induce BBB failure (Seiffert et al., 2004; Pavlovsky et al., 2005; Oby et al., 2006; Marchi et al., 2007a; Fabene et al., 2008; Ransohoff, 2009; Ivens et al., 2010). The unanswered question remains: why does BBB disruption (BBBD) lead to seizures? Candidate mechanisms linking BBBD to abnormal neuronal firing include extravasation of potassium, albumin, or cells of the immune system (see Table 1 and Fig. 2) (Seiffert et al., 2004; Fabene et al., 2008; David et al., 2009a). Other studies propose that seizures induce proinflammatory changes limited to the brain, but in these studies peripheral leukocyte activation has not been measured and thus its role cannot be ruled out (Vezzani & Granata, 2005). Unquestionably glial, microglial, and endothelial cells are all capable of reacting to seizures by producing typical proinflammatory molecules such as cytokines and adhesion molecules (e.g., (Librizzi et al., 2007). Recently, toll-like receptors (e.g., TLR-4) were associated with the perpetuating seizures (Maroso et al., 2010). These receptors are widely expressed in myeloid cells, thus it is unclear whether a peripheral response is involved.
Table 1.
Summary of mechanisms that may affect neuronal behavior after BBB disruption
| Mechanism or molecule | Role of BBB in mechanism | Evidence for | Evidence against |
|---|---|---|---|
| Extracellular K+ | Serum [K]>Brain [K] (Davson & Segal, 1995) | Kir knock-out mice exhibit low seizure threshold (Djukic et al., 2007) Extracellular potassium increases excitability (Janigro et al., 1997; Emmi et al., 2000; Hinterkeuser et al., 2000; Schroder et al., 2000; Kofuji & Newman, 2004; Tate & Sisodiya, 2007; Olsen & Sontheimer, 2008; Zhang & Verkman, 2008; Steinhauser & Seifert, 2010; Stewart et al., 2010; Pardini et al., 2011) |
Osmotic BBBD in rats did not cause increased [K+]out (Somjen et al., 1991) |
| Negligible K+ permeability across BBB (Stanness et al., 1996) | Potassium causes seizures when directly applied (Traynelis & Dingledine, 1988; Trombin et al., 2011) | ||
| Loss of potassium homeostasis in human epileptic brain (Bordey & Sontheimer, 1998; Heinemann et al., 2002; Steinhauser & Seifert, 2010) | |||
| Magnesium | Serum [Mg]<Brain [Mg] | Low magnesium causes epileptiform activity; high serum Mg is neuroprotective (Amtorp & Sorensen, 1974; Zhang et al., 1995) | |
| Serum albumin in astrocytes | Albumin quotient (serum>brain) | TGF-β receptor-mediated albumin uptake in neocortical epileptogenesis (Ivens et al., 2007; Cacheaux et al., 2009; David et al., 2009b) | Present in neurons less so in glia in human epilepsy (Marchi et al., 2010b,c) |
| Glutamate | Serum levels<brain levels (Smith, 2000) | Link with TGF-β and albumin (Ivens et al., 2007; Cacheaux et al., 2009; David et al., 2009b) | |
| Causes seizures and is elevated in brain of epileptics |
Figure 2.
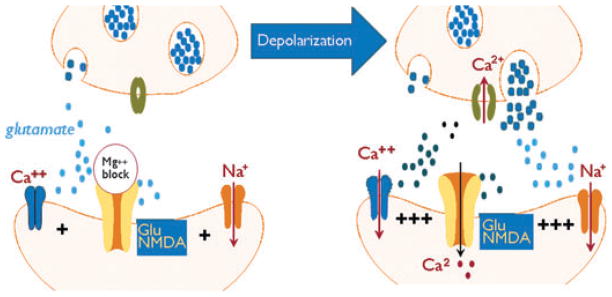
Predicted changes in synaptic excitability and synaptic changes after BBBD. A glutamatergic synaptic is depicted, but it is likely that all neurotransmitter release mechanisms are similarly affected by depolarization. In these examples the left panel shows normal synaptic transmission where the NMDA receptor is blocked by internal magnesium and synaptic transmission is limited to activation of 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl) propanoic acid (AMPA) receptors. After cell depolarization neural transmitter release is increased, blockade of NMDA receptors is removed and synaptic hyper-excitability is predicted. Changes in astrocytes (not shown) are perhaps the best documented after disruption of the BBB. Both glutamate uptake and potassium buffering are reduced by yet unknown mechanisms perhaps involving albumin. These, together with the changes shown in the right panel will synergistically increase both neuronal cell excitability and synchronization.
If BBB damage is a key factor in the etiology of CNS diseases, then BBB protective agents or drugs that improve BBB function should prevent the downstream cascade that affects neurons. These protective agents may also be of use when the BBB is compromised by events initiated in the parenchyma; therefore, this class of therapeutics may be of value to treat CNS diseases caused by either brain or blood-borne triggers. Drugs used to repair the BBB range from steroids to membrane sealing agents (Marchi et al., 2009, 2011a). An important correlate of the “BBB hypothesis of disease” is the lessening of issues of drug penetration into the brain, which is commonly recognized as a major if not insurmountable obstacle for CNS therapies (Loscher & Potschka, 2005).
Early Mechanisms of Neuronal Changes after Blood–Brain Barrier Disruption
The functional stability and reliability of mammalian neuronal networks is made possible thanks to a tightly controlled brain homeostasis. The intracellular and extracellular milieu of the CNS is regulated by metabolic, ionic, and transcriptional mechanisms, including buffering of extracellular ions such as potassium, control of pH and ATP levels (see Table 1 and Fig. 1), as well as constant synthesis of enzymes, proteins, and phospholipids that maintain the structural integrity of cells. Even a slight dysregulation of these mechanisms causes altered neuronal function, which in turn may affect behavior.
Figure 1.

Quantitative gradients across the BBB and their predicted effect on neuronal excitability. The size of the molecules and ions depicted on the left side of the figure are roughly proportional to their trans-BBB concentrations. The brain concentration changes indicated by arrows is a semiquantitative means of showing what expected after BBB disruption. The predicted effect on neuronal excitability is also shown. BBB “openings” of different duration and extent and occurring in different regions of the brain may have distinctly different effects.
Based on available data, the main candidates for ictogenic sequelae of blood–brain barrier disruption can be divided roughly into three categories: (1) ions, such as potassium and ion channel modulators (e.g., albumin (Ivens et al., 2007; David et al., 2009a,b); (2) neurotransmitters (adenosine, glutamate, and ATP); and (3) metabolic products (again adenosine, glucose, pH, CO2). The following paragraphs summarize the possible role of a few among these candidates.
Potassium ions are crucial for normal action potential generation in all excitable tissue. Therefore, in cardiac myocytes, neurons, vascular smooth muscle, and retinal effector cells, extracellular potassium concentration ([K+]out) is a key regulator of resting membrane potential and repolarization. The biophysical properties of most excitable cells are designed to adapt to a specific range of potassium concentrations. Therefore, cardiac action potential properties are well suited for a relatively broad range of [K+]out, whereas neurons are far less tolerant and require a stricter potassium homeostasis. In brain, potassium is maintained at levels that are significantly lower than in blood (Fig. 2 and Table 1), and the generation of fast action potentials typical of CNS neurons requires an equally rapid repolarizing potassium current. In addition, the size of brain extracellular space is reduced compared to that of other excitable tissues (Nicholson & Sykova, 1998). This exaggerates the amplitude of ionic changes following transmembrane fluxes. Finally, in addition, the frequency of neuronal firing far exceeds the cardiac rate; therefore, extracellular potassium changes following a single neuronal action potential need to be rapidly restored (Ransom et al., 1992; Pappas & Ransom, 1994). All of these factors have led to the development of a highly specialized glioneuronal system to buffer extracellular potassium (Kofuji & Newman, 2004).
Under conditions of preserved BBB integrity, the local maintenance of ion homeostasis is sufficient to maintain [K+]out within narrow ranges. However, when cerebrovascular events alter BBB integrity, a surge in potassium from the blood to brain will overcome glial potassium buffering. In addition, potassium ions regulate cerebral blood flow (Nguyen et al., 2000). Although modest changes in [K+]out (up to 12 mM) cause dilation of arteries, increased levels of K+ induce massive, potentially ischemic, reduction in blood flow. This may further affect homeostasis by decreasing metabolic support of neurons and glia (cerebral hypoxia).
The considerations made for potassium ions can be expanded to maintenance of brain pH, but the extent of changes caused by protons and their effect on neuronal firing is different. The brain is slightly acidic compared to blood, and protons have an inhibitory effects on sodium currents; a negative pH shift will decrease excitability (Benitah et al., 1997). Seizures are accompanied by a large drop in pH, which may ultimately contribute to seizure termination. In addition, low pH is a mechanism of vasodilation used by the brain to couple neuronal activity to cerebral blood flow (Aaslid, 2006). When the BBB is breached, these equilibria are lost and intrinsic controllers of activity are impaired, leading to increased neuronal firing.
Adenosine and glutamate have opposing effects on overall brain activity (Dunwiddie & Fredholm, 1989). Glutamate is more concentrated in blood, leading to an increase in neuronal firing and excitotoxic potential after BBB disruption. Adenosine is short-lived due to enzymatic activity, but its metabolic production is another efficient mechanism of neurovascular control. When the BBB is breached, a complex synergy of adenosine (Parkinson et al., 2003) and glutamate (Smith, 2000; Grant et al., 2003) transporters and catalytic enzymes is altered, leading to an overall drop in adenosine available to curb neuronal firing and increased glutamate; the result is a synergistic increase in neuronal excitability. As in the case of potassium and protons (pH), adenosine is also a powerful controller of cerebral blood flow (Ko et al., 1990); a reduced level of CNS adenosine will compromise autoregulation and exacerbate the consequences of BB disruption.
These examples demonstrate how BBB disruption may alter the intrinsic mechanisms that control of neuronal excitability. A direct effect on cell metabolism, resting membrane potential, or membrane conductance may occur in parallel with an indirect effect involving cerebral blood flow. Of interest, the lag time to seizure development after acute osmotic BBB opening is consistent with a rapid ionic (or neurotransmitter) changes. Therefore, after BBB disruption, seizures occur within minutes from the event (Marchi et al., 2007a, 2009, 2011b), suggesting that transcriptional or other molecular mechanisms are not involved. Whether this also applies to spontaneous epileptic seizures is not known.
Long-Term Mechanisms and Consequences of Blood–Brain Barrier Disruption
Epilepsy is a chronic disorder. Therefore, although the mechanisms described so far may explain how BBB disruption leads to individual seizures, these do not address the process of epileptogenesis and/or maintenance of an epileptic pathology. Friedman and Kaufer (Ivens et al., 2007; David et al., 2009a,b) have proposed that albumin extravasated across a “leaky” BBB may produce long-lasting effects, in particular directed toward astrocytic spatial buffering of K+ and glutamate transporters (David et al., 2009a). The process seems to depend on complex molecular machinery initiated by signaling at the transforming growth factor α (TGF-β) receptor. This model may explain posttraumatic epilepsy, where seizures originate, with a variable delay, in the same regions where the BBB was breached at time of head injury (Korn et al., 2005). Other chronic BBB-related events leading to seizures are glucose transporter 1 (GLUT-1) deficiency (DeVivo et al., 1991), and, albeit indirectly, multiple drug resistance to antiepileptic drugs (Dombrowski et al., 2001; Loscher & Potschka, 2005). The latter has been discussed elsewhere (Ghosh et al., 2010; Marchi et al., 2010a; Ghosh et al, 2011a,b).
Because it is generally agreed that “seizures begot seizures,” and that BBB disruption can cause a long-lasting, generalized seizure in humans and animal models, it is possible that seizures induced by BBB “opening” cause brain permeability changes that encourage future ictal events. In addition, the quantitative and qualitative morphologic or radiologic properties of seizure-inducing BBB permeability changes in acute, provoked, or chronic seizures are not clear. Data comparing morphologic aspects of BBB disruption in acute (mannitol, rodent brain) or chronic (rodent, human) seizures have shown a surprising similarity (Marchi et al., 2010b,c, 2011b,c). Both conditions are characterized by a regional, patchy or diffuse leakage from capillaries or venules, with perivascular accumulation of immunoglobin G (IgG) or extravasated albumin. In neither condition, in human (Marchi et al., 2011b,c), porcine (Marchi et al., 2007a, 2010c), or rodent brain (Marchi et al., 2007b, 2009, 2011c) have leukocytes been seen in the brain parenchyma, with the exception of one report using postmortem tissue (Fabene et al., 2008). However, when seizures are induced by a chemoconvulsants directly injected into the hippocampus (kainate), a robust leukocyte recruitment is seen (Zattoni et al., 2011). The clinical significance of these findings is not clear, but cell infiltration in CNS does not seem to be necessary for seizure generation (Marchi et al., 2011c). An important caveat in human studies using resected tissue is the fact that these samples are almost invariably “inflamed” by the chronic recording devices (grids, electrodes, etc.). The recording electrodes may cause reactive changes independent from seizures that can be considered artifacts.
Neurological Diseases, Leukocytes, and the BBB
The previous paragraphs summarized the consequences of BBB dysfunction on neuronal behavior. Most of the data were obtained from experimental models, and even when human tissue is used, methodological confounders exist (see above). To translate science to therapy, the real issue is to understand whether comparable cerebrovascular damage occurs in epilepsy and whether BBB leakage actively causes real life seizures. In particular, the participation of white blood cells (discussed briefly in preceding text) is important for therapeutic translation.
Direct evidence of a contribution of circulating leukocytes to human epilepsy requires specifically designed studies; indirect evidence is provided in a recent study (Marchi et al., 2011b) showing that antiinflammatory drugs effectively decrease seizure burden. The neuroanatomic correlates for leukocyte role in generic generation of these are important: (1) If systemic inflammation is a target to treat seizures, then these antiepileptic drugs do not require penetration across the BBB; (2) if conversely the crucial target is microglia or extravasated leukocytes, then the issue of CNS penetration remains a concern.
Pathologic leukocyte–BBB interactions that facilitate seizures are not dissimilar from those involved in MS (Fabene et al., 2008, 2010; Sotgiu et al., 2010) or ischemic stroke (Hurn et al., 2007; Subramanian et al., 2009). However, the extent and molecular or pathophysiologic overlap between epilepsy and recognized inflammatory brain diseases remains to be defined. For instance, although the interplay between activated leukocytes-BBB and associated BBB damage contributes to seizures, the occurrence and relevance of leukocyte brain extravasation is a matter of controversy (Fabene et al., 2008; Ravizza et al., 2008; Marchi et al., 2010b). Brain invasion of immune cells is a trademark of specific forms of epilepsy such as West syndromes or epilepsies triggered by viral infections (Vezzani & Granata, 2005); however, in the majority of the epilepsies a frank immunologic brain response has yet to be proven. This suggests that, after causing BBB damage, leukocytes may exhaust their role, highlighting the role of BBB damage as a seizure-promoting factor (Fabene et al., 2008, 2010).
Muscarinic Regulation of T-Cell Function
Why and how systemic inflammation becomes active before an ictal event is unknown. Basic science studies have shed some light here thanks to the use of receptorspecific convulsive agents such as pilocarpine, a muscarinic agent. Kainate acting on a subclass of glutamatergic synapses has also been employed. In addition to their known actions on neurons, these molecules activate the innate immune system by action on T or B cells (Marchi et al., 2007a, 2010b, 2011c; Sturgill et al., 2011). Because most of our knowledge derives from pilocarpine, we focus here on the effects of this drug.
In contrast to what is widely believed, pilocarpine does not promptly cross the BBB; pilocarpine’s seizurepromoting potency does, however, depend on BBB disruption (see below). Studies in which pilocarpine’s levels were measured at time of seizures or in animals before seizure onset have demonstrated that the permeability of pilocarpine is comparable to that of sucrose, this despite pilocarpine’s favorable oil-to-water partition coefficient (Marchi et al., 2007b). In addition, the levels of pilocarpine needed to cause seizures were not consistent with the high affinity that the drug has for its receptors (Dehaye et al., 1984). Finally, the combined effects of lithium (given before a low dose of pilocarpine) were shown to be due to lithium’s inflammatory action (Marchi et al., 2009). It was also shown that pilocarpine acts at least in part by a massive activation of leukocytes, in a manner comparable to that of lithium. In fact, pilocarpine and lithium caused seizures regardless of the order of administration, or the interval between the exposures of these two drugs. Others then demonstrated that pilocarpine has a profound effect on leukocyte adhesion to the endothelium (Fabene et al., 2008).
Pilocarpine acts on leukocytes that express cholinergic nicotinic and muscarinic receptors. Although activation of nicotinic receptors suppresses immune/inflammatory responses (Rosas-Ballina & Tracey, 2009), the role of muscarinic receptors in immunity is unclear. Muscarinic receptors play a role in the generation of CD8+ cytolytic T lymphocytes. Analysis of mice with targeted deletions of each of the known muscarinic receptors (M1–M5) showed that CD8+ T cells from M1 receptor–deficient mice had a defect in the ability to differentiate into cytolytic T lymphocytes (Razani-Boroujerdi et al., 2008). These findings are consistent with results linking pilocarpine-induced seizures to CD8+ T cell activation and mobilization from the spleen (Marchi et al., 2011c). The sequence of effects of cholinergic agonists leading to seizures can be thus summarized as follows: (1) Pilocarpine, injected systemically, activates T cells to acquire a cytotoxic (CD8+) phenotype; (2) These events occurring in the spleen, or being therein amplified, translate into an inflammatory response that with yet unknown mechanisms causes BBB leakage in regions that are highly epileptogenic (limbic system); several reports have described the pattern of BBBD before pilocarpine-induced status epilepticus ensues; (3) In addition to T cells, other cell types are involved (see below and Sandberg, 1994). Consistent with its inflammatory action, pilocarpine-induced seizures are prevented by antagonists of interleukin 1β (IL-1β) or steroids (Marchi et al., 2009, 2011b).
In addition to T cells, other cell types are involved in leukocytosis induced by convulsive agents. Perforin is a cytolytic protein found exclusively in the granules of CD8+ T cells and natural killer (NK) cells. Upon degranulation, perforin inserts itself into the target cell’s plasma membrane, forming a pore. Mice that are genetically deficient in perforin have severe immunodeficiency and impaired protection against viruses and tumors, because perforin is required to deliver granzymes into the cytosol of the target cell (Bischofberger et al., 2009; Voskoboinik et al., 2010). Recently published results have shown that perforin-deficient mice have a greatly reduced sensitivity to pilocarpine-induced seizures (Marchi et al., 2011c). However, how immune effector proteins utilized by NK cells contribute to CNS vascular permeability remains poorly understood (Suidan et al., 2008). For example, it is not known whether (1) perforin acts directly on endothelial cell membrane to cause BBB disruption; (2) perforin has direct effects on neuronal excitability; or if (3) other factors such as vascular endothelial growth factor (VEGF) released by neurons are involved.
These data may provide a novel therapeutic venue if it is confirmed that seizures are preceded by activation of T cells, acting together with NK cells to cause BBB disruption. A recent report has shown that pediatric patients affected by multidrug-resistant seizures benefit from antiinflammatory treatment, namely steroids (Marchi et al., 2011b). The positive effects of these therapeutic interventions were correlated with improved BBB function, suggesting that as seen in pilocarpine seizures, human epilepsy has an etiologic cerebrovascular component. A recent report has listed these antiinflammatory approaches for treating status epilepticus (Beghi & Shorvon, 2011 and Commentary); our data suggest that steroidal treatment can be used as an add-on therapy in a much broader range of epilepsies (Marchi et al., 2011b).
Splenectomy in Neurologic Diseases
The involvement of the immune system in brain disease is a well-accepted reality, and several therapies aimed at the maintenance of a normal immune response have been proven effective in MS and other neuroimmune diseases. These concepts have recently been expanded to encompass seizure disorders and stroke. The spleen is a chief controller of immune function and, in particular, it determines the fate of CD8+ T cells, which, as discussed earlier, are involved in pilocarpine seizures (Marchi et al., 2009, 2011c). Stroke researchers have shown that splenectomy has a profound ameliorating effect on postischemic function (Ajmo et al., 2008; Lee et al., 2008), and the same was reported for pilocarpine-induced seizures (Marchi et al., 2011c). All of these diseases share in common a leaky BBB and abnormal neuronal function or apoptosis. These results, taken together, strongly support a role for spleen-mediated activation of T cells as in precipitant of experimental seizures.
When comparing BBBD-triggered seizures to diseases with an acknowledged BBB component, it was reported that cell extravasation often seen in brain of MS is virtually absent in brains from epileptics or in rat brain at the time of status epilepticus induced by pilocarpine. Experiments in different animal models confirmed that leaky BBB does not always translate into presence of parenchymal leukocytes (Marchi et al., 2007a,b, 2009, 2010b, 2011c). Therefore, white blood cell extravasation is a rare event in the parenchyma of animals with disrupted BBB, or after seizures induced by pilocarpine or in patients with chronic epilepsy. Lymphocytes extravasated only into the Virchow-Robin space or in the subdural-pial space. However, widespread leakage of the BBB was nevertheless associated with increased leukocyte adhesion to the vascular wall. No extravasation was observed in the same regions. Consequently, adhesion of activated CD8+ T cells or perforin itself released by NK cells is sufficient to disrupt local BBB integrity without cell extravasation.
Conclusions
Models and strategies to study neurologic diseases constantly adapt to novel hypotheses and tools used to probe the CNS. The pairing of microanatomy with electrophysiology was an important step used to demonstrate how neuroanatomic detail marries function. New methods and instruments have been developed to study the brain, but understanding the anatomic detail of cellular processes remains important. For example, whether entry of leukocytes into the brain is important for seizure generation will likely be answered by careful anatomic studies. An accurate experimental design in basic research still requires a strong neuroanatomic component, as demonstrated by the continuous growth of neuroimaging in clinical neurosciences.
Acknowledgments
This issue of Epilepsia honors one of the most influential CNS neuroanatomists of our time. I was fortunate to have the honor to work with Dr. Jürgen Wenzel, for a short yet productive period of time. At that time, Dr. Wenzel was applying electron microscopy to hippocampal excitability and function. Unlike many other neuroanatomic studies, Jürgen’s work was dynamic in the sense that most of the cells or tissues he described were first characterized electrophysiologically. This approach allowed investigators to better understand what a neuron really does, since either technique (electrophysiology and morphology) used alone could not illustrate the consequences of neuronal activity. By using state-of-theart microscopic techniques on cell labeled during recordings, many new connections between principal cells and interneurons were discovered. This seminal work is still cited today by those studying interneurons or hippocampal circuits. In an article coauthored with Wenzel and colleagues, we demonstrated that, similar to neurons, astrocytes are topographically segregated across different hippocampal regions (D’Ambrosio et al., 1998). Jürgen used unprecedented methodologic rigor and tenacity to unveil unsuspected complexities in the neuroanatomy of these different populations of hippocampal astrocytes. In one of his superbly executed immunodetection studies he described the relationship between the BBB and hippocampal astrocytic processes (Emmi et al., 2000). This brain–vascular interface became an unexpected trigger of many neurologic diseases and the focus of this short review.
Footnotes
Disclosures
The author has no conflicts of interest to disclose.
I confirm that I have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Aaslid R. Cerebral autoregulation and vasomotor reactivity. Front Neurol Neurosci. 2006;21:216–228. doi: 10.1159/000092434. [DOI] [PubMed] [Google Scholar]
- Ajmo CT, Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to strokeinduced neurodegeneration. J Neurosci Res. 2008;86:2227–2234. doi: 10.1002/jnr.21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amtorp O, Sorensen SC. The ontogenetic development of concentration differences for protein and ions between plasma and cerebrospinal fluid in rabbits and rats. J Physiol. 1974;243:387–400. doi: 10.1113/jphysiol.1974.sp010759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asconape JJ, Penry JK. Poststroke seizures in the elderly. Clin Geriatr Med. 1991;7:483–492. [PubMed] [Google Scholar]
- Beghi E, Shorvon S. Antiepileptic drugs and the immune system. Epilepsia. 2011;52:40–44. doi: 10.1111/j.1528-1167.2011.03035.x. [DOI] [PubMed] [Google Scholar]
- Benitah J, Balser JR, Marban E, Tomaselli GF. Proton inhibition of sodium channels: mechanism of gating shifts and reduced conductance. J Membr Biol. 1997;155:121–131. doi: 10.1007/s002329900164. [DOI] [PubMed] [Google Scholar]
- Bischofberger M, Gonzalez MR, van der Goot FG. Membrane injury by pore-forming proteins. Curr Opin Cell Biol. 2009;21:589–595. doi: 10.1016/j.ceb.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Wenzel J, Schwartzkroin PA, McKhann GM, Janigro D. Functional specialization and topographic segregation of hippocampal astrocytes. J Neurosci. 1998;18:4425–4438. doi: 10.1523/JNEUROSCI.18-12-04425.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009a;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David Y, Flores LP, Ivens S, Heinemann U, Kaufer D, Friedman A. Astrocytic potassium and glutamate buffering controls synaptic responses in a frequency-dependent manner. Epilepsia. 2009b;50:86. [Google Scholar]
- Davson H, Segal MB. Blood–brain–CSF interactions. Physiology of the CSF and blood–brain barriers. CRC Press; Boca Raton, FL: 1995. [Google Scholar]
- Dehaye JP, Winand J, Poloczek P, Christophe J. Characterization of muscarinic cholinergic receptors on rat pancreatic acini by N-[3H]methylscopolamine binding. Their relationship with calcium 45 efflux and amylase secretion. J Biol Chem. 1984;259:294–300. [PubMed] [Google Scholar]
- DeVivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behma RA, Harik SI. Defective glucose transport across the blood–brain barrier as a cause for persistent hypoglycorrhachia, seizure and developmental delay. N Engl J Med. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced shortterm synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski SM, Desai SY, Marroni M, Cucullo L, Goodrich K, Bingaman W, Mayberg MR, Bengez L, Janigro D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Fredholm BB. Adenosine A1 receptors inhibit adenylate cyclase activity and transmitter release and hyperpolarize pyramidal neurons in the rat hippocampus. J Pharmacol Exp Ther. 1989;249:31–37. [PubMed] [Google Scholar]
- Emmi A, Wenzel HJ, Schwartzkroin PA, Taglialatela M, Castaldo P, Bianchi L, Nerbonne J, Robertson GA, Janigro D. Do glia have heart? Expression and functional role for ether-a-go-go currents in hippocampal astrocytes. J Neurosci. 2000;20:3915–3925. doi: 10.1523/JNEUROSCI.20-10-03915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabene PF, Navarro MG, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, Osculati A, Marzola P, Nicolato E, Homeister JW, Xia L, Lowe JB, McEver RP, Osculati F, Sbarbati A, Butcher EC, Constantin G. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabene PF, Bramanti P, Constantin G. The emerging role for chemokines in epilepsy. J Neuroimmunol. 2010;224:22–27. doi: 10.1016/j.jneuroim.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Ghosh C, Gonzalez-Martinez J, Hossain M, Cucullo L, Fazio V, Janigro D, Marchi N. Pattern of P450 expression at the human blood– brain barrier: roles of epileptic condition and laminar flow. Epilepsia. 2010;51:1408–1417. doi: 10.1111/j.1528-1167.2009.02428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh C, Marchi N, Desai NK, Puvenna V, Hossain M, Gonzalez-Martinez J, Alexopoulos AV, Janigro D. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia. 2011a;52:562–571. doi: 10.1111/j.1528-1167.2010.02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh C, Puvenna V, Gonzalez-Martinez J, Janigro D, Marchi N. Blood–brain barrier P450 enzymes and multidrug transporters in drug resistance: a synergistic role in neurological diseases. Curr Drug Metab. 2011b;12:742–749. doi: 10.2174/138920011798357051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GA, Meno JR, Nguyen TS, Stanness KA, Janigro D, Winn RH. Adenosine-induced modulation of excitatory amino acid transport across isolated brain arterioles. J Neurosurg. 2003;98:554–560. doi: 10.3171/jns.2003.98.3.0554. [DOI] [PubMed] [Google Scholar]
- Hauser WA. Epidemiology of acute symptomatic seizures. In: Engel G, Pedley TA, editors. Epilepsy: a comprehensive textbook. Lippincott Williams and Wilkins; Philadelphia, PA: 2008. pp. 71–75. [Google Scholar]
- Heinemann U, Eilers A, Gabriel S, Jandova K, Jauch R, Meencke HJ, Njunting M, Pasler D, Schulze K, Lehmann TN. Changes of glial functions in epileptic brain tissue: alteration of K+-buffering. Klinische Neurophysiologie. 2002;32:128–136. [Google Scholar]
- Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J, Steinhauser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–2096. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Ivens S, Gabriel S, Greenberg G, Friedman A, Shelef I. Blood– brain barrier breakdown as a novel mechanism underlying cerebral hyperperfusion syndrome. J Neurol. 2010;257:615–620. doi: 10.1007/s00415-009-5384-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janigro D, Gasparini S, DAmbrosio R, McKhann G, DiFrancesco D. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci. 1997;17:2813– 2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko KR, Ngai AC, Winn HR. Role of adenosine in regulation of regional cerebral blood flow in sensory cortex. Am J Physiol. 1990;259:H1703–H1708. doi: 10.1152/ajpheart.1990.259.6.H1703. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn A, Golan H, Melamed I, Pascual-Marqui R, Friedman A. Focal cortical dysfunction and blood–brain barrier disruption in patients with Postconcussion syndrome. J Clin Neurophysiol. 2005;22:1–9. doi: 10.1097/01.wnp.0000150973.24324.a7. [DOI] [PubMed] [Google Scholar]
- Krizanac-Bengez L, Mayberg MR, Janigro D. The cerebral vasculature as a therapeutic target for neurological disorders and the role of shear stress in vascular homeostatis and pathophysiology. Neurol Res. 2004;26:846–853. doi: 10.1179/016164104X3789. [DOI] [PubMed] [Google Scholar]
- Lee ST, Chu K, Jung KH, Kim SJ, Kim DH, Kang KM, Hong NH, Kim JH, Ban JJ, Park HK, Kim SU, Park CG, Lee SK, Kim M, Roh JK. Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain. 2008;131:616–629. doi: 10.1093/brain/awm306. [DOI] [PubMed] [Google Scholar]
- Librizzi L, Regondi MC, Pastori C, Frigerio S, Frassoni C, de Curtis M. Expression of adhesion factors induced by epileptiform activity in the endothelium of the isolated guinea pig brain in vitro. Epilepsia. 2007;48:743–751. doi: 10.1111/j.1528-1167.2007.01047.x. [DOI] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. Seizurepromoting effect of blood–brain barrier disruption. Epilepsia. 2007a;48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Oby E, Fernandez N, Uva L, de Curtis M, Batra A, Santaguida S, Barnes V, van Boxel A, Najm I, Janigro D. In vivo and in vitro effects of pilocarpine: relevance to epileptogenesis. Epilepsia. 2007b;48:1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Fan QY, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Gonzalez-Martinez J, Nguyen MT, Granata T, Janigro D. Transporters in drug-refractory epilepsy: clinical significance. Clin Pharmacol Ther. 2010a;87:13–15. doi: 10.1038/clpt.2009.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Teng Q, Ghosh C, Fan Q, Nguyen MT, Desai NK, Bawa H, Rasmussen P, Masaryk TK, Janigro D. Blood–brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Res. 2010b;1353:176–186. doi: 10.1016/j.brainres.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Teng QS, Nguyen MT, Franic L, Desai NK, Masaryk T, Rasmussen P, Trasciatti S, Janigro D. Multimodal investigations of trans-endothelial cell trafficking under condition of disrupted blood–brain barrier integrity. BMC Neurosci. 2010c;11:34. doi: 10.1186/1471-2202-11-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Granata T, Freri E, Ciusani E, Ragona F, Puvenna V, Teng QS, Alexopolous A, Janigro D. Efficacy of anti-inflammatory therapy in a model of acute seizures and in a population of pediatric drug resistant epileptics. PLoS ONE. 2011b;6(3):e18200. doi: 10.1371/journal.pone.0018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Johnson A, Puvenna V, Tierney W, Ghosh C, Cucullo L, Fabene PF, Janigro D. Modulation of peripheral cytotoxic cells and ictogenesis in a model of seizures. Epilepsia. 2011c;52:1627–1634. doi: 10.1111/j.1528-1167.2011.03080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TS, Winn HR, Janigro D. ATP-sensitive potassium channels may participate in the coupling of neuronal activity and cerebrovascular tone. Am J Physiol Heart Circ Physiol. 2000;278:H878–H885. doi: 10.1152/ajpheart.2000.278.3.H878. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- Oby E, Caccia S, Vezzani A, Moeddel G, Hallene K, Guiso G, Said T, Bingaman W, Marchi N, Baumgartner C, Pirker S, Czech T, Lo RG, Janigro D. In vitro responsiveness of human-drug-resistant tissue to antiepileptic drugs: insights into the mechanisms of pharmacoresistance. Brain Res. 2006;1086:201–213. doi: 10.1016/j.brainres.2006.02.068. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K(+) buffering to cell differentiation. J Neurochem. 2008;107:589–601. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas CA, Ransom BR. Depolarization-induced alkalinization (DIA) in rat hippocampal astrocytes. J Neurophysiol. 1994;72:2816–2826. doi: 10.1152/jn.1994.72.6.2816. [DOI] [PubMed] [Google Scholar]
- Pardini J, Bailes JE, Maroon JC. Mild traumatic brain injury in adults and concussion in sports. In: Winn HR, editor. Youmans neurological surgery. Elsevier; Philadelphia, PA: 2011. pp. 3380–3389. [Google Scholar]
- Parkinson FE, Friesen J, Krizanac-Bengez L, Janigro D. Use of a three-dimensional in vitro model of the rat blood–brain barrier to assay nucleoside efflux from brain. Brain Res. 2003;980:233–241. doi: 10.1016/s0006-8993(03)02980-9. [DOI] [PubMed] [Google Scholar]
- Pavlovsky L, Seiffert E, Heinemann U, Korn A, Golan H, Friedman A. Persistent BBB disruption may underlie alpha interferoninduced seizures. J Neurol. 2005;252:42–46. doi: 10.1007/s00415-005-0596-3. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. Immunology barrier to electrical storms. Nature. 2009;457:155–156. doi: 10.1038/457155a. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Walz W, Davis PK, Carlini WG. Anoxia-induced changes in extracellular K+ and pH in mammalian central white matter. J Cereb Blood Flow Metab. 1992;12:593–602. doi: 10.1038/jcbfm.1992.83. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Razani-Boroujerdi S, Behl M, Hahn FF, Pena-Philippides JC, Hutt J, Sopori ML. Role of muscarinic receptors in the regulation of immune and inflammatory responses. J Neuroimmunol. 2008;194:83–88. doi: 10.1016/j.jneuroim.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Tracey KJ. Cholinergic control of inflammation. J Intern Med. 2009;265:663–679. doi: 10.1111/j.1365-2796.2009.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg G. Leukocyte mobilization from the guinea pig spleen by muscarinic cholinergic stimulation. Experientia. 1994;50:40–43. doi: 10.1007/BF01992047. [DOI] [PubMed] [Google Scholar]
- Schroder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, Steinhauser C. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. 2000;41:S181–S184. doi: 10.1111/j.1528-1157.2000.tb01578.x. [DOI] [PubMed] [Google Scholar]
- Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829– 7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith QR. Transport of glutamate and other amino acids at the blood–brain barrier. J Nutr. 2000;130:1016S–1022S. doi: 10.1093/jn/130.4.1016S. [DOI] [PubMed] [Google Scholar]
- Somjen GG, Segal MB, Herreras O. Osmotic-hypertensive opening of the blood–brain barrier in rats does not necessarily provide access for potassium to cerebral interstitial fluid. Exp Physiol. 1991;76:507–514. doi: 10.1113/expphysiol.1991.sp003516. [DOI] [PubMed] [Google Scholar]
- Sotgiu S, Murrighile MR, Constantin G. Treatment of refractory epilepsy with natalizumab in a patient with multiple sclerosis. Case report. BMC Neurol. 2010;10:84. doi: 10.1186/1471-2377-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanness KA, Guatteo E, Janigro D. A dynamic model of the blood–brain barrier “in vitro”. Neurotoxicology. 1996;17:481–496. [PubMed] [Google Scholar]
- Steinhauser C, Seifert G. Astrocyte dysfunction in temporal lobe epilepsy. Epilepsia. 2010;51:54. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, D’Ambrosio R. Chronic dysfunction of astrocytic inwardly rectifying K(+) channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. 2010;104:3345–3360. doi: 10.1152/jn.00398.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill JL, Mathews J, Scherle P, Conrad DH. Glutamate signaling through the kainate receptor enhances human immunoglobulin production. J Neuroimmunol. 2011;233:80–89. doi: 10.1016/j.jneuroim.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Zhang B, Kosaka Y, Burrows GG, Grafe MR, Vandenbark AA, Hurn PD, Offner H. Recombinant T cell receptor ligand treats experimental stroke. Stroke. 2009;40:2539–2545. doi: 10.1161/STROKEAHA.108.543991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suidan GL, Mcdole JR, Chen Y, Pirko I, Johnson AJ. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS ONE. 2008;3(8):e3037. doi: 10.1371/journal.pone.0003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate SK, Sisodiya SM. Multidrug resistance in epilepsy: a pharmacogenomic update. Expert Opin Pharmacother. 2007;8:1441–1449. doi: 10.1517/14656566.8.10.1441. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in rat hippocampal slices. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Trombin F, Gnatkovsky V, de Curtis M. Changes in action potential features during focal seizure discharges in the entorhinal cortex of the in vitro isolated guinea pig brain. J Neurophysiol. 2011;106:1411– 1423. doi: 10.1152/jn.00207.2011. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev. 2010;235:35–54. doi: 10.1111/j.0105-2896.2010.00896.x. [DOI] [PubMed] [Google Scholar]
- Zattoni M, Mura ML, Deprez F, Schwendener RA, Engelhardt B, Frei K, Fritschy JM. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J Neurosci. 2011;31:4037– 4050. doi: 10.1523/JNEUROSCI.6210-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci. 2008;37:1–10. doi: 10.1016/j.mcn.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Dreier JP, Heinemann U. Paroxysmal epileptiform discharges in temporal lobe slices after prolonged exposure to low magnesium are resistant to clinically used anticonvulsants. Epilepsy Res. 1995;20:105–111. doi: 10.1016/0920-1211(94)00067-7. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]