

Abstract
A multitude of cytotactic cues direct cell migration in development, cancer metastasis and wound healing. However, our understanding of cell motility remains fragmented partially because current migration devices only allow the study of independent factors. We developed a cell motility assay that allows competitive recruitment of a given cell population simultaneously by gradients of multiple cytotactic cues, observable under real-time imaging. Well-defined uniform gradients of cytotactic cues can be independently generated and sustained in each channel. As a case study, bone marrow mesenchymal stem/stromal cells (MSCs) were exposed to 15 cytokines that are commonly present in arthritis. Cytokines that induced robust recruitment of MSCs in multiple groups were selected to ‘compete’ in a final round to yield the most chemotactic factor(s) based on cell migration numbers, distances, migration indices and motility over time. The potency of a given cytokine in competition frequently differed from its individual action, substantiating the need to test multiple cytokines concurrently due to synergistic or antagonistic effects. This new device has the rare capacity to screen molecules that induce cell migration in cancer therapy, drug development and tissue regeneration.
Keywords: Stem cells, migration, microfluidic, microfabrication, cell homing, cytokines
Introduction
Cells are constantly exposed to multiple cytotactic cues, concurrently or in succession, during fundamental biological processes such as development, wound healing or cancer metastasis. Closely regulated signals are necessary for the maintenance of stem cell quiescence and immune balance in postnatal homeostasis [1]. In either trauma or cancer metastasis, locally present or systemically circulating endogenous cells migrate to and from the tissues of interest [2, 3]. Cancer cells secrete abundant amounts of cytokines and chemokines that invite the sprouting of blood and lymphatic vessels in ways that benefit metastasis. Interventional approaches for tissue regeneration rely on transplanted cells and/or host endogenous cells that, when aggregated, begin to reorganize and regenerate tissues. However, our understanding of cell motility, one of the most fundamental processes in cell biology, remains fragmented. Proteins, peptides, and chemical compounds that regulate cell motility may target a broad range of therapeutic purposes [4]. Whereas cell therapy has long been practiced as blood transfusion and bone marrow transplantation, recent interest has focused on the delivery of cells in numerous disease models involving virtually all tissues and organs [2]. Bone marrow mesenchymal stem/stromal cells (MSCs) are the most widely delivered adult, non-hematopoietic cells in a broad range of experimental models, preclinical and patient studies. In several disease and trauma models, MSC delivery shows various efficacy that remains to be further evaluated [5–8]. However, the putative therapeutic potential of MSCs, which is still superficially understood for the majority of tissues and organs, is frequently attributed to the effects of MSCs on immune modulation, chemotaxis, and regulation of inflammation and angiogenesis, rather than necessarily serving as replacement cells for tissue repair [5–8]. Recently, we showed that host endogenous cells were recruited by TGFβ3 into microchanneled biomaterial scaffolds and led to the regeneration of the entire cartilage layer of a synovial joint in vivo [9]. However, the roles played by TGFβ3 could potentially be enhanced by other factors [9]. Cytotactic cues that are most efficacious in the regeneration of a given tissue are largely elusive.
Common cell migration assays including the Boyden chamber, scratch assay and under agarose assay are based on numerous reproducible experiments, but are invariably of low efficiency, typically allowing the study of one factor and one cell type at a time [10–12]. The Boyden chamber allows the observation of one factor on a single cell type at a time, and does not allow the monitoring of cell migration in real time. The scratch assay is designed to monitor the movement of confluent cells that migrate across a gap, but does not allow for the formation of a cytokine gradient that is typically responsible for cell motility in vivo [13]. The conventional under-agarose assay is conducted by allowing cell migration underneath a polymerized layer of agarose gel towards a chemoattractant gradient, but nonetheless is problematic for studying gradients formed by multiple cytokines that may cross-contaminate through the porous agarose. In addition, the conventional under-agarose assay allows cell migration in all directions, making it difficult to monitor cell motility patterns in real time. Cell migration devices utilizing microfluidic technology offer advantages of reproducibility, precision, minimal volume of reagent requirements, well-characterized culture environments and quantifiable gradient generation [14].
Several existing microfluidic devices generate concentration gradients based on either convective flow or molecular diffusion [15–22]. Laminar flow based devices offer the advantage of rapid gradient onset and the ability to generate continuous and switchable long-term gradients [16, 19, 22]. However, high intrinsic shear stress in these devices could wash away chemokines or cytokines that are produced by the cells, and may negatively affect cell survival and migration [15, 17]. Diffusion based devices have a limited gradient duration, but their stable conditions help promote cell viability and make them advantageous over laminar flow based devices for use in studying mammalian cell migration [17, 18, 23].
Various designs of microfluidic diffusion based gradient generators have been developed [14, 15, 24, 25]. In an effort to inhibit convective flows, porous polyethylene and polycarbonate membranes have been incorporated that allow for the diffusion of small molecules, but are difficult to fabricate [15, 25]. Alternatively, hydrogels are advantageous as a diffusion gradient generator due to ease of use, biocompatibility, comparable diffusion coefficients to water and ability to generate both short range (<1 mm) and long range concentration gradients (>1 cm) [24]. In particular, agarose has been used as a microfluidic gradient generator because it can form a stable hydrogel of high porosity, which can readily promote the diffusion of chemokines, gases and nutrients [20, 23, 24, 26]. Current agarose gradient generating migration devices can only test the effects of two opposing chemotactic signals on mammalian cell migration [20, 23]. While agarose migration devices have been developed for assaying the effects of various chemicals on bacterial cell migration, existing microfluidic systems cannot assay competitive recruitment of a given mammalian cell population simultaneously in response to multiple cytotactic cues in one single system [15, 26]. In particular, current devices do not allow for the simultaneous assay of multiple cell types against a chemotactic cue. Moreover, cells exposed to multiple cytokines that yield synergistic and/or antagonistic effects cannot be revealed by current agarose-based mammalian cell migration assays.
In this report, we developed a novel cell migration assay that allows simultaneous assay of competitive mammalian cell recruitment by multiple cytotactic cues, representing another step towards mimicking in vivo microenvironments. The device has six microfluidic channels, each with separate inlets for cell seeding and cytokine infusion. Given that cell migration is confined to microfluidic channels, the process of migration can be imaged in real time using time-lapse microscopy as we demonstrate below. Gradients generated within the agarose filled channels have reverse directionality, allowing the effects of a single cytokine to be concurrently tested on 6 different cell types as well. The following data demonstrate the utility of this high-throughput multi-channel cell migration assay and its potential broad applications in studying tissue development, cancer metastasis, wound healing and tissue regeneration.
Methods
Design and fabrication of the microfluidic cell migration system
A transparency mask containing the design of the device was made using AutoCAD and printed with high resolution (CADArt, Bandon, OR). Using standard photolithography, a silicon master was fabricated with a layer of SU-8 containing a negative imprint of the device pattern and then silanized by evaporation of trichloro (1,1,2,2-perfluoocytl) silane (Sigma, St. Louis, MO). Subsequently, using soft lithography, the master was replicated in polydimethylsiloxane (PDMS) to obtain a positive imprint of the pattern. Briefly, Sylgard 184 silicone elastomer base (Ellsworth Adhesives, Germantown, WI) was mixed at a 1:10 ratio with the curing agent, degassed in a vacuum chamber to remove any air bubbles, and cured in a dry oven at 60°C for 2 hrs. Biopsy punches with a diameter of 1 or 6 mm were used to create the inlets. Device channels were all 5 mm in length, 100 μm in height, and 250 μm in diameter. PDMS devices were permanently adhered to pre-cleaned glass slides (25×75 mm) using plasma bonding. Subsequently, the channels were filled with distilled water to maintain a hydrophilic environment.
Isolation of primary human MSCs
Human bone marrow MSCs were isolated from fresh adult donor samples (AllCells, Emeryville, CA) using our prior methods [27–29]. Briefly, the bone marrow sample was incubated with the RosetteSep® Human Mesenchymal Stem Cell Enrichment Cocktail (Stem Cell Technologies, Vancouver, Canada), placed on top of the Ficoll-Paque™ and centrifuged to separate the layers by density. Mononucleated cells were collected from the Ficoll-Paque™ interface, washed with PBS containing 2% FBS and 1 mM EDTA, and then plated. Mononucleated and adherent cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Carlsbad, CA), 10% FBS (Gibco, Carlsbad, CA) and 5% Antibiotics/Antimicrotics, and only expanded to the second passage for all experiments. These mononucleated and adherent cells are consistent with typical culture of MSCs in the literature [5, 30], although their heterogeneity is recognized by several studies including our recent clonal analysis [31].
Cytokine diffusion profile and agarose contractility characterization
To determine if this microfluidic assay system would be amenable to testing a variety of cytokines, fluorescein conjugated dextrans with molecular weights ranging from 3–70kDa were infused in the inlets of the channels to generate a concentration gradient and PBS was placed in the channel outlets. The channels were filled with 0.5% agarose (Invitrogen, Carlsbad, CA) mixed with Fluoresbrite® rhodamine carboxylate microspheres with a diameter of 6 μm (Polysciences, Warrington, PA) to determine if the agarose underwent any significant contraction over 12 hrs. Time-lapse microscopy captured images every 10 min for the calculation of microsphere position displacement and cytokine diffusion profiles. The fluorescent intensity profiles throughout the channel were determined using ImageJ software.
Separate diffusion experiments were conducted to determine if uniform cytokine diffusion can occur from the center chamber towards the peripheral chambers. Dextran conjugated with rhodamine B (10 kDa; Invitrogen, Carlsbad, CA) was infused into the center chamber, with 0.5% agarose in the channels and ultrapure water in the peripheral wells. Images were taken after 5 and 21 hrs of diffusion. The average pixel intensity in the six channels was calculated to assess the suitability of the device for testing the cytotactic effects of one cytokine in the center chamber on six different cell types that can be readily seeded in the peripheral wells. Additional experiments were conducted to determine if any fluorescent contamination occurs between the six channels when cytokines are diffused from the peripheral wells towards the center chamber. Dextran (10 kDa) conjugated with rhodamine B or fluorescein was infused into alternating peripheral wells, and imaged after 5 and 21 hrs. Consistency of the diffusion gradients was evaluated by diffusing dextran conjugated (10 kDa) with fluorescein from the outlets of three separate devices. After the tested 21 hrs, one channel in each device was imaged and the fluorescent intensity was determined in 15 locations within each channel. The average and standard deviation of the fluorescent intensities were calculated.
Cell migration by competitive effects of multiple cytokines
Microfluidic devices were sterilized for 10 min by flowing 70% ethanol through all the channels. Devices were washed once with PBS, followed by fibronectin coating (75 μg/mL; Sigma, St Louis, MO) for 2 hrs at 37°C, and then washed twice with PBS. Sterile 1% agarose was diluted 1:2 with cell culture medium and injected into the center inlet, which distributed throughout the channel due to the equal pressure differential between the agarose inlet and side wells. Immediately preceding the inlet to the side wells were two cubes (edge=50 μm) designed to increase the surface tension between the agarose and the channel and prevent the agarose from contracting. Following solidification, any excess agarose in the channel outlets was aspirated. The center chamber of the device was seeded with 40,000 MSCs. Cytokines listed in Figure 3a, each at a dose of 200 ng, were placed in the peripheral wells. PBS served as a control for each group and the final round. The following cytokines were selected for their high prevalence in the synovial joint in health and arthritis: TGFβ3 (Cell Sciences, Canton, MA); PDGFbb, Lymphotactin, Fractalkine, SDF1, IGF1, MIP1α, MIP1β, MCP1, IP10 (R & D Systems, Minneapolis, MN); TGFβ1, TNFα, VEGF (Peprotech, Rocky Hill, NJ); FGFb and IL6 (Invitrogen, Carlsbad, CA). A minimum of 3 separate experiments were conducted for each group and the final round, with devices maintained for 24 hrs at 37°C and 5% CO2 in an environmental chamber. Randomly selected experiments were time-lapse imaged. Subsequently, cells were fixed with 10% formalin and stained with DAPI to visualize nuclei. The total number of migrated cells, migration distances and migration indices were quantified using ImageJ.
Figure 3.

Cytokine selection for cell migration assays. a: Cytokines that are crucial for arthritis were randomly allocated into 3 groups with PBS as a control for each group. The ‘top performers’ from the first three groups (in red) were selected for the ‘final’ round, which yielded the final group capable of robust cell recruitment. b: Competitive cell migration of the selected, most robust cytotactic cues. Phase contrast and DAPI stained images at 24 hrs following exposure to MIP1α, TGFβ3, TNFα, lymphotactin, PDGFbb and PBS (control). DAPI images were subsequently used to quantify the number of migrated cells, migratory distance and migration index.
Cell migration by PBS only
To determine if cells are biased to migrate towards a certain channel in the device, a control experiment was conducted by filling all 6 outlets of competition devices with PBS (n=3). The center chambers were seeded with 40,000 MSCs and allowed to migrate for 24 hrs at 37°C and 5% CO2. All post-migration procedures were consistent with cytokine containing experiments including cell fixation, data acquisition and analysis.
Cell migration by PDGF only
A separate device was fabricated with individual channels that were identical in dimensions to each of the channels in the connected 6-channel devices. All experimental conditions were consistent with the connected channel device experiments, including seeding the MSCs at a density of 40,000 cells from the same donor, infusion of PDGFbb at a dose of 200 ng and all post-migration procedures including cell fixation, data acquisition and analysis.
Statistical analysis
All quantitative data for each condition were pooled from at least three independent experiments. Upon confirmation of a normal data distribution, One-way ANOVA with post hoc LSD tests was used with α values of p ≤ 0.05.
Results
We designed and fabricated a high-throughput microfluidic device (Fig. 1a,b) and tested the chemotactic effects of multiple cytokines on human mesenchymal stem cells (MSCs). The device requires minimal setup, including agarose injection, cell seeding and cytokine loading (Fig. 1c). The device was rigorously characterized by determining diffusion profiles and agarose contractility to ensure reproducibility between experiments.
Figure 1.
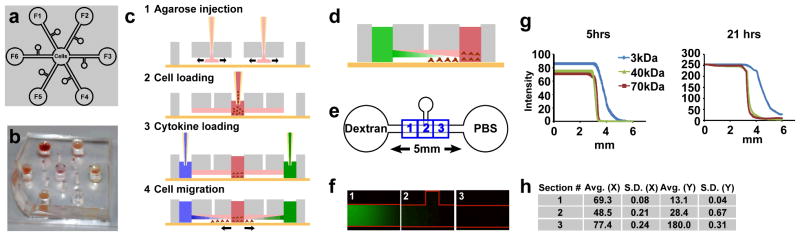
Design, experimental schematics and device characterization. a: Design of a novel microfluidic device for competitive cell migration assays. Cells are seeded in the center chamber, whereas cytotactic factors (F1–F6) are infused in peripheral wells. A channel (dia: 250 μm) connects the center chamber to each of the peripheral wells. b: Photograph of the fabricated microfluidic device. c: Schematic of agarose loading and cell seeding. 1. Agarose is injected into the chamber and allowed to flow and fill the entire microchannel. 2. Cells are seeded into the center chamber. 3. Cytotactic cues are loaded into the peripheral wells. 4. A gradient of cytotactic cues is established over time to form a descending concentration gradient towards the center chamber. d: Channels were filled with agarose and fluorescently tagged microspheres. The peripheral wells were infused with fluorescently tagged dextrans of varying molecular weights. e: Three consecutive regions centered in the channel, each with a length of 600 μm, were selected to measure agarose contractility. f: A gradient of fluorescence intensity (10 kDa) was formed throughout the three regions within the observed 70 min. g: Diffusion profiles of fluorescently tagged dextrans (3, 40, and 70 kDa) after 5 hrs and 21 hrs. Profiles are represented as pixel intensity versus distance h: Average difference (Avg.) in X or Y pixel coordinate location between two microspheres over time and the corresponding standard deviations (S.D.). The modest standard deviations of the average microsphere position displacement in each of the three regions indicate minimum agarose contraction over the observed 490 min.
Verification of diffusion gradient and minimum agarose contractility
To determine whether a cytokine gradient was formed, we first studied the diffusion profiles of various fluorescein conjugated dextrans in agarose filled channels (Fig. 1d,e). Dextrans with molecular weights of 3, 40 and 70 kDa were selected to represent the range of molecular weights of the cytokines to be studied in the present work. A gradient of decreasing fluorescence intensity was observed in the agarose over time (Fig. 1f). Despite similar diffusion patterns, dextrans with smaller molecular weights diffused more rapidly than those with larger molecular weights (Fig. 1g). The agarose was mixed with rhodamine carboxylate microspheres to determine agarose contractility, if any, by analysis of position displacement over time. Up to the observed 12 hrs, agarose underwent minimum contraction, as evidenced by the minimal standard deviations both along the X and Y directions (Fig. 1h). Thus the microfluidic agarose assay can be used without significant contraction and promotes relatively uniform diffusion parameters.
Separate diffusion experiments were conducted to determine whether a given cytokine may uniformly diffuse from the center well to the peripheral wells. This configuration would be used to test the effects of a single cytokine on competitively recruiting six different cell types (Sup. Fig. 1). Dextran conjugated with rhodamine B (10 kDa) was infused into the center chamber, whereas the peripheral wells were filled with ultrapure water. A dextran gradient was formed in each agarose (0.5% w/v) filled channel over 21 hrs (Sup. Fig. 1a). The average pixel intensity and associated standard deviations through all six channels were calculated (Sup. Fig. 1b). The minimum standard deviations of the pixel intensity after 5 hrs (3.4) and after 21 hrs (1.6) indicate the formation and maintenance of uniform diffusion patterns in each of the six channels.
To determine if any fluorescence contamination occurs between the device channels, dextran conjugated with rhodamine B or fluorescein was infused into alternating peripheral wells (Fig. 2), whereas the center chamber was filled with ultrapure water. The device was imaged after 5 and 21 hrs of diffusion. After 21 hrs, alternating gradients of fluorescence intensity could be readily visualized (Fig. 2a). We observed a continuous distribution of dextran (Fig. 2b and d), with sustained fluorescence intensity above zero throughout the channel’s entire length (Fig. 2c). Interestingly, the fluorescence intensity increases in the center chamber, demonstrating the potential of ample cytokine exposure to cells seeded in the center chamber (Fig. 2d). The center chamber imaged after 5 hrs displayed distinct regions of rhodamine B or fluorescein surrounding each channel inlet, with minimal crossover between channel inlets (Fig. 2e–h). In the observed 21 hrs, continuous concentric rings of rhodamine B or fluorescein had reached and filled the center chamber, demonstrating that cells seeded in the center chamber are exposed to multiple cytotactic factors infused in the peripheral wells and therefore may be recruited competitively by the most robust cytotactic cue(s) among the six factors infused in the peripheral wells (Fig. 2i–j).
Figure 2.

Gradients converge from peripheral wells to the center chamber. Dextran (10 kDa) conjugated with rhodamine B or fluorescein was infused into alternating peripheral wells. The channels were filled with 0.5% agarose. The center chamber was filled with ultrapure water. a: After 21 hrs, fluorescent gradients can be seen in each channel (a1–a6) with minimum fluorescent cross-over between channels. b: A single channel (a2) used to determine the intensity profile of dextran conjugated with fluorescein (10 kDa) over the entire channel length after 21 hrs. c: The intensity profile remains above zero over the entire channel length, indicating a continuous cytokine distribution over the entire channel. Interestingly, the fluorescence intensity increases in the center chamber, demonstrating robust cytokine exposure to cells when they are seeded in the chamber. d: Enlarged image of a2. e: Fluorescein presence at the center chamber after 5 hrs. f: Rhodamine B presence at the center chamber after 5 hrs. g: Merged fluorescence images after 5 hrs. Distinct regions of rhodamine B or fluorescein can be seen surrounding each channel inlet, with minimal cross-over between channel inlets. h: Schematic of the center region imaged for e–j. i: Rhodamine B presence at the center chamber after 21 hrs. j: Fluorescein presence at the center chamber after 21 hrs. Continuous rings of rhodamine B or fluorescein can be seen filling the center channel, indicating a uniformity in cytokine exposure to all potentially seeded cells in the center chamber and demonstrating competition among cytokines placed in the periphery.
Separate diffusion experiments were conducted to determine whether cytokine diffusion profiles were consistent between channels. Dextran conjugated with fluorescein (10 kDa) was infused into the peripheral wells of three separate channels, whereas the center chamber was filled with ultrapure water. After the tested 21 hrs, uniform gradients of fluorescence intensity were readily visualized in each channel (Supp. Fig. 2a). The average pixel intensity at 15 locations in each channel and associated standard deviations in all channels were calculated (Supp. Fig. 2b). No significant differences were noted between the average pixel intensities in each channel, indicating that cytokine diffusion profiles were consistent from channel to channel.
Competitive cell migration assay
As a case study, we tested the migration of MSCs in response to multiple simultaneous cytotactic cues. MSCs were chosen as they are the mostly widely used adult, non-hematopoietic stem cells in preclinical and patient models. The disease model we selected for this case study is arthritis, not only because of its high prevalence and our familiarity with this model, but also because of the excessive cost associated with arthritis treatment [32]. Over 65 cytokines have been shown to play important roles in rheumatoid arthritis [33]. We selected a subset of these cytokines based on their known molecular characteristics [33–35]: TGFβ3, TGFβ1, PDGFbb, lymphotactin, fractalkine, SDF1, IGF1, MIP1α, MIP1β, MCP1, IP10, TGFβ1, TNFα, VEGF, bFGF and IL6 (Fig. 3a). Cytokines were randomly allocated into Groups 1, 2 or 3 (Fig. 3a), and tested for their efficacy in cell recruitment. Top performer(s) of each group were selected to ‘compete’ in a final round (Final in Fig. 3a).
MSCs migrated under the agarose gel towards a cytokine gradient over the observed 4, 8 and 24 hrs (Supp. Fig. 3). For example, an abundant number of MSCs had migrated after only 4 hrs of lymphotactin (LPTN) exposure in comparison to the control (Supp. Fig. 3g), consistent with its previously observed chemotactic effects on MSCs [36]. By 24 hrs, TGFβ3 induced marked cell migration (Supp. Fig. 3c) as compared to the control (Supp. Fig. 3i), consistent with previous reports [9, 29]. Likely due to the presence of serum (10%) in the cell-seeded well, MSCs could be seen migrating in the PBS control group (Supp. Fig. 3), but to a lesser extent compared to the cytokine-containing groups.
A separate control experiment was conducted to determine if MSCs were biased to migrate into a particular channel within the device (Supp. Fig. 4). All peripheral chambers of the microfluidic devices were filled with PBS. MSCs seeded in the center chamber were allowed to migrate for the tested 24 hrs (Supp. Fig. 4a). Each channel was allocated with a number (Supp. Fig. 4b) and the average number of MSCs that migrated into each channel was quantified (Supp. Fig. 4c). No significant differences were found between the averages in each channel, indicating that cells were not inherently biased to migrate into a particular channel in the device.
The migratory effects of multiple cytokines were concurrently monitored in real time (Supp. Movie 1), representing an advantage over conventional cell migration assays that only allow collection of endpoint data. MSCs seeded in the center chamber can be seen continuously migrating towards an increasing concentration of IGF1, as an example, in the peripheral well on the right side over the observed 12 hrs (Supp. Movie 1).
Following each cell migration experiment, cell nuclei were stained with DAPI and imaged with phase contrast and fluorescence microscopy to view cell migration in response to each factor (Fig. 3b). DAPI stained nuclei were quantified to determine the total number of cells that had migrated into each channel (Fig. 4a–d), the migration index (Fig. 4e–h) and the migratory distance (Fig. 5). Significantly more MSCs were recruited by TGFβ3 in Group 1 (147 cells), TNFα in Group 2 (128 cells) and MIP1α in Group 3 (132 cells) than the control by 24 hrs (Fig. 4a, b and c, respectively). In the ‘final’ round with top performers of cell recruitment selected from Groups 1, 2 and 3, significantly more MSCs were recruited by PDGFbb (115 cells), TGFβ3 (120 cells), and TNFα (125 cells) (Fig. 4d) compared to the control. Additionally, cell migration index (MI) was quantified by multiplying cell numbers by migration distances (Fig. 4e–h). SDF1 and TGFβ3 induced significantly greater MI (26 and 33 respectively) than the PBS control (13) in Group 1 (Fig. 4e). TNFα and VEGF induced significantly greater MI (23 and 16 respectively) than the control in Group 2 (Fig. 4f). LPTN and MIP1α induced significantly greater MI (18 and 20 respectively) than the control in Group 3 (Fig. 4g). MIP1α, PDGFbb, LPTN, and TNFα induced significantly greater MI (27, 26, 20 and 29 respectively) than the control in the final round (Fig. 4h).
Figure 4.

Quantification of migrated cells and Migration Index (MI). a–d: The total number of migrated cells was quantified for the first three groups of cytotactic cues, and also for the final round of the most robust cues. Significantly more cells migrated than the control in response to TGFβ3 in Group 1, TNFα in Group 2, MIP1α in Group 3 and TNFα, TGFβ3, and PDGFbb in the final. e–h: The cell migration index was derived by multiplying the total number of cells migrated into the channel by the migratory distance. Significantly greater MI was induced than controls by SDF1 and TGFβ3 in Group 1, TNFα and VEGF in Group 2, LPTN and MIP1α Group 3 and MIP1α, PDGFbb, LPTN, and TNFα in the final. Values shown are means ± SE. *: p<0.05.
Figure 5.
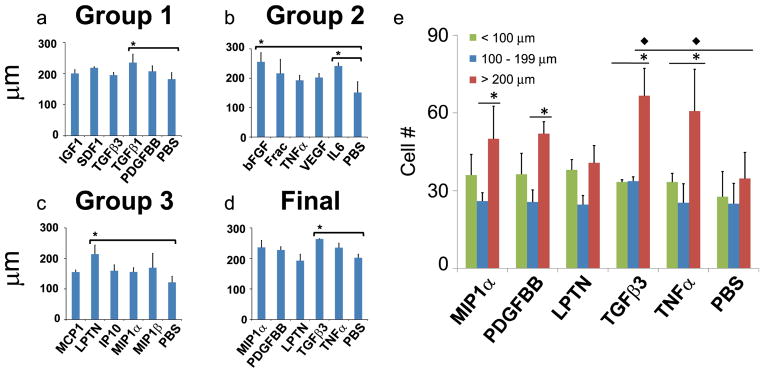
Quantification of cell migration distance. a–d: Cells migrated significantly longer distances than controls in response to TGFβ1 in Group 1, bFGF and IL6 in Group 2, LPTN in Group 3, and TGFβ3 in the final. *: p<0.05. e: Cell migratory distances were stratified into categories of <100 μm, 100–200 μm and >200 μm. Values shown are means ± SE. * indicates significance (p<0.05) over other distances for a given one factor. ◆ indicates significance (p<0.05) of one factor over the control group for the same distance value. *: p<0.05.
The distance over which cells migrate is of interest in stem cell biology, neutrophil motility, tissue regeneration and cancer metastasis. The most robust cytokines that induced significantly longer migratory distances were TGFβ1 (235 μm) in Group 1; bFGF (255 μm) and IL6 (241 μm) in Group 2, lymphotactin (214 μm) in Group 3, and TGFβ3 (264 μm) in the ‘final’ round (Fig. 5a–d). Remarkably, TGFβ3 promoted both significant cell recruitment and induced the longest migration distance of MSCs, which might account for our recent in vivo finding that TGFβ3 not only recruited an abundant number of cells, but also promoted the regeneration of the entire articular cartilage surface of a synovial joint [27, 29, 37]. However, further experimentation is necessary to elucidate the mode by which TGFβ3 was able to promote the regenerative effects observed in our previous in vivo study. To further demonstrate the robustness of this system, the cell migration distance in the final round was stratified into categories of <100 μm, 100–200 μm and >200 μm (Fig. 5e). MIP1α and PDGFbb induced significantly more cells that migrated distances greater than 200 μm compared to the number of cells that migrated distances ranging between 100–200 μm (Fig. 5e). TGFβ3 and TNFα were particularly robust for inducing cell migration for distances greater than 200 μm (Fig. 5e).
Given that fluorescent dextran diffusion in each channel reached the center chamber (Fig. 2), cell migration data substantiate the possibility of competitive recruitment of cells by multiple cytokines in each group. For example, MIP1α in Group 3 induced significantly more MSCs to migrate than the control (Fig. 4c), but failed to induce significantly more MSCs to migrate in the final round (Fig. 4d), suggesting that other cytokines in the final round, PDGFbb, LPTN, TGFβ3 and/or TNFα, may have exerted even stronger cytotactic effects than MIP1α. A similar pattern was observed for MSC migratory distances in response to TNFα, which showed modest effects on migratory distance in Group 2 (Fig. 5b), but enhanced cytotactic effects in the final round (Fig. 5d). These effects are similar to previous observations that TNFα enhances the effects of other chemokines [38]. Remarkably, TGFβ3 only had modest effects on MSC migratory distance in Group 1 (Fig. 5a), but induced a robust increase in migratory distance on MSCs in the final round (Fig. 5d), likely owing to a putative synergistic effect with other cytokines including MIP1α, PDGFbb, LPTN, and/or TNFα. When cells are exposed to multiple cytokines in vivo, chemotactic effects may be enhanced or reduced depending on the presence of synergistic or antagonistic factors. To this end, a separate migration experiment was conducted to determine the migratory effects of PDGFbb on MSCs in a non-competition setting (Supp. Fig. 5). After 24 hrs, PDGFbb alone in a non-competition setting induced significantly more MSCs to migrate (226 cells) than not only the control (88 cells) (p<0.05) (Supp. Fig. 5), but also compared to PDGFbb in a competition setting in Group 1 (122 cells) (Fig. 4a) or the final round (115 cells) (Fig. 4d). The chemotactic effects of PDGFbb on MSCs are also consistent with previous findings [38, 39]. Thus, the varied effects of PDGFbb on MSCs in a competition setting compared to a non-competition setting substantiate the need to test multiple cytokines concurrently.
Discussion
This novel microfluidic device enables the competitive recruitment of cells that are simultaneously exposed to multiple cytotactic cues under real-time imaging. Multiple chemotactic factors with gradients that converge on cells of interest generate complex synergistic and/or antagonistic effects on cell migration, thus simulating the in vivo milieu of chemotactic cues that act on cells in development, cancer metastasis, wound healing or tissue repair. A cytokine gradient is generated across the agarose-filled channels within minutes and maintained for the tested 24 hrs. Diffusion of dextrans of varying molecular weights and cell migration data demonstrate the effectiveness of this microfluidic device for testing the simultaneous actions of multiple chemotactic cues. Another conceptual innovation, to be investigated in future experiments, is to assay the effects of a given chemotactic cue on the migration of multiple cell populations, mimicking in vivo conditions whereby multiple cell types may be attracted or repelled by a given factor. These two features are not present in typical cell migration assays including the Boyden chamber, Transwell and scratch assays.
The observed competitive effects of multiple cytokines in the recruitment of MSCs further demonstrate that multiple cytokine gradients reach the center chamber where cells are seeded. Additional experiments that can be conducted with this migration system include chemokinesis, migration directionality and evaluating clonal fractions of a heterogeneous cell population. Determining which cytokine combinations are the most effective at maximizing cell homing may offer novel approaches for tissue regeneration with or without the need for cell delivery [9, 29]. Specifically, the device may help to determine the spatial location for placement of a cytotactic cue in a bioactive scaffold from a native cell source to induce effective cell migration. Besides tissue regeneration, this device may be used to better understand fundamental biological processes such as development, cancer cell aggregation and spread, motility of inflammatory cells and drug development.
High throughput is desirable for screening the migration of multiple cell populations or the effects of multiple chemotactic cues on the migration of a given cell type. A number of devices allow high throughput screening of cell migration such as the ChemoTx® system (Neuro Probe, Gaithersburg, MD). Microfluidic platforms have a number of advantages, over existing cell migration assays, including low cost, high reproducibility and minimal volume of required reagents. The present microfluidic cell migration assay can be readily scaled up by not only using multiple devices, but also by adding more channels to each device to allow for the screening of numerous cytotactic cues or testing the cytotactic effects of a given factor against multiple cell populations. Compared to the high cell number evaluated in other migration systems, the present device only allows a limited number of cells to access the channel entrance at a given time for motility analysis because of the small channel diameter (250 μm). Since only the cells migrating within the channel are quantified and not those showing motility within the central chamber, the total number of migrating cells in the channel is a subset of the total cell motility. To maintain consistency, a uniform concentration of 200 ng was selected to test all cytokines in the present work. It is conceivable that cell migration can be enhanced at higher or lower concentrations for a given cytokine. In summary, this microfluidic migration device has the unique advantage of allowing assays of cell migration competitively in response to multiple cytotactic cues with broad applications ranging from drug development to wound healing and tissue regeneration.
Supplementary Material
Acknowledgments
We would like to thank Brian M. Gillette, Hesam Parsa and Eric D. Frank for their technical assistance, and F. Guo for technical assistance. The present study is funded in part by NIH grants R01DE018248 and R01EB0062621 to J.J.M.
Footnotes
Author contribution
A.M. designed and performed the experiments, analyzed the data and drafted the manuscript; Y.C. participated in the design and performance of some experiments; K.P. assisted in experimental procedures; M.C., K.K., J.T. and Z.D. assisted with data analysis; S.K.S. contributed to data analysis; J.J.M. designed the experiments, analyzed the data and drafted the manuscript.
References
- 1.Doorn J, et al. Therapeutic Applications of Mesenchymal Stromal Cells; Paracrine Effects and Potential Improvements. Tissue Eng Part B Rev. doi: 10.1089/ten.TEB.2011.0488. [DOI] [PubMed] [Google Scholar]
- 2.Laird DJ, von Andrian UH, Wagers AJ. Stem cell trafficking in tissue development, growth, and disease. Cell. 2008;132(4):612–30. doi: 10.1016/j.cell.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 3.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–16. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Hannoush EJ, et al. Impact of enhanced mobilization of bone marrow derived cells to site of injury. J Trauma. 71(2):283–9. doi: 10.1097/TA.0b013e318222f380. discussion 289–91. [DOI] [PubMed] [Google Scholar]
- 5.Prockop DJ. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Mol Ther. 2009;17(6):939–46. doi: 10.1038/mt.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singer NG, Caplan AI. Mesenchymal stem cells: mechanisms of inflammation. Annu Rev Pathol. 6:457–78. doi: 10.1146/annurev-pathol-011110-130230. [DOI] [PubMed] [Google Scholar]
- 7.Shah S, et al. Mobilization of bone marrow cells to the site of injury is necessary for wound healing. J Trauma. 2009;67(2):315–21. doi: 10.1097/TA.0b013e3181a5c9c7. discussion 321–2. [DOI] [PubMed] [Google Scholar]
- 8.Fox JM, et al. Recent advances into the understanding of mesenchymal stem cell trafficking. Br J Haematol. 2007;137(6):491–502. doi: 10.1111/j.1365-2141.2007.06610.x. [DOI] [PubMed] [Google Scholar]
- 9.Lee CH, et al. Regeneration of the articular surface of the rabbit synovial joint by cell homing: a proof of concept study. Lancet. 376(9739):440–8. doi: 10.1016/S0140-6736(10)60668-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyden S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med. 1962;115:453–66. doi: 10.1084/jem.115.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson RD, Quie PG, Simmons RL. Chemotaxis under agarose: a new and simple method for measuring chemotaxis and spontaneous migration of human polymorphonuclear leukocytes and monocytes. J Immunol. 1975;115(6):1650–6. [PubMed] [Google Scholar]
- 12.Lauffenburger D, Rothman C, Zigmond SH. Measurement of leukocyte motility and chemotaxis parameters with a linear under-agarose migration assay. J Immunol. 1983;131(2):940–7. [PubMed] [Google Scholar]
- 13.Todaro GJ, Lazar GK, Green H. The initiation of cell division in a contact-inhibited mammalian cell line. J Cell Physiol. 1965;66(3):325–33. doi: 10.1002/jcp.1030660310. [DOI] [PubMed] [Google Scholar]
- 14.Kothapalli CR, et al. A high-throughput microfluidic assay to study neurite response to growth factor gradients. Lab Chip. 11(3):497–507. doi: 10.1039/c0lc00240b. [DOI] [PubMed] [Google Scholar]
- 15.Kim T, Pinelis M, Maharbiz MM. Generating steep, shear-free gradients of small molecules for cell culture. Biomed Microdevices. 2009;11(1):65–73. doi: 10.1007/s10544-008-9210-7. [DOI] [PubMed] [Google Scholar]
- 16.Englert DL, Manson MD, Jayaraman A. Flow-based microfluidic device for quantifying bacterial chemotaxis in stable, competing gradients. Appl Environ Microbiol. 2009;75(13):4557–64. doi: 10.1128/AEM.02952-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim BJ, Wu M. Microfluidics for mammalian cell chemotaxis. Ann Biomed Eng. 40 (6):1316–27. doi: 10.1007/s10439-011-0489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abhyankar VV, et al. Characterization of a membrane-based gradient generator for use in cell-signaling studies. Lab Chip. 2006;6(3):389–93. doi: 10.1039/b514133h. [DOI] [PubMed] [Google Scholar]
- 19.Kim D, Haynes CL. Neutrophil Chemotaxis within a Competing Gradient of Chemoattractants. Anal Chem. 84(14):6070–8. doi: 10.1021/ac3009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haessler U, et al. An agarose-based microfluidic platform with a gradient buffer for 3D chemotaxis studies. Biomed Microdevices. 2009;11(4):827–35. doi: 10.1007/s10544-009-9299-3. [DOI] [PubMed] [Google Scholar]
- 21.Shin Y, et al. In vitro 3D collective sprouting angiogenesis under orchestrated ANG-1 and VEGF gradients. Lab Chip. 11(13):2175–81. doi: 10.1039/c1lc20039a. [DOI] [PubMed] [Google Scholar]
- 22.Lin F, Butcher EC. T cell chemotaxis in a simple microfluidic device. Lab Chip. 2006;6(11):1462–9. doi: 10.1039/b607071j. [DOI] [PubMed] [Google Scholar]
- 23.Cheng SY, et al. A hydrogel-based microfluidic device for the studies of directed cell migration. Lab Chip. 2007;7(6):763–9. doi: 10.1039/b618463d. [DOI] [PubMed] [Google Scholar]
- 24.Kim M, Kim T. Diffusion-based and long-range concentration gradients of multiple chemicals for bacterial chemotaxis assays. Anal Chem. 82(22):9401–9. doi: 10.1021/ac102022q. [DOI] [PubMed] [Google Scholar]
- 25.Chueh BH, et al. Leakage-free bonding of porous membranes into layered microfluidic array systems. Anal Chem. 2007;79(9):3504–8. doi: 10.1021/ac062118p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Si G, et al. A parallel diffusion-based microfluidic device for bacterial chemotaxis analysis. Lab Chip. 12(7):1389–94. doi: 10.1039/c2lc21219f. [DOI] [PubMed] [Google Scholar]
- 27.Lee CH, et al. Tissue formation and vascularization in anatomically shaped human joint condyle ectopically in vivo. Tissue Eng Part A. 2009;15(12):3923–30. doi: 10.1089/ten.tea.2008.0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alhadlaq A, et al. Adult stem cell driven genesis of human-shaped articular condyle. Ann Biomed Eng. 2004;32(7):911–23. doi: 10.1023/b:abme.0000032454.53116.ee. [DOI] [PubMed] [Google Scholar]
- 29.Mendelson A, et al. Chondrogenesis by chemotactic homing of synovium, bone marrow, and adipose stem cells in vitro. Faseb J. 25(10):3496–504. doi: 10.1096/fj.10-176305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lennon DP, Caplan AI. Isolation of human marrow-derived mesenchymal stem cells. Exp Hematol. 2006;34(11):1604–5. doi: 10.1016/j.exphem.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Lee CH, et al. CTGF directs fibroblast differentiation from human mesenchymal stem/stromal cells and defines connective tissue healing in a rodent injury model. J Clin Invest. 120(9):3340–9. doi: 10.1172/JCI43230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yelin ECM, Foreman A, Pasta D, Murphy L, Helmick C. National and state medical expenditures and lost earnings attribute to arthritis and other rheumatic conditions -United States, 2003. MMWR. 2007;56(1):4–7. [PubMed] [Google Scholar]
- 33.Rohner E, et al. Inflammatory synovial fluid microenvironment drives primary human chondrocytes to actively take part in inflammatory joint diseases. Immunol Res. doi: 10.1007/s12026-011-8247-5. [DOI] [PubMed] [Google Scholar]
- 34.Iwamoto T, et al. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. Febs J. 2008;275(18):4448–55. doi: 10.1111/j.1742-4658.2008.06580.x. [DOI] [PubMed] [Google Scholar]
- 35.Cuellar JM, et al. Diagnostic utility of cytokine biomarkers in the evaluation of acute knee pain. J Bone Joint Surg Am. 2009;91(10):2313–20. doi: 10.2106/JBJS.H.00835. [DOI] [PubMed] [Google Scholar]
- 36.Endres M, et al. Chemokine profile of synovial fluid from normal, osteoarthritis and rheumatoid arthritis patients: CCL25, CXCL10 and XCL1 recruit human subchondral mesenchymal progenitor cells. Osteoarthritis Cartilage. 18(11):1458–66. doi: 10.1016/j.joca.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Thorpe SD, et al. The response of bone marrow-derived mesenchymal stem cells to dynamic compression following TGF-beta3 induced chondrogenic differentiation. Ann Biomed Eng. 38(9):2896–909. doi: 10.1007/s10439-010-0059-6. [DOI] [PubMed] [Google Scholar]
- 38.Ponte AL, et al. The in vitro migration capacity of human bone marrow mesenchymal stem cells: comparison of chemokine and growth factor chemotactic activities. Stem Cells. 2007;25(7):1737–45. doi: 10.1634/stemcells.2007-0054. [DOI] [PubMed] [Google Scholar]
- 39.Mishima Y, Lotz M. Chemotaxis of human articular chondrocytes and mesenchymal stem cells. J Orthop Res. 2008;26(10):1407–12. doi: 10.1002/jor.20668. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.