

Abstract
Several nuclear receptors regulate diverse metabolic functions that impact on critical biological processes, such as development, differentiation, cellular regeneration, and neoplastic conversion. In the liver, some members of the nuclear receptor family, such as peroxisome proliferator-activated receptors (PPARs), constitutive androstane receptor (CAR), farnesoid X receptor (FXR), liver X receptor (LXR), pregnane X receptor (PXR), glucocorticoid receptor (GR), and others, regulate energy homeostasis, the formation and excretion of bile acids, and detoxification of xenobiotics. Excess energy burning resulting from increases in fatty acid oxidation systems in liver generates reactive oxygen species, and the resulting oxidative damage influences liver regeneration and liver tumor development. These nuclear receptors are important sensors of exogenous activators as well as receptor-specific endogenous ligands. In this regard, gene knockout mouse models revealed that some lipid-metabolizing enzymes generate PPARα-activating ligands, while others such as ACOX1 (fatty acyl-CoA oxidase1) inactivate these endogenous PPARα activators. In the absence of ACOX1, the unmetabolized ACOX1 substrates cause sustained activation of PPARα, and the resulting increase in energy burning leads to hepatocarcinogenesis. Ligand-activated nuclear receptors recruit the multisubunit Mediator complex for RNA polymerase II-dependent gene transcription. Evidence indicates that the Med1 subunit of the Mediator is essential for PPARα, PPARγ, CAR, and GR signaling in liver. Med1 null hepatocytes fail to respond to PPARα activators in that these cells do not show induction of peroxisome proliferation and increases in fatty acid oxidation enzymes. Med1-deficient hepatocytes show no increase in cell proliferation and do not give rise to liver tumors. Identification of nuclear receptor-specific coactivators and Mediator subunits should further our understanding of the complexities of metabolic diseases associated with increased energy combustion in liver.
Keywords: Liver regeneration, Mediator complex, Med1, Hepatocarcinogenesis
INTRODUCTION
Liver, a complex metabolic organ located strategically in the organism, orchestrates a variety of metabolic functions including the maintenance of fat and carbohydrate energy homeostasis, synthesis of serum proteins, formation and excretion of bile acids as products of cholesterol catabolism, and the detoxification of xenobiotics, including alcohol (1,2). Disturbances in these processes contribute invariably to acute or chronic liver injury. For example, the burgeoning pandemic of dietary obesity adversely impacts on hepatic energy homeostasis with a risk of developing nonalcoholic fatty liver disease (NAFLD), which leads to the development of cirrhosis of the liver and hepatocellular carcinoma (HCC), outcomes similar to those seen with alcoholic fatty liver disease (2,3). Liver is also the main target for the five most common hepatotropic viruses, namely, hepatitis A, hepatitis B, hepatitis C, hepatitis D, and hepatitis E (4). Of these, chronic hepatitis B virus and hepatitis C virus infections cause 75–80% of liver cancers diagnosed worldwide (5). Many of the chronic liver injury conditions create a microenvironment that is conducive to the confounding confluence of cell death, inflammation, fibrosis, and lingering hepatocyte regeneration, necessary for the development of end-stage liver disease of cirrhosis and HCC (3,6). The normal replicative capacity of hepatocytes, with an average life span of 200–300 days, and mitosis occurring at the rate of 1 in 20,000 cells, is low (7,8), but liver is evolutionarily endowed with enormous potential to regenerate, designed to restore the functional capacity of diminished liver (9). End-stage liver disease poses a major health challenge and points to the increasing importance of a deeper understanding of the molecular signaling mechanisms responsible for hepatocellular regeneration known to contribute to the carcinogenic process, although cell proliferation per se may not initiate cancer (10,11).
Liver regeneration is a predictable biological process triggered in response to many types of injury (7,9). Surgical removal of part of the liver, a process which emanates healing signals that instruct remaining mature hepatocytes to resume growth and division, has yielded fundamental clues governing liver regeneration (9,12). The predominant mode of liver regeneration entails normally functioning, quiescent, but conditionally dividing, differentiated hepatocytes that reenter cell cycle, multiply, and grow to replenish cells lost due to injury (9). Regeneration of differentiated hepatocytes is controlled by metabolic needs such that the process terminates once an appropriate liver to body weight ratio is achieved to prevent regenerating cells from running amok (13). A relatively minor mode of liver regeneration is the participation of putative liver stem cells that are activated only when mature hepatocytes can no longer engage in regeneration due to senescence or other constraints (9,12). Of interest is that during regeneration, liver cells, like all other proliferating cells, continue to perform critical metabolic functions, such as glucose and lipid homoeostasis and protein synthesis. A better understanding of how cell proliferation and metabolism are interconnected and coregulated may provide therapeutic leads (14).
Many of the important metabolic functions in liver are regulated by nuclear receptors (2). Activation of some nuclear receptors, such as peroxisome proliferator-activated receptor-α (PPARα) or constitutive androstane receptor (CAR), by their cognate ligands stimulates hepatocellular proliferation along with the characteristic metabolic effects. For example, Wy-14,643, ciprofibrate, and other peroxisome proliferators are potent hepatic mitogens as they activate PPARα (11,15,16). Likewise, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), a potent activator of CAR, is well recognized as a hepatocellular mitogen (17).
The biological processes regulated by nuclear receptors require coordinated assembly of transcription coactivator complexes on liganded nuclear receptors to facilitate chromatin remodeling and to recruit the Mediator complex for the expression of RNA polymerase II (Pol II)-transcribed genes (18). Recent evidence indicates a role for Med1, an important subunit of the Mediator complex in controlling the metabolic status and regeneration in liver (19,20). We focus this minireview on Med1 because it is an essential subunit of the multisubunit Mediator complex in regulating fatty acid oxidation and glucose metabolism (19). Furthermore, Med1 is also necessary for liver regeneration induced, in particular, by nuclear receptor PPARα and CAR activators (9,21). Recent evidence indicates that Med1 is also sufficient by itself in inducing hepatocellular proliferation (20).
NUCLEAR RECEPTORS IN LIVER REGENERATION
Nuclear receptor superfamily consists of 48 members in the human genome, and most of these receptors function as ligand-dependent transcription factors with a major role in development and metabolic homeostasis (2,22,23). In liver, several of these receptors serve as intracellular sensors of endogenous, naturally occurring, small molecules, such as lipid-soluble hormones and dietary lipid intermediates. Transcriptional activators that play a key role in the varied pathogenesis of NAFLD include three members of the PPAR subfamily of nuclear receptors (namely, PPARα, PPARδ/β, and PPARγ), CAR, farnesoid X receptor (FXR), liver X receptor-α (LXRα), pregnane X receptor (PXR), glucocorticoid receptor (GR), and others (2,22,23). These nuclear receptors and other transcriptional activators are associated with lipid sensing, lipid synthesis, and fatty acid oxidation (2). Nuclear receptors, such as PPARα and CAR, are also activated by exogenous molecules including several xenobiotics (2,11,22,24). They participate in the modulation of hepatocyte proliferation, and some of these receptors are implicated in hepatocarcinogenesis resulting from sustained activation by cognate exogenous or endogenous ligand(s) (11,25–28) (Table 1).
Table 1.
Nuclear Receptors in Liver Regeneration
| Nuclear Receptor | Nuclear Receptor Responsive Element | Natural Ligands | Synthetic Ligands | Function: Lipogenesis Versus Fatty Acid Oxidation | Function: Liver Regeneration |
|---|---|---|---|---|---|
| PPARα | 5′-AGGTCA(N)AGGTCA-3′ | Free fatty acids Fatty acid derivatives |
Fibrates Wy-14,643 Ciprofibrates |
Increases fatty acid oxidation | Increases |
| PPARγ | 5′-AGGTCA(N)AGGTCA-3′ | Free fatty acids Fatty acid derivatives |
Glitazones | Increases lipid storage/lipogenesis | Increases |
| LXRα,β | 5′-(A/G)G(G/T)T(C/T)Annnn-(A/G)G(G/T)T(C/T)A-3′ | Oxysterols | T0901317 GW3965 |
Increases lipogenesis | Decreases |
| FXR | 5′-AGAGCA(N)AGGGGA-3′ | Bile acids Oxysterols Polyunsaturated fatty acids |
GW4064 6ECDCA MFA1 Obeticholic acid |
Decreases lipogenesis | Increases |
| CAR | 5′-(A/G)GTTCAnnnn-(A/G)GTTCA-3′ | Xenobiotics Androstenol |
Phenobarbital TCPOBOP Antimalarial drug artemisinin |
Decreases lipogenesis/increases fatty acid oxidation | Increases |
| PXR | 5′-(A/G)GTTCAnnnn-(A/G)GTTCA-3′ | Bile acids Cholesterol derived 5-cholestane-3, 7, 12-triol |
Drugs Xenobiotics |
Decreases lipogenesis/fatty acid oxidation | Increases |
| TR | 5′-(A/G)GGTCAnnnn-(A/G)GGTCA-3′ | T3 and T4 | CO23,GC-1 KB-141 KB2115 MB07811 |
Increases fatty acid oxidation | Increases |
PPARα
PPARα is a lipid- and xenobiotic-sensing nuclear receptor expressed in tissues such as the liver, kidney, heart, and skeletal muscle with high capacity for fatty acid oxidation and energy burning (2,11,15,16). In liver, PPARα is responsible for the peroxisome proliferator-induced pleiotropic responses that include hepatocellular proliferation, increase in peroxisome population in hepatocytes, and enhanced fatty acid oxidation resulting in excess energy burning in liver (11,16,25). Accordingly, PPARα activation can modulate fatty acid oxidation and influence the progression of NAFLD (2,16). Synthetic PPARα agonists, called peroxisome proliferators, include structurally diverse compounds such as fibrates with hypolipidemic activity, phthalate ester plasticizers, industrial solvents, herbicides, food flavoring agents, and others (29,30). PPARα heterodimerizes with RXRα and transcriptionally regulates peroxisomal and mitochondrial β-oxidation and microsomal ω-oxidation of fatty acids and plays an essential role in burning energy, resulting in the generation of hydrogen peroxide and other reactive oxygen species (11,16,31). Sustained activation of PPARα, either by synthetic or endogenous ligands, results in the development of liver tumors, attributable in part to increased oxidative damage caused by fatty acid oxidation and to liver cell proliferation (11,26–28).
PPARα activators, in particular the peroxisome proliferators such as ciprofibrate and Wy-14,643, are powerful liver mitogens (29,32). Peroxisome proliferators were identified as primary mitogens and proposed that the effects are mediated by a receptor, which was subsequently identified as PPARα (33). The first indication for the formation and degradation of endogenous PPARα activators came from fatty acyl-CoA oxidase (ACOX1) null mice that display profound spontaneous peroxisome proliferation in hepatocytes with induction of genes that are activated by PPARα, implying that ACOX1 is essential for the inactivation of endogenous PPARα activators (16,28,34). PPARα agonists, depending on their potency, exert primary hepatomitogenic properties within the first week of treatment, as they are able to induce PPARα-mediated hepatocyte proliferation in the absence of liver injury (direct hyperplasia) (11,32,33). Chronic administration of these agents does not maintain a high level of sustained mitogenic response (31). Stimulation of PPARα represses the microRNA let-7, which degrades the c-myc oncogene and induces oncogenic mir-17 miRNA (35). Mice deficient in PPARα show a complete loss of cell proliferative response in liver following exposure to peroxisome proliferators, and these mice also show no induction of fatty acid oxidation systems and fail to develop liver tumors (36,37). These data contribute to the concept that energy burning-related increase in oxidative stress combined with increased liver cell proliferation contributes to liver cancer development by a PPARα-dependent mechanism (11,16,38). Species- and cell-specific differences in response to peroxisome proliferators, as reviewed recently (11), appear mostly quantitative but not qualitative in nature. Adenovirally directed expression of human PPARα in PPARα−/− mice induced liver cell proliferation similar to that in magnitude seen with mouse PPARα (39,40). The other two members of PPAR subfamily, PPARδ/β and PPARγ, are also involved in lipid metabolism and energy homeostasis in that PPARδ/β, which is ubiquitously expressed, participates in fatty acid oxidation and energy burning, and PPARγ is a major factor responsible for adipogenesis and energy conservation (16,22).
CAR
The constitutive androstane receptor (CAR) is most abundantly expressed in liver and regulates the transcription of drug-metabolizing enzymes and transporters to minimize the toxicity of harmful chemicals (41–43). Like PPARs, CAR also plays a role in the regulation of energy homeostasis by influencing glucose and lipid metabolism. Activation of CAR results in the reduction of blood glucose by suppressing the expression of hepatic gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6P) in mouse liver (44). Activation of CAR inhibits hepatic lipogenesis and induces fatty acid oxidation, and these appear beneficial in reducing hepatic steatosis (44). In liver, endogenous CAR resides in the cytoplasm of hepatocytes, and upon exposure to its agonist phenobarbital or 1,4-bis[2-(3,5-dichloropyridyloxy)] benzene (TCPOBOP), CAR translocates from the cytoplasm to the nucleus and triggers the transcription of target genes (43). Activation of CAR by TCPOBOP leads to profound hepatomegaly that involves both hypertrophic and hyperplastic responses (42,45). CAR agonists also translocate adenovirally expressed exogenous CAR from hepatocyte cytoplasm to the nucleus (46). Of interest is that PPARα ligands, which induce fatty acid oxidation enzymes, have been shown to drive CAR into the hepatocyte nucleus, but the functional significance of PPARα ligand-mediated translocation of CAR to the nucleus is unclear (47). Mice deficient in CAR show complete loss of hyperplastic response of hepatocytes to CAR activators (42). Of interest is that PPARα−/− mice show enhanced hepatocyte proliferation in response to CAR agonist TCPOBOP (48). CAR activation by phenobarbital is known to induce liver tumor promotion (49). Recently, it has been shown that the growth arrest and the DNA damage-inducible 45-β (Gadd45β) gene are most strongly induced by TCPOBOP during early liver regeneration (50).
PXR
PXR, also called steroid X receptor (SXR), like CAR, is a xenobiotic sensor. It is localized in the cytoplasm of hepatocytes and, when activated, it translocates to the nucleus (2,44,51). In the nucleus, PXR heterodimerizes with RXRα, and this heterodimer binds to xenobiotic-responsive enhancer module-bearing target genes to enhance their transcription (52). PXR is activated by several drugs and xenobiotics, which include pregnenolone-16-carbonitrile (PCN), taxol, rifampicin, and clotrimazole, among others (53). The major endogenous activators of PXR include bile acids and cholesterol-derived 5-cholestane-3,7,12-triol (triol) (54). PXR controls the expression of phase I and phase II drug-metabolizing enzymes as well as some members of the drug-transporter family (55). PXR reduces hepatic fatty acid oxidation by downregulating the expression of carnitine palmitoyltransferase 1α (CPT1α) and mitochondrial 3-hydroxy-3-methylglutarate-CoA synthase 2, the enzymes involved in mitochondrial β-oxidation of fatty acids (56). CPT1α is required for the carnitination of fatty acids, which is essential for entry of fatty acids into the mitochondria for β-oxidation (25).
PXR also plays a role in hepatocyte regeneration by virtue of its role in fatty acid metabolism as it increases de novo lipogenesis and inhibits fatty acid oxidation. Mice lacking PXR reveal a reduction of hepatocyte proliferation at 36 h following partial hepatectomy (57). PXR ligand PCN produced hepatomegaly in the wild-type mice but not in the PXR-KO mice (58). PCN also increased both the number of proliferating cell nuclear antigen immunopositive nuclei and apparent cell size in the wild-type mice but not in the PXR-KO mice (59).
FXR
Farnesoid X receptor (FXR) is the primary nuclear receptor for sensing bile acids (59,60). Bile acids are important products of cholesterol metabolism and are excreted in bile as byproducts of metabolism by liver. Bile acid levels are tightly regulated, as they serve as activators of FXR and also other xenobiotic nuclear receptors, CAR and PXR (22,58,61,62). FXR controls the synthesis and transport of bile acids in the liver and gut (60–65). Upon activation by bile acids, FXR positively regulates a number of genes that decrease cellular levels of toxic bile acids. FXR induces the small heterodimer partner (SHP) in liver that downregulates Cyp7a1 and Cyp8b1 genes encoding enzymes that synthesize bile acids from cholesterol. FXR is also known to inhibit hepatic lipogenesis by repressing SREBP-1c (2,22). FXR agonists decrease serum triglyceride levels. Activation of FXR by elevated bile acid levels accelerates liver regeneration, whereas decreased bile acid levels and absence of FXR inhibit liver growth (60,62). FXR appears to exert a dual functioning role, first by inhibiting cholesterol 7α-hydroxylase (CYP7a1) to reduce bile acid stress and then enhance hepatocellular proliferation by activating Foxm1b and FGF15 (62). Of note is that although the absence of FXR inhibits liver growth, FXR null mice spontaneously develop liver tumors as they age, asserting that bile acid-induced DNA damage in FXR null mice may be critical in liver tumor development even if FXR absence limits liver regeneration (63,64).
LXR
Liver X receptors α and β regulate cholesterol, glucose, and fatty acid homeostasis and are highly expressed in the liver (65–68). LXRs are endogenously activated by various oxygenated cholesterol derivatives or oxysterols, such as 22(R)-hydroxycholesterol, 24(S)-hydroxycholesterol, 24(S),25-epoxycholesterol, 27-hydroxycholesterol, and cholestenoic acid (65–68). LXRs function as important regulators of cholesterol catabolism by inducing the transcription of genes that participate in the conversion of cholesterol into bile acids and enhancing reverse cholesterol transport from peripheral sources to the liver (69). Activation of LXRs by endogenous oxysterols and synthetic ligands, such as T0901317 and GW3965, mediates increased transcription of lipogenic genes and cholesterol export genes (2). Hepatic steatosis is promoted by LXRs in a CD36-dependent manner (70). In addition, LXR-regulated cholesterol metabolism has implications in liver cell proliferation. Proliferating cells require excess cholesterol, and LXR activation affects cell proliferation (71). Also, the specific synthetic ligands for LXRs are known to inhibit cell proliferation. Hepatocyte proliferation resulting from partial hepatectomy is accompanied by the suppression of LXR-driven pathways to ensure increased intracellular cholesterol levels required for dividing cells. Reactivation of LXR pathways by synthetic ligands hampers the liver regenerative capacity by decreasing the hepatic cholesterol content (72).
TR
Thyroid hormone (T3) influences metabolism, growth, and development, and these effects are mediated by thyroid hormone receptors (TRs). Like CAR and PPARα activators, T3 is a strong inducer of liver cell proliferation (73). T3-mediated hepatocyte mitogenic response is mediated by PKA-dependent β-catenin activation (73).
MEDIATOR COMPLEX AND NUCLEAR RECEPTOR FUNCTION
Mediator is a large multisubunit complex composed of up to at least 31 subunits in all eukaryotes (18,74). This complex acts as a molecular bridge between gene-specific transcription factors and the RNA polymerase II machinery (74). The Mediator complex was first isolated from Saccharomyces cerevisiae and was shown to be necessary for transcriptional activation (75,76). Mammalian Mediator was then isolated as thyroid hormone receptor-associated protein (TRAP) complex (18,74). Subsequently, Mediator complexes were isolated using specific nuclear receptors and designated as ARC (activator recruited cofactor), DRIP (vitamin D receptor-interacting protein), and others (77,78). The Mediator subunits are functionally conserved throughout the evolution from yeast to human, and the current nomenclature is based on the original yeast MED proteins (79). Yeast and human Mediator subunits are organized in a similar core structure comprised of a head, middle, and tail module (80,81). The middle and head modules interface with the pol II basal transcription machinery (82). The tail and middle modules are mainly targeted by gene-specific activators. The Mediator complex associates with the C-terminal domain of the largest subunit of RNA Pol II and is considered essential for basal and regulated expression of most of the RNA polymerase II-dependent genes (82). Depletion of human Mediator from nuclear extracts abolishes transcription by pol II (83). Some Mediator subunits govern the expression of many genes, whereas others appear necessary for a specific gene (84). In this review, we focus on the emerging role of Med1 subunit in liver function.
Med1 AS KEY SUBUNIT OF THE MEDIATOR COMPLEX
Transcription coactivator Med1 is regarded as a key subunit of the mammalian mediator complex. Med1, also called PBP/PPARBP/TRAP220/DRIP205/RB18A, contains two LXXLL motifs located at amino acids 589–593 and 630–634 (85). These signature motifs of Med1 are necessary for the binding of a variety of cofactors, including SRC family members, PGC-1 family members, p300/CBP, and RIP140 (86). Med1 is a pivotal component of the TRAPs, DRIP complex, and ARC complex (87–89). PPARα-interacting cofactor complex (PRIC) also contains Med1 (78). Med1 also interacts with several transcription factors and cofactors, such as PRIP, PIMT, C/EBPβ, SRC, GATA family members, CBP/P300, PGC-1, and tumor-suppressor P53 (90–94). These interactions signify that Med1 has a major role in nuclear receptor-regulated metabolic functions, cellular proliferation, and differentiation. Med1 is expressed in various tissues of adult mice, including brain, heart, liver, lung, kidney, adipose tissues, and testis (85).
Deletion of Med1 Results in Embryonic Lethality
Med1 null mutation in mice is embryonically lethal at midgestation (day 11.5 postcoitum; E11.5), illustrating that Med1 is an essential and nonredundant coactivator and is necessary for embryonic development (95). Embryonic lethality is attributed to impaired development of placental vasculature and defects in the heart, eye, vascular, and hematopoietic system (21,96). The phenotypic changes in Med1 null mice are somewhat similar to those observed in mice deficient in members of GATA transcription factor family (91). Since Med1 interacts with GATA factors, it is possible that these are involved in similar developmental pathways (91). Med1 null fibroblasts failed to show PPARγ-stimulated adipogenesis, as they do not express adipogenic genes in response to PPARγ stimulation. These observations establish that Med1 is required for PPARγ regulated transcription (97).
Med1 Is Required for Liver Regeneration After Partial Hepatectomy
The embryonic lethality of Med1 null mutants necessitated the generation of conditional null mice using the Cre-loxP strategy for elucidating the cell- and gene-specific roles of Med1. Med1 liver conditional null mice were used to determine the role of Med1 in mouse liver regeneration (98). Liver regeneration is regulated by a complex network of signals involving cytokines, chemokines, growth factors, nuclear receptors, and cofactors. Disruption of Med1 in liver impairs liver regeneration with low survival after partial hepatectomy (98). In general, Med1 null hepatocytes are smaller in size compared with hepatocytes in littermate control (98). In wild-type mice, Med1 mRNA expression levels begin to increase 2 h after partial hepatectomy with maximal at 18 and 24 h (Fig. 1). The liver to body weight ratio increased progressively in wild-type mice, whereas similar increases were not observed in conditional Med1 liver null mice (98). In wild-type mice, DNA synthesis, as measured by BrdUrd incorporation, was prominent, with a peak labeling at 36 and 48 h after partial hepatectomy. In contrast, Med1 null liver demonstrated minimal labeling at all times after partial hepatectomy (98). Over 25 genes are upregulated more than sixfold 3 h after partial hepatectomy in wild-type mice compared to Med1 liver null mice (98). Most of these genes are related to cell cycle, cell growth, apoptosis, and signal transduction, such as insulin-like growth factor 1 (Igf1), IGF-binding protein 1 (IGFBP1), E2F transcription factor, growth arrest, and DNA damage-inducible 45γ (GADD45 γ) (98). The reduced expression of these cell cycle genes and cell growth regulatory factors in Med1 null liver indicates that Med1 null hepatocyte fails to respond to partial hepatectomy. Impaired liver regeneration in Med1 null livers suggests a defect in exit from quiescence and diminished entry into G2/M phase (98). Med1 null hepatocytes also failed to respond to hepatocyte growth factor/scatter factor, implying that hepatic Med1 deficiency affects c-met signaling. Taken together, Med1 plays a critical role in stimulation of liver regeneration following partial hepatectomy.
Figure 1.
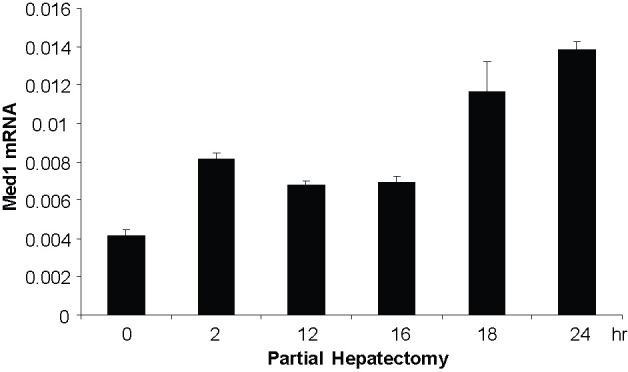
Increased Med1 expression following partial hepatectomy, as revealed by real-time RT-PCR. Livers were harvested at time points as indicated following hepatectomy.
Med1 Is Essential for PPARα-Regulated Gene Expression
PPARα is activated by a structurally diverse array of synthetic chemicals that are potent hepatic mitogens and carcinogens in mice and rats (24,99). In PPARα null mice, the pleiotropic responses induced by peroxisome proliferators such as Wy,14,643, ciprofibrate, and others fail to occur, and these mice also do not develop hepatocellular carcinomas in response to peroxisome proliferators (36). Thus, it is now well established that PPARα is necessary for peroxisome proliferator-induced liver cell proliferation. Recent evidence indicates that Med1 subunit of the Mediator complex is essential for PPARα signal transduction in that Med1 null hepatocytes are similar to PPARα null liver cells in their inability to respond to PPARα ligands (21). Med1 deficiency in liver parenchymal cells results in the abrogation of peroxisome proliferative response and PPARα target gene transcription mimicking the absence of PPARα (21). No DNA synthesis was noted in Med1 null hepatocytes in response to PPARα activators, whereas scattered residual Med1-positive hepatocytes that escaped Cre-mediated excision of floxed alleles in Med1 null livers showed DNA synthesis and were markedly hypertrophic with peroxisome proliferation in response to PPARα ligands (21). Med1 null hepatocytes fail to respond to PPARα ligand-induced peroxisome proliferation (Fig. 2). Moreover, in Med1 null mouse livers, the rare Med1-positive hepatocytes exhibit dramatic increases in peroxisome proliferation and clonal expansion when chronically treated with PPARα ligands such as Wy-14,643 (Fig. 2). Interestingly, the Med1 liver null mice develop liver tumors on long-term exposure to PPARα ligand, but all these tumors are derived from residual Med1-positive cells, but none developed from Med1 null hepatocytes (100). These results imply that Med1 plays a pivotal role in PPARα ligand-induced liver tumor development. In essence, the absence of Med1 in hepatocytes in vivo mimics the absence of PPARα (21,100), indicating that both PPARα and Med1 are essential for PPARα-regulated gene expression including hepatocyte proliferation and liver tumor development.
Figure 2.

Effects of Med1 null on PPARα ligand induced responses in liver. Med1 liver null mice treated with Wy-14,643 (0.125% w/w) for 2 weeks (A, B, and C) and for 6 months (D, E, and F). Immunohistochemical localization of Med1 in Med1 liver conditional null mouse demonstrating absence of Med1 nuclear staining in hepatocytes except for positive staining for Med1 in few hepatocytes (A) and expanding colonies of escaped Cre deletion of large hepatocytes (D). These expanding colonies also reveal abundant cytoplasmic expression of L-PBE (F).
Med1 Is Necessary for CAR-Regulated Gene Expression
Nuclear receptor CAR mediates the hypertrophic and hyperplastic effects in liver upon treatment with CAR activators, phenobarbital, and TCPOBOP. As a nuclear receptor, CAR interacts with the two nuclear receptor-interacting LXXLL motifs in Med1 in a ligand-dependent manner. The unliganded CAR is located in the cytoplasm in hepatic parenchymal cells. It is rapidly translocated to the nucleus in response to activation by CAR agonists (17). In Med1-deficient hepatocytes, CAR fails to translocate to the nucleus in response to activation by phenobarbital or TCPOBOP (21). Adenoviral reconstitution of Med1 in Med1 null mouse livers restores Med1-mediated nuclear translocation of CAR as well as the overexpression of CAR-regulated genes of CYP1A2, CYP2B10, CYP3A11, and CYP7A1 (21). Accordingly, Med1 is considered essential for the translocation, retention, and/or concentration of CAR in the nucleus (101,102). CAR-mediated induction of hepatic CYP3A11, CYP2B10, and CYP1A2 in Med1 null livers is reduced significantly when compared to wild-type mouse livers (21). In wild-type mice, cell cycle proteins cyclin A and cyclin D1 are upregulated in liver at 48 and 96 h after TCPOBOP injection, but not in Med1 null mice. Taken together, the Med1 is considered essential for the function of CAR (21,46). Consistent with these findings is that Med1 deficiency in liver abrogates acetaminophen hepatotoxicity (21). Of interest is striking abrogation of CCl4-induced hepatocyte proliferation and hepatotoxicity in Med1 null livers (98). Furthermore, the diethylnitrosamine (DEN) initiation–phenobarbital promotion (CAR activation) experiment demonstrated that no tumors developed from Med1 null hepatocytes, and all tumors were Med1 positive (100). These observations suggested that all tumors were derived from residual hepatocytes, which escaped Cre-mediated deletion with intact Med1 gene, and hepatocytes deficient in Med1 were not susceptible to neoplastic transformation (100). Med1(fl/fl) HCC cell line generated from these tumors expressed Med1, and deletion of Med1(fl/fl) allele by adeno-Cre in situ injection into tumors led to necrosis of tumor cells. These data illustrate that Med1 is essential for the development of HCC in the mouse (100). In summary, Med1 is required for CAR-regulated gene expression, liver cell proliferation, and hepatocarcinogenesis. These effects may be due to the fact that CAR translocation is Med1 dependent and that CAR target gene expression requires nuclear CAR.
Med1 Overexpression Induces Hepatocyte Proliferation
In earlier studies, Med1 was shown to function as estrogen receptor (ER) coactivator, and Med1 gene was found to be amplified in breast cancers (101). Med1 is also overexpressed in prostate cancer cells, implying that Med1 may play a pivotal role in neoplastic and non-neoplastic cell proliferation (102). Since Med1 null hepatocytes failed to respond to PPARα and CAR activators, and Med1 null hepatocytes did not give rise to tumors, it was hypothesized that Med1 is essential for nuclear receptor signal transduction in liver or that this coactivator per se might be a hepatomitogen. Liver cell proliferative response, if any, that is directly related to Med1 expression was assessed by using adenovirally driven expression of Med1 in mouse liver (20). Overexpression of Med1 in liver cells led to the induction of a broad spectrum of genes as well as hepatocyte proliferation (20). Microarray analysis revealed that Med1 upregulates many genes, including those belonging to initiation and elongation of DNA replication and cell cycle progression, those related to cell growth and mitosis (20). Induction of genes regulated by nuclear receptors PPARα, FXR, CAR, HNF4, and LXR as well as Wnt signaling pathways and genes related to NF-κB regulation was also noted in with Med1 overexpression (20). Interestingly, among these genes is the Foxm1, a key transcription factor for liver regeneration regulated by FXR (103). Foxm1 is upregulated during early cancer development and is involved in tumorigenesis due to its role in cell cycle progression and proliferation (103). Gene expression profiling data revealed that most genes involved in liver regeneration are induced significantly during Med1 overexpression (20). These include early response genes Egr1, JunB, Fos, and C-myc-binding protein (Mycbp). Med1 overexpression also dramatically induced several genes, including cyclins (B1, D1, and E1), Cdks (Cdk1, 2, 4), Cdca8, Cdc16, Cdc20, polo-like kinase 1 (PLK1), centromere proteins (CENPs), E2F family members (E2F1, 4, 6), and survivin. Almost all of these induced genes are involved in G2/M transition. Some genes related to cell growth and mitosis are also induced significantly (greater than fourfold) and include IGFBP1, IGF2, FGF21, GADD45, Gab1 (growth factor receptor bound protein 2-associated protein 1), Mapk 14/p38, Mapk6, AKT1s1 (AKT1 substrate 1), and Gsk3α. It is suggested that Med1 is a key regulator for G1/S and G2/M transition and M phase progression, indicating that Med1 has a critical role in cell cycle progression and cell proliferation. Some important genes involved in a variety of liver functions include Foxm1, FoxO1, nuclear protein 1, interleukin-6-dependent-binding protein, GATA-binding protein 6 (Gata6), transcriptional enhancer factor 1(Tef1), chREBP, and C/EBP. These were induced greater than fourfold, indicating that Med1 has a pivotal role in regulating several transcriptional pathways. Interestingly, due to Med1 overexpression, ∼15 subunits of the Mediator complex were also induced greater than twofold (Med25) (20), implying that Mediator complex formed in these cells may have been changed with respect to their capacity to activate transcription. Med1 overexpression induced PPARα greater than twofold along with the upregulation of approximately eight peroxisomal proteins. Our microarray data also showed induction of the DNA repair and DNA damage response-related genes as well as several apoptosis-related genes, indicating that some of the Med1-overexpressing liver cells may undergo apoptosis.
As discussed above, Med1 null hepatocytes fail to respond to peroxisome proliferator and abrogated PPARα function (19). Of interest is that Med1 overexpression-induced liver cell proliferation does not depend on PPARα. It is well recognized that Med1 is needed for PPARα-regulated gene expression, and available data also points the essential role of Med1 in the activation of several other nuclear receptors, such as CAR, FXR, TR, and GR (21,103–105). These receptors also induce liver cell proliferation. According to our microarray data, Med1 could induce hepatocyte proliferation by amplifying the signaling of various nuclear receptors and transcription factors.
Med1 Is Required for GR Function
Glucocorticoid receptor (GR) agonist dexamethasone (Dex) induces hepatic steatosis and also increases CAR receptor expression (105). Med1 is needed for GR- and CAR-mediated transcriptional activation; it suggests that Med1 deficiency would result in the attenuation of Dex-induced hepatic steatosis. Med1 null livers exhibited reduced levels of GR- and CAR-regulated mRNA compared to wild-type mouse livers (21,105). Administration of glucocorticoids resulted in diminished liver regeneration, in part attributable to GR-induced hepatic steatosis.
PHOSPHORYLATION OF Med1
Phosphorylation augments functional diversity of nuclear receptors and cofactors and provides the basis for a combinatorial code required for specific gene activation. Phosphorylation is the most common post-translational modification that dynamically regulates the molecular properties of coactivators and endows complexity to nuclear receptor-dependent gene expression and associated physiological processes. Evolutionarily, cells have developed several means to respond to internal and external stimuli that signal imbalances in metabolic processes and energy utilization. These include rapid responses, such as phosphorylation events, as well as relatively latent effects on gene transcription (20). PPAR and other nuclear receptors are phosphoproteins, and their transcriptional activity is affected by crosstalk with kinases and phosphatases. Phosphorylation by various kinases such as ERK-/p38-MAPK, PKA/PKC, AMPK, and glycogen synthase kinase-3 (GSK3) that are activated by growth factors and cellular ATP levels regulate PPAR transcriptional activity in a context-dependent manner (106). The activity and specificity of coactivators is subject to regulation by phosphorylation. Coactivators such as SRCs, CBP/p300, PGC-1, and Med1 are phosphorylated by kinases that are involved in diverse cellular signaling (107,108).
Med1 protein has several motifs containing potential serine and threonine residues that are well conserved across the species, suggesting that Med1 is a phosphoprotein. Using in vitro phosphorylation assays, mouse Med1/PBP/TRAP220 by MAPK (ERK1 and ERK2), PKA, and PKC (109) was investigated. These studies revealed an exclusive protein kinase A (PKA) phosphorylation site at serine 656, two protein kinase C (PKC) sites at serine 796 and serine 1345, a common PKA/PKC site at serine 756, and two extracellular signal-regulated kinase 2 sites of the mitogen-activated protein kinase (MAPK) family at threonine 1017 and threonine 1444 (109). Later, it was demonstrated that ERK phosphorylates human TRAP220/Med1 in vivo (HeLa cells) at two specific sites: threonine 1032 and threonine 1457 (110). It is important to note that the motif containing threonine 1017 and 1444 in mouse Med1 is the same as human motif-containing threonine 1032 and 1444. The difference is due to the number of amino acids in mouse (1560) and human (1581) Med1 protein. Phosphorylation at these two sites by ERK stabilizes and increases the intrinsic activity of Med1/TRAP220 (109). The external stimulus for the activation of MAPK–ERK for the phosphorylation of Med1 was shown to be thyroid and gonadal hormone (dihydrotestosterone). This phosphorylation in Hela cells is required for the nuclear receptor MED1 association with the Mediator (109). Functional significance of Med1 phosphorylation at threonine 1032 was addressed in the castration-resistance prostate cancer cell growth (102). UBEC2 overexpress in many types of solid tumors, including androgen receptor (AR)-negative castration-resistant prostate cancer (CRPC) cells. PI3K/AKT-mediated Med1 phosphorylation causes interaction with proteins bound to the promoter element (FoxA1, RNA polymerase II, and TATA-binding protein) of the UBE2C oncogene with the far upstream enhancer making UBE2C locus chromatin looping to stimulate transcription (111). Phosphorylation of Med1 by ERK and/or AKT in prostate cancer cells augments androgen receptor transcriptional activity, which in turn enhances Med1 overexpression and upregulation of genes involved in inflammation, cell cycle progression, and survival (102). Persistent activation of ERK/MAPK leads to Med1 overexpression, resulting in ER-positive breast cancer cells resistant to tamoxifen (112). Phosphorylated Med1 exhibits nuclear accumulation, increased recruitment of ER–Med1 complex on the promoter of ER-responsive upon tamoxifen treatment (112). Recently, phosphorylation of Med1 by energy-sensing kinase AMP-activated protein kinase (AMPK) was demonstrated (20).
AMPK interacts with and directly phosphorylates Med1 in vitro at serine 656, serine 756, and serine 796. AMPK also phosphorylates Med1 in vivo in mouse liver and in cell lines. Of interest is that PPARα activators such as fenofibrate and Wy-14,643 increase AMPK-mediated Med1 phosphorylation in vivo (20). Furthermore, inhibition of AMPK by compound C decreases hepatocyte proliferation induced by Med1 and by PPARα activators implying a link between energy sensing, AMPK phosphorylation of Med1, and hepatocyte proliferation induced by nuclear receptors in concert with the metabolic perturbations.
PERSPECTIVE
Nuclear receptors and coactivators modulate the expression of many RNA polymerase II (Pol II)-transcribed genes involved in metabolic homeostasis and cell proliferation in liver. Perturbations of metabolic functions and changes in cell proliferation appear tightly regulated by nuclear receptors such as PPARα, CAR, FXR, and others. Of the many transcription coactivators identified to date, the Med1 subunit of the Mediator complex appears essential for the PPARα, CAR, and GR-regulated gene expression. Med1 null hepatocytes do not respond to the hepatocyte proliferative effects of PPARα activators. Med1-deficient hepatocytes do not respond to CAR activators, and this is because, in the absence of Med1, CAR remains in the cytoplasm and fails to translocate to the nucleus in response to CAR agonists. Since these two receptors also regulate fat metabolism by elevating fatty acid oxidation systems, absence of Med1 also affects energy metabolism. Mediator contains nearly 30 proteins, but as of now, there is insufficient knowledge as to the role of individual subunits of the Mediator complex in the nuclear receptor-regulated metabolic functions (113–115). Although there is interest in Med1 subunit regulated functions, it is unclear as to the role of other subunits of Mediator complex in relaying the transcriptional signal, for example, from the PPARα and Med1 relay. It is possible that other coactivators and other components of the Mediator may be required for Med1 to transmit the signal to transcriptional machinery. These issues can be explored as mice lacking other subunits of the Mediator become available.
REFERENCES
- 1. Huang J, Borensztajn J, Reddy JK. Hepatic lipid metabolism. In: Monga SPS, editor. Molecular pathology of liver diseases. Springer; 2011. P. 133–146. [Google Scholar]
- 2. Vluggens A, Reddy JK. Nuclear receptors and transcription factors in the development offatty liver disease. Curr Drug Metab 2012; 13:1422–1435. [DOI] [PubMed] [Google Scholar]
- 3. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: Old questions and new insights. Science 2011; 332:1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heather MC, Abigail EM, National Research Council. Hepatitis and liver cancer: A national strategy for prevention and control of hepatitis B and C. Washington, DC: The National Academies Press; 2010. [PubMed] [Google Scholar]
- 5. World Health Organization–International Agency for Research on Cancer. IARC Monograph on the evaluation of carcinogenic risks to humans: Volume 59 Hepatitis Viruses. Geneva: IARC Press; 1994. [Google Scholar]
- 6. Park EJ, Lee JH, Yu GY, He G, Ali DR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010; 140:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michalopoulos GK. Liver regeneration. J Cell Physiol 2007; 213:286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Terpstera OT, Malt RA, Bucher NL. Negligible role of adrenal hormones in regulation of DNA synthesis in livers of partially hepatectomized rats. Proc Soc Exp Bio Med 1979; 161:326–331. [DOI] [PubMed] [Google Scholar]
- 9. Riehle KJ, Dan YY, Campbell JS, Fauto N. New concepts in liver regeneration. J Gastroenterol Hepatol 2011; 26:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perra A, Kowalik MA, Pibiri M, Ledda-Columbano GM, Columbano A. Thyroid hormone receptor ligands induce regression of rat preneoplastic liver lesions causing their reversion to a differentiated phenotype. Hepatology 2009; 49:1287–1296. [DOI] [PubMed] [Google Scholar]
- 11. Misra P, Reddy JK. Peroxisome proliferator-activated receptor-α activation and excess energy burning in hepatocarcinogenesis. Biochimie 2014; 98:63–74. [DOI] [PubMed] [Google Scholar]
- 12. Michalopoulos GK. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am J Pathol 2010; 176:2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davenport RJ. What controls organ regeneration? Science 2005; 309:84. [DOI] [PubMed] [Google Scholar]
- 14. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2005; 324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michalik L, Auwerx J, Berger JP, Chatterjee VK, Gonzalez FJ, Grimaldi PA, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 2006; 58:726–741. [DOI] [PubMed] [Google Scholar]
- 16. Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: Energy combustion, hypolipidemia, inflammation and cancer. Nuclear Receptor Signaling 2010; 8:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol 2000; 20:2951–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci 2005; 30:235–239. [DOI] [PubMed] [Google Scholar]
- 19. Jia Y, Qi C, Kashireddi P, Suraureddi S, Zhu YJ, Rao MS, et al. Transcription coactivator PBP, the peroxisome proliferator-activated receptor (PPAR)-binding protein, is required for PPARα-regulated gene expression in liver. J Biol Chem 2004; 279:24427–24434. [DOI] [PubMed] [Google Scholar]
- 20. Viswakarma N, Jia Y, Bai L, Gao Q, Lin B, Zhang X, et al. The Med1 subunit of the mediator complex induces liver cell proliferation and is phosphorylated by AMP kinase. J Biol Chem 2013; 288:27898–27911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jia Y, Guo GL, Surapureddi S, Sarkar J, Qi C, Guo D, et al. Transcription coactivator peroxisome proliferator-activated receptor-binding protein/mediator 1 deficiency abrogates acetaminophen hepatotoxicity. Proc Natl Acad Sci USA 2005;102:12531–12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sonoda J, Pei L, Evans RM. Nuclear receptors: Decoding metabolic disease. FEBS Lett 2008; 582:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology 2011; 53:1023–1034. [DOI] [PubMed] [Google Scholar]
- 24. Reddy JK, Krishnakantha TP. Hepatic peroxisome proliferation: Induction by two novel compounds structurally unrelated to clofibrate. Science 1975; 190:787–789. [DOI] [PubMed] [Google Scholar]
- 25. Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu Rev Nutr 2001; 21:193–230. [DOI] [PubMed] [Google Scholar]
- 26. Reddy JK, Rao S, Moody DE. Hepatocellular carcinomas in acatalasemic mice treated with nafenopin, a hypolipidemic peroxisome proliferator. Cancer Res 1976; 36:1211–1217. [PubMed] [Google Scholar]
- 27. Reddy JK, Azarnoff DL, Hignite CE. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 1980; 283:397–398. [DOI] [PubMed] [Google Scholar]
- 28. Fan CY, Pan J, Usuda N, Yeldandi AV, Rao MS, Reddy JK. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem 1998; 273:15639–15645. [DOI] [PubMed] [Google Scholar]
- 29. Reddy JK, Lalwani ND. Carcinogenesis by hepatic peroxisome proliferators: Evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. Crit Rev Toxicol 1983; 12:1–58. [DOI] [PubMed] [Google Scholar]
- 30. Maloney EK, Waxman DJ. Trans-activation of PPARα and PPARγ by structurally diverse environmental chemicals. Toxicol Appl Pharmacol 1999; 161:209–218. [DOI] [PubMed] [Google Scholar]
- 31. Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat Res 2000; 448:159–177. [DOI] [PubMed] [Google Scholar]
- 32. Reddy JK, Rao MS, Azarnoff DL, Sell S. Mitogenic and carcinogenic effects of a hypolipidemic peroxisome proliferator, [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio]acetic acid (Wy-14, 643), in rat and mouse liver. Cancer Res 1979; 39:152–161. [PubMed] [Google Scholar]
- 33. Yeldandi AV, Milano M, Subbarao V, Reddy JK, Rao MS. Evaluation of liver cell proliferation during ciprofibrate-induced hepatocarcinogenesis. Cancer Lett 1989; 15:21–27. [DOI] [PubMed] [Google Scholar]
- 34. Schupp M, Lazar MA. Endogenous ligands for nuclear receptors: Digging deeper. J Biol Chem 2010; 285:40409–40415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor α regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol 2007; 27:4238–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 1995; 15:3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peters JM, Cattley RC, Gonzalez FJ. Peroxisome proliferator Wy-14,643. Carcinogenesis 1997; 18:2029–20233. [DOI] [PubMed] [Google Scholar]
- 38. Gonzalez FJ, Shah YM. PPARα: Mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology 2008; 246:2–8. [DOI] [PubMed] [Google Scholar]
- 39. Yu S, Cao WQ, Kashireddy P, Meyer K, Jia Y, Hughes DE, et al. Human peroxisome proliferator-activated receptor alpha (PPARα) supports the induction of peroxisome proliferation in PPARα-deficient mouse liver. J Biol Chem 2001; 276:42485–42491. [DOI] [PubMed] [Google Scholar]
- 40. Morimura K, Cheung C, Ward JM, Reddy JK, Gonzalez FJ. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor α to Wy-14,643-induced liver tumorigenesis. Carcinogenesis 2006; 27:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Forman BM, Tzameli I, Choi HS, Chen J, Simha D, Seol W, et al. Androstane metabolites bind to and deactivate the nuclear receptor CAR-β. Nature 1998; 395:612–615. [DOI] [PubMed] [Google Scholar]
- 42. Wei P, Zhang J, Egan-Hafley M, Liang S, Moore D. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 2000; 407:920–923. [DOI] [PubMed] [Google Scholar]
- 43. Swales K, Negishi M. CAR, driving into the future. Mol Endocrinol 2004; 18:1589–1598. [DOI] [PubMed] [Google Scholar]
- 44. Gao J, Xie W. Pregnane X receptor and constitutive androstane receptor at the crossroads of drug metabolism and energy metabolism. Drug Metab Dispos 2010; 38:2091–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Costa RH, Kalinichenko VV, Tan Y, Wang IC. The CAR nuclear receptor and hepatocyte proliferation. Hepatology 2005; 42:1004–1008. [DOI] [PubMed] [Google Scholar]
- 46. Guo D, Sarkar J, Ahmed MR, Viswakarma N, Jia Y, Yu S, et al. Peroxisome proliferator-activated receptor (PPAR)-binding protein (PBP) but not PPAR-interacting protein (PRIP) is required for nuclear translocation of constitutive androstane receptor in mouse liver. Biochem Biophys Res Commun 2006; 347:485–495. [DOI] [PubMed] [Google Scholar]
- 47. Guo D, Sarkar J, Suino-Powell K, Xu Y, Matsumoto K, Jia Y, et al. Induction of nuclear translocation of constitutive androstane receptor by peroxisome proliferator- activated receptor alpha synthetic ligands in mouse liver. J Biol Chem 2007; 282:36766–76. [DOI] [PubMed] [Google Scholar]
- 48. Columbano A, Ledda-Columbano G, Pibiri M, Concas D, Reddy JK, Rao MS. Peroxisome proliferator-activated receptor-α-/- mice show enhanced hepatocyte proliferation in response to the hepatomitogen 1,4-bis[2-(3,5-dochloropyridyloxy)]benzene, a ligand of constitutive androstane receptor. Hepatology 2001; 34:262–266. [DOI] [PubMed] [Google Scholar]
- 49. Dragani TA, Manenti G, Galliani G, Della Porta G. Promoting effects of 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene in mouse hepatocarcinogenesis. Carcinogenesis 1985; 2:225–228. [DOI] [PubMed] [Google Scholar]
- 50. Tian T, Huang H, Hoffman B, Liebermann DA, Ledda-Columbano GM, Columbano A, et al. Gadd45β is an inducible coactivator of transcription that facilitates rapid liver growth in mice. J Clin Invest 2011; 121:4491–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matic M, Mahns A, Tsoli M, Corradin A, Polly P, Robertson GR. Pregnane X receptor:Promiscuous regulator of detoxification pathways. Int J Biochem Cell Biol 2007; 39:478–483. [DOI] [PubMed] [Google Scholar]
- 52. Squires EJ, Sueyoshi T, Negishi M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J Biol Chem 2004; 279:49307–29314. [DOI] [PubMed] [Google Scholar]
- 53. Di Masi A, De Marinis E, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol Aspects Med 2009; 30:297–343. [DOI] [PubMed] [Google Scholar]
- 54. Dussault I, Yoo HD, Lin M, Wang E, Fan M, Batta AK, Salen G, Erickson SK, Forman BM. Identification of an endogenous ligand that activates pregnane X receptor-mediated sterol clearance. Proc Natl Acad Sci USA 2003; 100:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wada T, Gao J, Xie W. PXR and CAR in energy metabolism. Trends Endocrinol Metab 2009; 20:273–279. [DOI] [PubMed] [Google Scholar]
- 56. Nakamura K, Moore R, Negishi M, Sueyoshi T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J Biol Chem 2007; 282:9768–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dai G, He L, Bu P, Wan YJ. Pregnane X receptor is essential for normal progression of liver regeneration. Hepatology 2008; 47:1277–1287. [DOI] [PubMed] [Google Scholar]
- 58. Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos 2001; 29:1467–1472. [PubMed] [Google Scholar]
- 59. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999; 284:1365–1368. [DOI] [PubMed] [Google Scholar]
- 60. Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear receptor-pendent bile acid signaling is required for normal liver regeneration. Science 2006; 312:233–236. [DOI] [PubMed] [Google Scholar]
- 61. Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem 2004; 279:49517–49522. [DOI] [PubMed] [Google Scholar]
- 62. Zhang L, Wang YD, Chen WD, Wang X, Lou G, Liu N, et al. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology 2012; 56:2336–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007; 28:940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 2007; 67:863–867. [DOI] [PubMed] [Google Scholar]
- 65. Vacca M, Degirolamo C, Massafra V, Polimeno L, Mariani-Costantini R, Palasciano G, et al. Nuclear receptors in regenerating liver and hepatocellular carcinoma. Mol Cell Endocrinol 2013; 368:108–119. [DOI] [PubMed] [Google Scholar]
- 66. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev 2000; 14:2819–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tontonoz P, Mangelsdorf DJ. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol 2003; 17:985–993. [DOI] [PubMed] [Google Scholar]
- 68. York AG, Bensinger SJ. Subverting sterols: Rerouting an oxysterol-signaling pathway to promote tumor growth. J Exp Med 2013; 210:1653–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, et al. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000; 289:1524–1529. [DOI] [PubMed] [Google Scholar]
- 70. Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008; 134:556–567. [DOI] [PubMed] [Google Scholar]
- 71. Mehrotra A, Kaul D, Joshi K. LXR-α selectively reprogrammes cancer cells to enter into apoptosis. Mol Cell Biochem 2011; 349:41–55. [DOI] [PubMed] [Google Scholar]
- 72. Lo Sasso G, Celli N, Caboni M, Murzilli S, Salvatore L, Morgano A, Vacca M, Pagliani T, Parini P, Moschetta A. Down-regulation of the LXR transcriptome provides the requisite cholesterol levels to proliferating hepatocytes. Hepatology 2010; 51:1334–1344. [DOI] [PubMed] [Google Scholar]
- 73. Fanti M, Singh S, Ledda-Columbano GM, Columbano A, Monga SP. Triiodothyronine induces hepatocyte proliferation by protein kinase A-dependent-catenin activation in rodents. Hepatology. in press. DOI: 10.1002/hep.26775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen W, Roeder RG. Mediator-dependent nuclear receptor function. Semin Cell Dev Biol 2011; 22:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kelleher RJ 3rd, Flanagan PM, Kornberg RD. A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 1990; 61:1209–1215. [DOI] [PubMed] [Google Scholar]
- 76. Flanagan PM, Kelleher RJ 3rd, Sayre MH, Tschochner H, Kornberg RD. A mediator required for activation of RNA polymerase II transcription in vitro. Nature 1991; 350:436–438. [DOI] [PubMed] [Google Scholar]
- 77. Fondell JD, Ge H, Roeder RG. Ligand induction of a transcriptionally active thyroid hormone receptor coactivator complex. Proc Natl Acad Sci USA 1996; 93:8329–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Surapureddi S, Yu S, Bu H, Hashimoto T, Yeldandi AV, Kashireddy P, et al. Identification of a transcriptionally active peroxisome proliferator-activated receptor α-interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc Natl Acad Sci USA 2002; 99:11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Myers LC, Kornberg RD. Mediator of transcriptional regulation. Annu Rev Biochem 2000; 69:729–749. [DOI] [PubMed] [Google Scholar]
- 80. Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 2010; 11:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Guglielmi B, van Berkum NL, Klapholz B, Bijma T, Boube M, Boschiero C, et al. A high resolution protein interaction map of the yeast Mediator complex. Nucleic Acids Res 2004; 32:5379–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Soutourina J, Wydau S, Ambroise Y, Boschiero C, Werner M. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science 2011; 331:1451–1454. [DOI] [PubMed] [Google Scholar]
- 83. Gustafsson CM, Samuelsson T. Mediator-a universal complex in transcriptional regulation. Mol Microbiol 2001; 41:1–8. [DOI] [PubMed] [Google Scholar]
- 84. Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 1998; 95:717–728. [DOI] [PubMed] [Google Scholar]
- 85. Zhu Y, Qi C, Jain S, Rao MS, Reddy JK. Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J Biol Chem 1997; 272:25500–25506. [DOI] [PubMed] [Google Scholar]
- 86. Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, et al. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: Discovery of peptide antagonists of estrogen receptors alpha and beta. Mol Cell Biol 1999; 19:8226–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yuan CX, Ito M, Fondell JD, Fu ZY, Roeder RG. The TRAP220 component of a thyroid hormone receptor- associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc Natl Acad Sci USA 1998;95:7939–7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Naar AM, Beaurang PA, Zhou S, Abraham S, Solomon W, Tjian R. Composite co-activator ARC mediates chromatin-directed transcriptional activation. Nature 1999; 398:828–832. [DOI] [PubMed] [Google Scholar]
- 89. Rachez C, Lemon BD, Suldan Z, Bromleigh V, Gamble M, Naar AM, et al. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 1999; 398:824–828. [DOI] [PubMed] [Google Scholar]
- 90. Roeder RG. Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett 2005; 579:909–915. [DOI] [PubMed] [Google Scholar]
- 91. Crawford SE, Qi C, Misra P, Stellmach V, Rao MS, Engel JD, et al. Defects of the heart, eye, and megakaryocytes in peroxisome proliferator activator receptor-binding protein (PBP) null embryos implicate GATA family of transcription factors. J Biol Chem 2002; 277:3585–3592. [DOI] [PubMed] [Google Scholar]
- 92. Lottin-Divoux S, Barel M, Frade R. RB18A enhances expression of mutant p53 protein in human cells. FEBS Lett 2005; 579:2323–2326. [DOI] [PubMed] [Google Scholar]
- 93. Gordon DF, Tucker EA, Tundwal K, Hall H, Wood WM, Ridgway EC. MED220/thyroid receptor-associated protein 220 functions as a transcriptional coactivator with Pit-1 and GATA-2 on the thyrotropin-beta promoter in thyrotropes. Mol Endocrinol 2006; 20:1073–1089. [DOI] [PubMed] [Google Scholar]
- 94. Li H, Gade P, Nallar SC, Raha A, Roy SK, Karra S, et al. The Med1 subunit of transcriptional mediator plays a central role in regulating CCAAT/enhancer-binding protein-beta-driven transcription in response to interferon-gamma. J Biol Chem 2008; 283:13077–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhu Y, Qi C, Jia Y, Nye JS, Rao MS, Reddy JK. Deletion of PBP/PPARBP, the gene for nuclear receptor coactivator peroxisome proliferator-activated receptor-binding protein, results in embryonic lethality. J Biol Chem 2000; 275:14779–14782. [DOI] [PubMed] [Google Scholar]
- 96. Ito M, Yuan CX, Okano HJ, Darnell RB, Roeder RG. Involvement of the TRAP220 component of the TRAP/SMCC coactivator complex in embryonic development and thyroid hormone action. Mol Cell 2000; 5:683–693. [DOI] [PubMed] [Google Scholar]
- 97. Ge K, Guermah M, Yuan CX, Ito M, Wallberg AE, Spiegelman BM, et al. Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature 2002; 417:563–567. [DOI] [PubMed] [Google Scholar]
- 98. Matsumoto K, Yu S, Jia Y, Ahmed MR, Viswakarma N, Sarkar J, et al. Critical role for transcription coactivator peroxisome proliferator-activated receptor (PPAR)-binding protein/TRAP220 in liver regeneration and PPARalpha ligand-induced liver tumor development. J Biol Chem 2007; 282:17053–17060. [DOI] [PubMed] [Google Scholar]
- 99. Rao MS, Dwivedi RS, Subbarao V, Reddy JK. Induction of peroxisome proliferation and hepatictumours in C57BL/6N mice by ciprofibrate, a hypolipidaemic compound. Br J Cancer 1988; 58:46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Matsumoto K, Huang J, Viswakarma N, Bai L, Jia Y, Zhu YT, et al. Transcription coactivator PBP/MED1-deficient hepatocytes are not susceptible to diethylnitrosamine-induced hepatocarcinogenesis in the mouse. Carcinogenesis 2010; 31:318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R 3rd, Atkins GB, et al. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc Natl Acad Sci USA 1999; 96:10848–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jin F, Irshad S, Yu W, Belakavadi M, Chekmareva M, Ittmann MM, et al. ERK and AKT signaling drive MED1 overexpression in prostate cancer in association with elevated proliferation and tumorigenicity. Mol Cancer Res 2013; 11:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Borude P, Edwards G, Walesky C, Li F, Ma X, Kong B, et al. Hepatocyte-specific deletion of farnesoid X receptor delays but does not inhibit liver regeneration after partial hepatectomy in mice. Hepatology 2012; 56:2344–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fondell JD. The Mediator complex in thyroid hormone receptor action. Biochim Biophys Acta 2013; 1830:3867–3875. [DOI] [PubMed] [Google Scholar]
- 105. Jia Y, Viswakarma N, Fu T, Yu S, Rao MS, Borensztajn J, et al. Conditional ablation of mediator subunit MED1 (MED1/PPARBP) gene in mouse liver attenuates glucocorticoid receptor agonist dexamethasone-induced hepatic steatosis. Gene Expr 2009; 14:291–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Burns KA, VandenHeuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 2007; 1771:952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rowan BG, Weigel NL, O’Malley BW. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem 2000; 275:4475–4483. [DOI] [PubMed] [Google Scholar]
- 108. Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell 2001; 8:971–982. [DOI] [PubMed] [Google Scholar]
- 109. Misra P, Owuor ED, Li W, Yu S, Qi C, Meyer K, et al. Phosphorylation of transcriptional coactivator peroxisome proliferator-activated receptor (PPAR)-binding protein (PBP). Stimulation of transcriptional regulation by mitogen-activated protein kinase. J Biol Chem 2002; 277:48745–48754. [DOI] [PubMed] [Google Scholar]
- 110. Belakavadi M, Pandey PK, Vijayvargia R, Fondell JD. MED1 phosphorylation promotes its association with mediator: Implications for nuclear receptor signaling. Mol Cell Biol 2008; 28:3932–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen Z, Zhang C, Wu D, Chen H, Rorick A, Zhang X, et al. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO J 2011; 30:2405–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Nagalingam A, Tighiouart M, Ryden L, Joseph L, Landberg G, Saxena NK, et al. Med1 plays a criticalrole in the development of tamoxifen resistance. Carcinogenesis 2012; 33:918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Stumpf M, Yue X, Schmitz S, Luche H, Reddy JK, Borggrefe T. Specific erythroid-lineage defect in mice conditionally deficient for Mediator subunit Med1. Proc Natl Acad Sci USA 2010; 107:21541–21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen W, Zhang X, Birsoy K, Roeder RG. A muscle-specific knockout implicates nuclear receptor coactivator MED1 in the regulation of glucose and energymetabolism. Proc Natl Acad Sci USA 2010; 107:10196–101201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Krebs P, Fan W, Chen YH, Tobita K, Downes MR, Wood MR, et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc Natl Acad Sci USA 2011; 108:19678–19682. [DOI] [PMC free article] [PubMed] [Google Scholar]