

ABSTRACT
Human metapneumovirus (HMPV) is a leading cause of respiratory disease in infants, children, and the elderly worldwide, yet no licensed vaccines exist. Live-attenuated vaccines present safety challenges, and protein subunit vaccines induce primarily antibody responses. Virus-like particles (VLPs) are an attractive alternative vaccine approach because of reduced safety concerns compared with live vaccines. We generated HMPV VLPs by expressing viral proteins in suspension-adapted human embryonic kidney epithelial (293-F) cells and found that the viral matrix (M) and fusion (F) proteins were sufficient to form VLPs. We previously reported that the VLPs resemble virus morphology and incorporate fusion-competent F protein (R. G. Cox, S. B. Livesay, M. Johnson, M. D. Ohi, and J. V. Williams, J. Virol. 86:12148–12160, 2012), which we hypothesized would elicit F-specific antibody and T cell responses. In this study, we tested whether VLP immunization could induce protective immunity to HMPV by using a mouse model. C57BL/6 mice were injected twice intraperitoneally with VLPs alone or with adjuvant and subsequently challenged with HMPV. Mice were euthanized 5 days postinfection, and virus titers, levels of neutralizing antibodies, and numbers of CD3+ T cells were quantified. Mice immunized with VLPs mounted an F-specific antibody response and generated CD8+ T cells recognizing an F protein-derived epitope. VLP immunization induced a neutralizing-antibody response that was enhanced by the addition of either TiterMax Gold or α-galactosylceramide adjuvant, though adjuvant reduced cellular immune responses. Two doses of VLPs conferred complete protection from HMPV replication in the lungs of mice and were not associated with a Th2-skewed cytokine response. These results suggest that nonreplicating VLPs are a promising vaccine candidate for HMPV.
IMPORTANCE Human metapneumovirus (HMPV) is a leading cause of acute respiratory infection in infants, children, and the elderly worldwide, yet no licensed vaccines exist. Live-attenuated vaccines present safety challenges, and protein subunit vaccines induce primarily antibody responses. Virus-like particles (VLPs) are an attractive alternative vaccine approach. We generated HMPV VLPs by expressing the viral matrix (M) and fusion (F) proteins in mammalian cells. We found that mice immunized with VLPs mounted an F-specific antibody response and generated CD8+ T cells recognizing an F protein-derived epitope. VLP immunization induced a neutralizing-antibody response that was enhanced by the addition of either TiterMax Gold or α-galactosylceramide adjuvant. Two doses of VLPs conferred complete protection against HMPV replication in the lungs of mice and were not associated with a Th2-skewed cytokine response. These results suggest that nonreplicating VLPs are a promising vaccine candidate for HMPV.
INTRODUCTION
Human metapneumovirus (HMPV) is a leading cause of acute lower respiratory tract infection worldwide, with high prevalence in pediatric, elderly, and immunocompromised patients (1–12). There are no licensed vaccines against HMPV. Several strategies to develop live-attenuated HMPV vaccines have been explored, including cold passage, gene deletion, and chimeric viruses (13–17). While live-virus vaccines elicit humoral and cellular responses, they also pose safety risks. Attenuated virus strains have the potential to revert to a wild-type phenotype and cause disease or be transmitted to nonimmune individuals. For these reasons, live attenuated vaccines are often contraindicated for immunocompromised patients, individuals who are at risk for severe HMPV infections. Moreover, it is often difficult to find the correct balance between attenuation and immunogenicity. Many years of research on respiratory syncytial virus (RSV) live attenuated vaccines attest to the challenges (18–22).
Subunit protein vaccines against HMPV targeting mainly the fusion (F) protein have been effective in rodent models by inducing B cell responses only (23, 24). Experience with formalin-inactivated (FI) RSV and HMPV vaccines in humans and animals further raises concern about imbalanced immunity (25–30). Studies demonstrate that the generation of antibodies to a denatured F protein or low-affinity nonneutralizing antibodies is associated with enhanced respiratory disease (31–36). Thus, a safe and effective vaccine should induce both potent neutralizing antibodies and cytotoxic T cell responses.
A vaccination strategy combining elements of both live virus and subunit vaccines is virus-like particles (VLPs). VLPs are formed by the self-assembly of viral structural proteins but lack the virus genome and thus are not able to replicate. VLPs are an attractive vaccine candidate because they are noninfectious, but the particles mimic virus structure and are capable of inducing protective humoral and cellular immune responses. Viral antigens can be presented in a native conformation, similar to viral protein contained in infectious virus particles. VLPs are similar in size to infectious virus and present viral antigens in a repetitive and ordered fashion, making it likely that the immune response to the antigen will be similar to the response induced by infectious virus. Furthermore, VLP-based particles can be designed to incorporate a limited number of viral proteins, thus focusing the humoral and cellular immune responses on protective antigens. Several VLP-based vaccine strategies are currently being developed for a variety of nonenveloped and enveloped viruses (reviewed in reference 37). For example, VLP vaccines for hepatitis C virus (38), influenza virus (reviewed in references 39 and 40), and HIV-1 (reviewed in reference 41) are being developed and tested in preclinical trials. Furthermore, VLP vaccine formulations for hepatitis B virus (42) and human papillomavirus (43) have been licensed.
There are three HMPV surface glycoproteins, the fusion (F) protein, the glycoprotein (G), and the small hydrophobic (SH) protein (Fig. 1A). Only the F protein is required for virus assembly, fusion, and entry (13, 44). Furthermore, the HMPV F protein is the primary target of neutralizing antibodies (13, 23, 45–48). In contrast, while HMPV G is immunogenic in rodents, G-specific antibodies do not neutralize HMPV and G vaccines provide no protection in animals (47–49). Thus, we hypothesized that stimulation of immune responses with VLPs bearing the F protein would be sufficient to induce protective immunity against HMPV infection.
FIG 1.
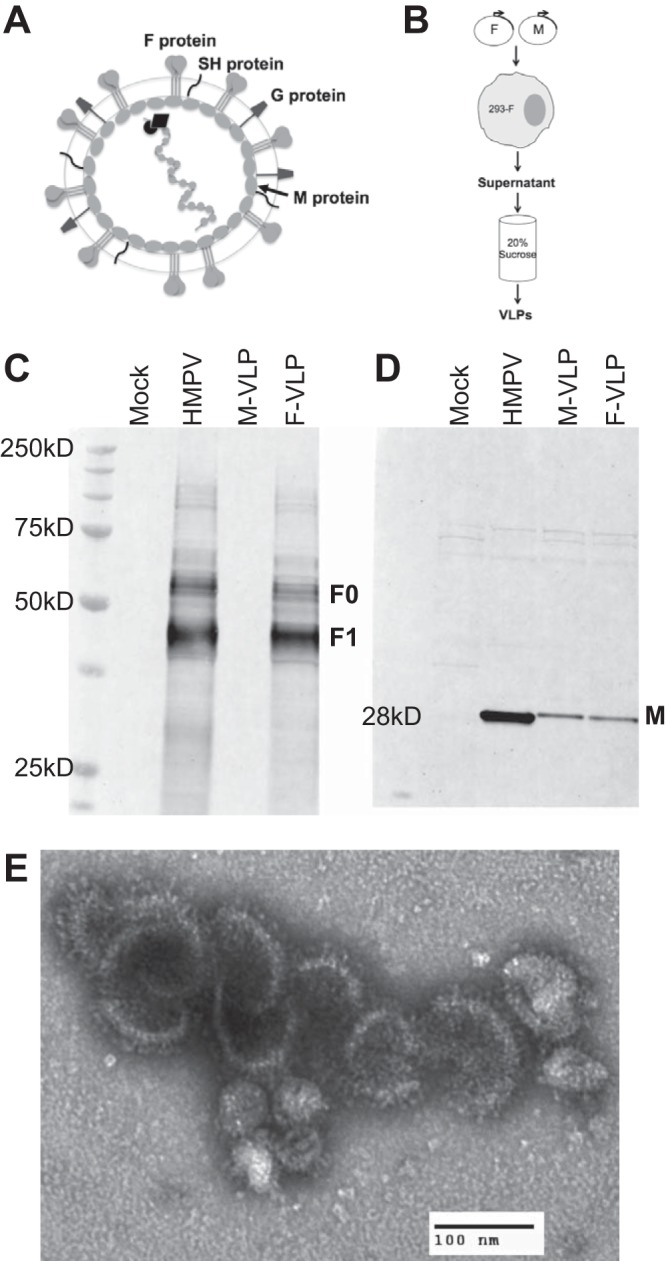
HMPV VLPs resemble virus in morphology and incorporate the F and M proteins. (A) Schematic representation of an HMPV virion. The F, G, and SH proteins are depicted at the virion surface. The M protein lines the inner leaflet of the virus membrane. Encapsidated within the viral envelope is the RNA genome that is associated with the nucleoprotein (N), viral polymerase (L), phosphoprotein (P), and matrix 2 (M2) proteins, which are not labeled. (B) Schematic showing the procedure used to generate VLPs. 293-F cells were transfected with HMPV M or M-plus-F protein expression plasmids. At 4 days posttransfection, cell supernatants were harvested and purified through 20% sucrose and VLP pellets were resuspended in PBS. (C, D) Particles released from untransfected (mock VLPs), HMPV-infected (HMPV), M-transfected (M-VLP), and M-plus-F-transfected (F-VLP) cells were harvested, purified, and analyzed by Western blot assay to verify their composition. The same amount (20 μg total protein of either VLP or virus) of each preparation was separated by denaturing, reducing SDS-PAGE and immunoblotted with anti-F MAb (C) or polyclonal anti-HMPV M antiserum (D). Molecular mass markers are shown to the left of the protein ladder. Bands corresponding to F0 (uncleaved F), F1 (cleaved F), and M are indicated. (E) F-VLPs released from cells transfected with HMPV M plus F were purified and analyzed by electron microcopy. An electron micrograph of a negatively stained sample showing a cluster of F-VLPs is shown. Magnification, ×28,000. F glycoprotein spikes are visible on the VLP surface.
The HMPV F protein is a trimeric type I membrane glycoprotein that must be proteolytically cleaved to convert the inactive precursor, F0, into a fusion-competent, disulfide-linked F1–F2 heterodimer. After cleavage, HMPV F exists in a metastable, prefusion structural conformation that mediates virus entry into cells during infection. We used a mammalian expression system to generate HMPV VLPs that display the F protein in a membrane-anchored trimeric native conformation. Particle-based entry assays previously reported by our group demonstrated that the F protein on the surface of VLPs is functional in mediating particle binding and fusion (50).
In this study, we tested whether immunization with HMPV F-bearing VLPs could induce humoral and cellular immunity in a mouse model of HMPV infection. We show that the VLPs are immunogenic in mice, inducing F-specific antibodies and a neutralizing-antibody response. Immunization with VLPs induced T cell migration to the lungs after HMPV inoculation, and F-epitope CD8+ T cells were identified in the lungs and spleens of immunized mice. Finally, we demonstrate that immunization with HMPV VLPs restricts HMPV replication in the upper respiratory tract and completely protects mice from HMPV replication in the lungs. These results suggest that VLPs containing the F protein represent a promising vaccine candidate for the prevention of HMPV infection.
MATERIALS AND METHODS
Cell culture and HMPV preparation.
LLC-MK2 (ATCC CCL-7) cells were maintained in Opti-MEM I medium (Invitrogen) containing 2% fetal bovine serum–2 mM l-glutamine–50 μg/ml gentamicin–2.5 μg/ml amphotericin. Suspension 293-F cells were maintained as recommended by the manufacturer (Freestyle 293 Expression System; Invitrogen). HMPV TN/94-49, a clinical isolate passaged five to seven times and thrice plaque purified in LLC-MK2 cells, was propagated by using suspension 293-F cells in 293 Freestyle Expression medium supplemented with 5 μg/ml trypsin-EDTA (both from Invitrogen). Briefly, 293-F cells were infected at 0.1 PFU/cell. Four days postinfection, supernatant virus was collected and infected cells were rapidly frozen and thawed three times to release cell-associated virus. The pooled virus preparation was clarified by centrifugation at 300 × g for 5 min and pelleted through a 20% sucrose cushion via ultracentrifugation at 100,000 × g for 90 min at 4°C. The virus pellet was resuspended in phosphate-buffered saline (PBS), snap-frozen in a dry ice-alcohol bath, and stored at −80°C. Virus was titrated on LLC-MK2 cells as previously described (51).
Generation of HMPV VLPs.
HMPV VLPs were generated in suspension 293-F cells by transient expression of the HMPV matrix (M) and/or fusion (F) proteins. Full-length M and F viral protein sequences were derived from pathogenic clinical HMPV (genotype A2) isolates TN/94-49 and TN/92-4, respectively. Expression plasmids for HMPV F and M were optimized for mammalian expression as previously described (23, 50). Two types of VLPs were prepared, M-VLPs (containing the HMPV matrix protein) and F-VLPs (containing the HMPV matrix and fusion proteins). Suspension 293-F cells were transfected with pcDNA3.1-M (40 μg) and either pcDNA3.1 (20 μg) or pcDNA3.1-F (20 μg) with 293fectin transfection reagent (60 μl) as recommended by the manufacturer and grown in serum-free medium (Invitrogen). Eighteen hours posttransfection, the growth medium was changed and 5 μg/ml trypsin was added. Three days posttransfection, the supernatant was collected, clarified by centrifugation at 300 × g for 5 min, and pelleted through 20% sucrose as described above for virus. VLP pellets were resuspended in PBS, snap-frozen in a dry ice-alcohol bath, and stored at −80°C. A mock particle preparation was generated from the supernatant collected from 293-F cells. HMPV was also prepared as described for VLPs, except that cells were incubated for 4 days before harvesting of the supernatant virus for purification.
Characterization of VLPs. (i) Protein content.
The protein content of particle preparations was determined with the DC Protein Assay (Bio-Rad). Particle preparations contained similar levels of total protein, mock preparation (1.19 ± 0.07 mg/ml), HMPV (2.39 ± 0.08 mg/ml), M-VLP (2.32 ± 0.04 mg/ml), and F-VLP (2.08 ± 0.14 mg/ml).
(ii) Electron microscopy.
Sucrose-purified F-VLPs were stained with uranyl formate (0.75%) and prepared for transmission electron microscopy as described previously (52). Samples were imaged on a FEI Morgagni electron microscope operated at an acceleration voltage of 100 kV. Images were recorded at a magnification of ×22,000 and collected with a charge-coupled device camera (AMT) at a 1,000-by-1,000 resolution.
(iii) Western blot analysis.
Purified virus or VLPs (20 μg total protein) were lysed with 1% SDS, boiled in NuPAGE LDS Sample Buffer (Invitrogen) containing 5% β-mercaptoethanol (Sigma), separated on 10% NuPAGE Bis-Tris gels (Invitrogen), and transferred to polyvinylidene fluoride membranes. Membranes were blocked for 30 min with 5% milk in PBS plus 0.1% Tween (PBS-T; blocking buffer). An Armenian hamster anti-F monoclonal antibody (MAb; mAb1017, kindly provided by Nancy Ulbrandt [46]) and rabbit anti-M serum were diluted in blocking buffer and incubated with membranes overnight at 4°C. Cy5-conjugated anti-Armenian hamster (Jackson ImmunoResearch) or LI-COR IRDye 800CW anti-rabbit secondary antibodies were diluted in blocking buffer and incubated with membranes for 1 h at room temperature. Membranes were washed three times in PBS-T and twice in PBS to remove residual detergent and dried. Bands were imaged with an Odyssey infrared imaging system (LI-COR).
(iv) HMPV F protein content.
F protein content was determined for virus and F-VLP particles by Western blot assay. Recombinant F protein lacking the transmembrane domain (FΔTM) (23) was used to generate a standard curve in the linear range of the fluorescent signal obtained under the anti-F blotting conditions described above. HMPV, F-VLPs, and F protein standards were analyzed on the same membrane, and bands were quantified with the Odyssey infrared imaging software (LI-COR). HMPV particles contained 396 ± 55 ng/μl and F-VLPs contained 136 ± 13 ng/μl F protein.
Immunization of animals.
Six-week-old female C57BL/6 (B6) mice were purchased from Jackson Laboratory. Animals were anesthetized with ketamine-xylazine prior to virus inoculation. Mice in groups of five were immunized by intraperitoneal (i.p.) injection with 100 μl of VLPs adjuvanted 1:1 with PBS, TiterMax Gold (TMG; Sigma), or α-galactosylceramide (αGC; Funakoshi). TMG and αGC were chosen as adjuvants on the basis of our experience with their use and immunogenicity in rodents (23, 49, 53, 54). The i.p. route was chosen over the intramuscular route on the basis of the National Institutes of Health Office of Animal Care recommendations for adjuvant use in rodents (http://oacu.od.nih.gov). HMPV-infected animals were anesthetized with ketamine-xylazine and infected intranasally (i.n.) with 1.5 × 106 PFU of HMPV in a 100-μl volume. Serum was collected from mice by submandibular bleeding prior to immunization and before boost immunization.
HMPV challenge.
Mice were inoculated on day 28 i.n. with 108 PFU of HMPV in a 100-μl volume. Five days later, mice were euthanized by CO2 asphyxiation and exsanguinated. Briefly, nasal turbinates (NT) and lung tissues were harvested and virus titers were measured by plaque titration on LLC-MK2 cells as previously described (51). HMPV neutralizing-antibody titers in mouse serum samples were determined by plaque reduction assay as previously described (51).
Immunohistochemistry of mouse lung specimens.
The lower half of the left lung was inflated with 10% formalin, fixed overnight, and processed as previously described (51). Lungs were stained with anti-mouse CD3 (Santa Cruz Biotechnology) for 60 min at room temperature. A trained pathologist who was blinded to the immunization group scored the stained lung sections on the basis of the level of CD3+ cell infiltrate present in the alveolar septa, which was defined as follows: none, 0; some, 1; moderate, 2; marked, 3. Stained slides also were converted to digital images with an Aperio ScanScope CS2 (magnification, ×20) and analyzed with Aperio Image Scope software (v11.2.0780), and CD3+ cells were quantified by using a color deconvolution algorithm. Ten random regions of interest across the entire lung section were chosen, and the CD3+ area was calculated.
ELISA.
Flat-bottom microtiter 96-well plates (Immulon 2HB; Thermo) were coated with 100 ng of HMPV FΔTM protein (23). The plates were blocked with 5% milk in PBS-T (blocking buffer) for 2 h at room temperature. Serial 2-fold dilutions of each mouse serum sample, starting at 1:40, were prepared in blocking buffer and added to FΔTM-coated plates for 1 h of incubation at room temperature on an orbital rocker. The plates were washed thrice with PBS-T and then incubated with an anti-mouse Ig (H+L) horseradish peroxidase-coupled secondary antibody (Southern Biotech) for 1 h at room temperature on an orbital shaker. The plates were washed four times with PBS-T and developed with Pierce Ultra-TMB colorimetric substrate. Color development was stopped with 2 M sulfuric acid, and absorbance at 450 nm was measured with a plate reader (Chameleon V; Hidex). Nonlinear regression analyses (sigmoidal, variable slopes) were performed to fit dose-response curves to enzyme-linked immunosorbent assay (ELISA) absorbance values and used to calculate the HMPV F-specific antibody titer at the half-maximum response (50% inhibitory concentration [IC50]).
Flow cytometry staining and gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) assay.
Lymphocytes were isolated from the lung and/or spleen and tetramer stained or restimulated for intracellular cytokine staining (ICS) as previously described (55). Briefly, isolated lymphocytes were first stained with LIVE/DEAD dye violet (Invitrogen), Fc receptors were blocked with anti-CD16/32 (BD Biosciences) antibody at 1 μg/106 cells, and finally cells were incubated with allophycocyanin-labeled HMPV H2-Db-F528-536 (F528) tetramer (0.1 to 1 μg/ml), anti-CD8α (clone 53-6.7; BD Biosciences), anti-CD4 (clone RM4-5; BD Biosciences), and anti-CD19 (clone 1D3; iCyt) antibodies for 1 h at room temperature in the presence of 50 nM dasatinib (LC Laboratories). An irrelevant influenza virus-specific tetramer (H2-Db/NP366-374) was used for gating. Flow cytometric data were collected with an LSR II (BD Biosciences) and analyzed with FlowJo software (Tree Star).
For ICS, lung lymphocytes were restimulated in vitro for 6 h at 37°C with F528 or an irrelevant peptide (10 μM final concentration), with brefeldin A and monensin (BD Bioscience) added for the final 4 h of restimulation. Stimulation with 50 ng/ml phorbol myristate acetate plus 2 μg/ml ionomycin (Sigma) served as a positive control. After restimulation, cells were surface stained for CD3ε (clone 145-2C11), CD8α, and CD19, followed by fixation-permeabilization and staining for intracellular IFN-γ (clone XMG1.2) (all from BD Bioscience), and analyzed by flow cytometry. Background IFN-γ levels following restimulation with an irrelevant peptide were subtracted from each experimental value.
ELISPOT analysis was performed as previously described (55). Splenocytes were incubated with F528 peptide, an irrelevant peptide as a negative control, or concanavalin A (ConA; Sigma) as a positive control. The average number of spots in the negative-control wells was subtracted from each experimental value, and the number of spot-forming cells (SFC) per 106 lymphocytes was calculated and expressed as a percentage of the response of mock-immunized animals (set at 100%).
Real-time RT-PCR.
Lungs were collected on day 5 postchallenge and homogenized, and RNA was extracted with the MagNA Pure LC Total Nucleic Acid Isolation kit (Roche Applied Sciences) on a MagNA Pure LC. Real-time reverse transcription (RT)-PCR was performed with commercial gene probes (Life Technologies) and 25-μl reaction mixtures containing 5 μl of extracted RNA on an ABI StepOnePlus real-time PCR system (Life Technologies) with the AgPath-ID One-Step RT-PCR kit (Life Technologies). All values were normalized to the hypoxanthine phosphoribosyltransferase (HPRT) housekeeping gene, and experimental results are reported as fold differences (determined by the ΔΔCT method) from those of mice that were mock VLP immunized.
Statistical analysis.
To calculate mean serum neutralization titers, plaque counts (in the presence of serial dilutions of mouse serum) were normalized to an untreated control by calculating the percent plaque reduction. Percent plaque reduction was plotted versus serum dilution, and nonlinear regression curves assuming a sigmoidal dose-response curve, allowing variable slopes, with bottom = 0 and top = 100 constraints, were calculated. The mean IC50 for each curve was defined as the mean neutralizing-antibody titer. Multiple-group comparisons were performed by one-way analysis of variance (ANOVA) with Dunnett's multiple-comparison posttest for comparison of individual groups. Comparisons of two groups were performed with an unpaired two-tailed Student t test. For all analyses, P ≤ 0.05 was considered statistically significant.
Study approval.
All animals were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals and handled according to protocols approved by the Vanderbilt University Institutional Animal Care and Use Committee.
RESULTS
Development of HMPV VLPs.
Different viral structural proteins are necessary for paramyxovirus particle formation (reviewed in reference 56). Thus, we first explored which HMPV structural proteins (F, M, N, and/or P) are required to form VLPs by transient expression of viral proteins in cells. We chose to produce VLPs in suspension 293-F cells because these cells (i) have a high transfection efficiency; (ii) express large amounts of plasmid-encoded proteins; (iii) grow to very high densities (>3 × 106/ml) without loss of cell viability; (iv) propagate readily in serum-free medium, allowing the medium to be supplemented with trypsin, which is required to process the HMPV F protein; and (v) have been previously used to generate recombinant HMPV F and G proteins (23, 49, 57).
In initial experiments, we transfected plasmids encoding the HMPV M; F; F and M; or F, M, N, and P proteins into 293-F cells, and analyzed the cell supernatants at 72 h posttransfection by Western blotting (data not shown). We determined that HMPV VLPs could be reproducibly recovered when 293-F cells were transfected with plasmids encoding HMPV M alone (M-VLP) or the M and F proteins (F-VLP) (Fig. 1B). We purified and concentrated the VLPs by ultracentrifugation through a 20% sucrose cushion and analyzed the particle pellets. We confirmed the incorporation of HMPV F and M by Western blotting (Fig. 1C and D). The uncleaved F0 form of the F protein was detected at a molecular mass of ∼60 kDa, and the trypsin-cleaved, active F1 form was detected at ∼47 kDa. HMPV M was detected at the expected molecular mass of 28 kDa. The levels of F protein were similar in virus and F-VLP preparations, while less M protein was incorporated into F-VLPs than into virus particles. However, the levels of M protein were similar in M-VLP and F-VLP preparations (Fig. 1D). We examined the F-VLPs by electron microscopy and found enveloped particles with surface glycoprotein spikes (Fig. 1E) resembling HMPV virions. A detailed characterization of the morphological similarities between F-VLPs and HMPV has been previously published (50). Briefly, F-VLPs range in diameter from 54 to 240 nm (mean ± standard error of the mean [SEM], 123 ± 10 nm) and F glycoprotein spikes measuring 13.3 nm are visible on the F-VLP and HMPV virion surfaces. We previously reported that F incorporated into F-VLPs is folded into the prefusion conformation and is functional in binding and fusion assays (50). We have found that VLP preparations stored at −80°C retain their fusion capacity for >12 months.
HMPV VLPs induce neutralizing humoral immune responses.
To test whether the VLPs were immunogenic in a mouse model of HMPV respiratory infection, we conducted preliminary experiments with B6 mice. Groups of mice were either immunized i.p. with F-VLPs alone on days 0 and 35 or infected i.n. with HMPV on days 0 and 35. The serum neutralizing-antibody titers of both groups were determined on days 35 (preboost) and 42 (postboost). The serum neutralizing-antibody titer in F-VLP-immunized mice at 35 days (preboost) was 5.0 ± 0.5 log2, which was significantly lower (P < 0.01) than the F-VLP postboost (8.4 ± 0.6 log2), HMPV preboost (7.9 ± 0.8 log2), and HMPV postboost (8.7 ± 0.3 log2) titers, none of which differed from one another. Thus, a single dose of F-VLPs without adjuvant yielded lower serum neutralizing-antibody titers than HMPV infection, but a boost increased titers to a level indistinguishable from that of infected mice.
On the basis of these results, we designed an experiment where we could monitor humoral immune responses and determine protective efficacy against HMPV infection. A schematic of the experimental protocol is shown in Fig. 2A. We administered the VLPs by using a prime-boost strategy, where mice received two doses of the same HMPV particle preparation with a 2-week rest between doses. We injected groups of B6 mice i.p. with M-VLPs or F-VLPs alone, M-VLPs or F-VLPs in suspension with TMG, or M-VLPs or F-VLPs in suspension with αGC. A mock VLP preparation was used as a negative control, and one group was infected i.n. with HMPV on day 0 as a positive control.
FIG 2.

Immunization with HMPV VLPs induces F-specific antibodies. (A) Schematic showing the immunization protocol. B6 mice (five per group) were immunized two times, 14 days apart, by i.p. injection with VLPs containing either HMPV M (M-VLP) or M plus F (F-VLP) protein. VLPs were administered alone, with TMG adjuvant, or with αGC adjuvant. Negative-control mice were injected with purified supernatant from untreated 293-F cells (mock VLPs). Positive-control mice were infected i.n. with HMPV on day 0. Blood was collected at the time points indicated and at euthanasia to determine the production of F-specific and neutralizing antibodies. (B to J) HMPV F-specific antibody levels in serum samples were measured by ELISA. Preimmune serum was collected on day 0. Postinfection and postimmunization serum samples were collected 5 days after an HMPV challenge. The absorbance resulting from serum antibody binding to plates coated with recombinant F protein is shown for each vaccination group as follows: panel B, mock VLPs; panel C, HMPV; panel D, M-VLP; panel E, M-VLP plus TMG; panel F, M-VLP plus αGC; panel G, F-VLP; panel H, F-VLP plus TMG; panel I, F-VLP plus αGC. Absorbance data are shown as the mean ± the SEM for five mice from three independent ELISAs. Nonlinear regression analyses were performed for groups with F-specific antibodies and used to calculate reciprocal serum titers at half-maximal absorbance (IC50), shown in panel J. Data points represent individual mice, and the bar depicts the mean IC50 titer of each immunization group. Comparisons of multiple groups were made by one-way ANOVA for the P values shown in panel J.
Mouse serum was sampled before immunization (preimmune) and 14 days after the first injection, when a second injection was given. Mice were challenged i.n. with HMPV on day 28, and blood was collected 5 days postinfection, when all of the animals were euthanized (postimmunization). To analyze the humoral immune responses of immunized mice, we first measured the HMPV F-specific IgG antibody response to recombinant F protein by ELISA (Fig. 2B to J). Mock-immunized mice had no detectable levels of HMPV F-specific antibody (Fig. 2B), and mice that had been previously infected with HMPV had elevated titers of F-specific antibodies (Fig. 2C). As expected, immunization with M-VLPs alone or with adjuvant did not induce F-specific antibody responses (Fig. 2D to F). Immunization with F-VLPs induced F-specific IgG antibody responses in all of the immunized animals (Fig. 2G to I). F-VLP immunization resulted in F-specific antibody titers that were similar to the antibody levels of previously infected mice, while mice immunized with F-VLPs in the presence of either adjuvant had significantly higher levels of F-specific IgG antibodies (Fig. 2J). These results demonstrate that immunization with HMPV F-VLPs induces a serum IgG antibody response to the F protein.
To determine whether the F-specific serum antibodies were capable of neutralizing HMPV infection, we measured antibody function with a plaque reduction assay. Serum samples collected from mice immunized with F-VLPs significantly inhibited HMPV infection (Table 1). F-VLPs administered with TMG or αGC induced a significantly higher neutralizing-antibody titer than F-VLPs alone. Two doses of F-VLPs with either adjuvant were sufficient to induce a neutralizing-antibody response similar to that seen after HMPV infection. A neutralizing-antibody response in mock-immunized, HMPV-challenged mice was not detectable on day 5 postinfection, and M-VLP immunization with or without adjuvant did not induce a neutralizing-antibody response. Collectively, these results confirm that HMPV VLPs are immunogenic and induce a humoral neutralizing-antibody response in B6 mice.
TABLE 1.
Serum neutralizing-antibody titers of immunized groups of mice
| Vaccination typea | Adjuvant | Mean log serum neutralizing-antibody titer ± SEMb (statistical group[s])c |
||
|---|---|---|---|---|
| Preimmunization | 14 days postimmunization | 5 days postchallenge | ||
| Mock | None | <3.0 | <3.0 | <3.0 |
| M-VLP | None | <3.0 | <3.0 | <3.0 |
| M-VLP | TMG | <3.0 | <3.0 | ≤3.0 |
| M-VLP | αGC | <3.0 | <3.0 | 3.6 ± 0.6 (A) |
| F-VLP | None | <3.0 | 4.0 ± 0.2 (A) | 4.3 ± 0.3 (A) |
| F-VLP | TMG | <3.0 | 6.1 ± 0.7 (B) | 9.2 ± 0.7 (A, B, C) |
| F-VLP | αGC | <3.0 | 7.7 ± 0.7 (B, C) | 7.4 ± 0.3 (C, D) |
| HMPV | None | <3.0 | 9.0 ± 0.1 (C) | 8.8 ± 0.8 (B, D) |
Mice were inoculated in the i.p. cavity with vaccine preparations on days 0 and 14, except that the HMPV group was infected i.n. on day 0.
Serum samples were collected preimmunization, 14 days after primary immunization before boost inoculation, and 5 days after HMPV i.n. infection. Titers of neutralizing antibody against HMPV were determined with an HMPV plaque reduction assay. The neutralizing-antibody titer was calculated as the antibody dilution at which the HMPV plaque number was reduced by 50%, based upon nonlinear regression analysis.
Mean antibody titers were compared by one-way ANOVA and assigned to statistically similar groups by using Tukey's multiple-comparison test. Values within a column that have a letter in common do not differ significantly (P > 0.05).
Immunization with HMPV VLPs protects mice against HMPV infection.
Infection of B6 mice with the TN94-49 strain of HMPV leads to viral replication in the NT and lungs, with peak titers occurring on days 4 and 5 postinfection, respectively (55). To determine whether immunization with HMPV VLPs could protect against HMPV infection, we harvested the NT and lungs of HMPV-challenged mice and quantified the amount of replication-competent virus by plaque assay. HMPV infection led to replication in the NT and lungs of mock-immunized mice (Fig. 3A and B, triangles). Mice previously infected with HMPV had no detectable levels of replicating HMPV in the NT or lungs (Fig. 3A and B, diamonds). Immunization with M-VLPs with or without adjuvant did not protect from HMPV infection of the NT, and M-VLPs alone did not reduce the lung virus titer. However, mice that received M-VLPs plus either adjuvant exhibited lower levels of HMPV replication in the lungs, though this did not quite reach statistical significance (Fig. 3B, open and gray squares). Immunization with F-VLPs alone conferred a 1-log10 reduction in HMPV replication in the NT (Fig. 3A, circles), with a significantly greater virus titer reduction in mice vaccinated with F-VLPs plus either adjuvant (open and gray circles). F-VLP immunization in the absence or presence of adjuvant led to undetectable levels of replicating HMPV in the lungs of all of the mice (Fig. 3B, circles). In a replicate experiment, mice immunized twice with nonadjuvanted F-VLPs and subsequently challenged were completely protected against lung virus replication, in contrast to mock VLP-immunized mice (data not shown). These results demonstrate that immunization with two doses of F-VLPs confers complete protection against HMPV replication in the lungs of mice. Furthermore, when F-VLPs are delivered in the presence of immune adjuvants, they induce partial protection from HMPV replication in the upper respiratory tract.
FIG 3.
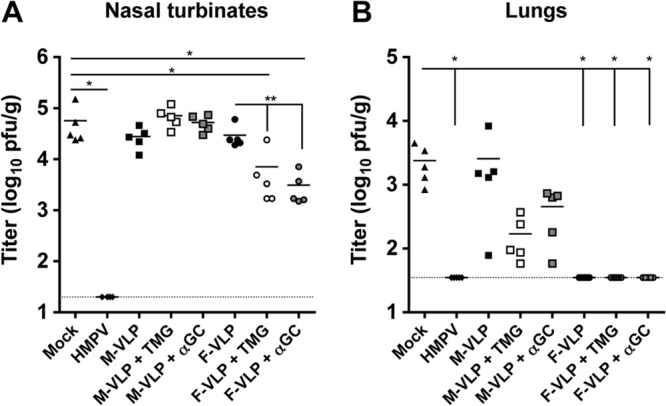
Immunization with HMPV VLPs protects mice against HMPV infection. The experimental protocol used is shown in Fig. 2A. Levels of replicating HMPV were quantified by plaque assay on day 33 (5 days after an HMPV challenge). (A) Nasal titers of HMPV. (B) Lung titers of HMPV. The dotted lines in panels A and B indicate the limits of detection (20 and 35 PFU/g, respectively); the bars represent the mean virus titers of groups of mice (n = 5). Comparisons of multiple groups were made by one-way ANOVA with Dunnett's posttest (*, P < 0.05). Comparisons of two groups were made with an unpaired Student t test (**, P < 0.05).
HMPV VLPs induces T cell migration into mouse lungs.
Interestingly, immunization with M-VLPs and adjuvant led to reduced HMPV replication in the lungs of infected mice. Because these mice had a negligible neutralizing humoral immune response, we reasoned that the modest level of protection might be cell-mediated immunity to HMPV. Thus, we sought to determine whether VLP immunization induces T cell responses. Lung samples from immunized and HMPV-challenged mice were collected 5 days postinfection, fixed, and stained for CD3+ T cells. Perivascular CD3+ T cells were present in all of the lung sections. Mice immunized with either M-VLPs or F-VLPs in the presence of TMG or αGC had a trend toward greater CD3+ cell infiltrate levels in the lung sections than primary infected mice (Mock), previously infected mice (HMPV), or mice that received only VLPs (Fig. 4A), though this did not reach statistical significance. We also quantified the CD3+ area of the lung in digital images of the entire lung sections by using a computer algorithm as described in Materials and Methods (Fig. 4B). Acute infection with HMPV, represented by the mock-immunized and HMPV-challenged mice (mock treatment group), was used as the background reference for this analysis, because the HMPV-specific T cell response in the lungs of infected mice begins on day 5 and does not peak until day 10 postinfection (55). Compared to mock-immunized mice, significantly higher levels of CD3+ T cell infiltrate were observed in mice that received M-VLPs with TMG adjuvant, F-VLPs, or F-VLPs with TMG adjuvant (Fig. 4B). Representative images from the lung sections stained for CD3+ T cells are shown (Fig. 4C to H). These results suggest that immunization with HMPV VLPs leads to increased T cell migration to the site of HMPV infection.
FIG 4.

HMPV VLPs induce a CD3+ T cell infiltrate in the lungs of immunized animals after an HMPV challenge. At day 33 (5 days after an HMPV challenge), slices were collected from the left lung of each vaccinated mouse. Lung samples were fixed, paraffin embedded, sectioned at a 5-μm thickness, and stained with anti-CD3 antibody. (A) Lung samples were analyzed and scored for CD3+ T cell infiltration on a scale of 0 to 3. Infiltrate scores (mean ± SEM) of two mice from each group are shown. Comparisons of multiple groups were made by one-way ANOVA with Dunnett's posttest. No significant differences were noted. (B) Digital images of the CD3-stained lung sections were generated with an Aperio ScanScope CS2, and a color deconvolution algorithm was used to quantify CD3 staining. Ten random regions, covering the entire lung area, from two mice per group were chosen, and the CD3+ area of each region was calculated. Results are presented as the mean ± the SEM. Comparisons of multiple groups were made by one-way ANOVA with Dunnett's posttest (*, P < 0.05). (C to H) Representative images of lung sections from immunized or infected mice at 5 days after an HMPV challenge. Magnification, ×20. CD3+ T cells are stained dark brown. Scale bars are shown in the lower left corners.
F-VLPs generate HMPV-specific CD8+ T cells in the respiratory tract and spleen.
Infection of mice with HMPV elicits a localized CD3+ T cell response (Fig. 4) (55), and vaccines that specifically elicit CD8+ T cells provide various degrees of protection against paramyxovirus infection (55, 58–60). To determine whether HMPV VLPs are capable of eliciting cellular immunity, mice were infected with HMPV or immunized with F-VLP without adjuvant and 7 days later splenic CD8+ T cells were tested for IFN-γ secretion in response to stimulation with the immunodominant H2-Db-restricted F528-536 (F528) epitope peptide by ICS (Fig. 5A). F-VLP immunization, even without adjuvant, was capable of inducing F528-specific CD8+ T cells at a higher level than primary HMPV infection on day 7. To test whether repeat VLP immunization could boost the T cell response, mice were either infected with HMPV or immunized on days 0 and 14 with F-VLP without adjuvant or mock VLP, and then all of the groups were challenged on day 28 with HMPV. Splenocytes were collected on day 5 postchallenge and stimulated with either F528 peptide or controls, and IFN-γ secretion was measured by ELISPOT assay. Both previously HMPV-infected and F-VLP-immunized groups mounted significantly greater IFN-γ responses than mock-immunized and challenged mice (Fig. 5B). To test whether VLP immunization was capable of inducing CD8+ T cell responses in the lung, we either immunized mice i.p. with F-VLPs plus adjuvant or infected mice i.n. with HMPV on day 0 and quantified the HMPV-specific CD8+ T cell response via tetramer staining on day 10. We found that F-VLPs induced CD8+ T cells specific for the F528 epitope at levels comparable to those induced by HMPV infection in both the lungs (2.7% for F-VLPs versus 5.0% for HMPV) and spleen (0.4% versus 0.6%) (Fig. 5C and D). These results confirm that VLP immunization generates cellular immunity.
FIG 5.

HMPV VLPs generate HMPV-specific CD8+ T cells. (A) B6 mice were infected with HMPV or immunized with F-VLP without adjuvant, and splenocytes were isolated 7 days later. Lymphocytes were stimulated with either an HMPV-specific F528-536 peptide or an irrelevant influenza virus-specific NP366-374 peptide as described in Materials and Methods and analyzed for IFN-γ secretion. The two groups were compared by using an unpaired Student t test. (B) B6 mice were infected with HMPV on day 0 or immunized on days 0 and 14 with F-VLP without adjuvant, and then all of the groups were challenged on day 28 with HMPV. Splenocytes were collected on day 5 postchallenge and tested by ELISPOT assay by using stimulation with an HMPV-specific F528-536 peptide, an irrelevant peptide, or ConA. The average number of spots in the negative-control wells was subtracted from each experimental value, and the number of SFC per 106 lymphocytes was calculated and is expressed as a percentage of the response of mock VLP-immunized animals (set at 100%). Comparisons of multiple groups were made by one-way ANOVA with Dunnett's posttest (*, P < 0.05). (C) B6 mice were infected with HMPV or immunized with F-VLP plus TMG, and spleen and lung lymphocytes were isolated 10 days later. Lymphocytes were tetramer stained with either an HMPV-specific F528-536 tetramer or an irrelevant influenza virus-specific NP366-374 tetramer and anti-CD8 antibody as described in Materials and Methods. The percentage of tetramer+ F-specific lung TCD8 or spleen TCD8 cells was calculated. Data (mean ± SEM) represent an independent experiment with three to five mice per group. (D) Representative flow cytometry plots showing F528-536 tetramer staining on the gated CD8+ T cell population from the lungs of HMPV-infected or F-VLP-immunized mice. The values under the quadrants are the percentages of HMPV-specific F528-536 tetramer+ CD8+ T cells.
F-VLP vaccination induces balanced Th1 and Th2 responses.
Enhanced respiratory disease was associated with FI RSV vaccine, one component of which was an exuberant Th2 response (19). To test whether VLP vaccination was associated with Th2 bias upon HMPV challenge, cytokine gene expression levels in lung homogenates were measured by real-time RT-PCR. Mice immunized with VLPs in conjunction with TMG adjuvant exhibited significantly greater transcription of both Th1 (IFN-γ; F-VLP plus TMG) and Th2 (interleukin-10 [IL-10]; M- and F-VLPs plus TMG) cytokines than mock-immunized and challenged mice, while groups immunized with αGC exhibited increased IL-4 expression (Fig. 6). There was a nonsignificant trend toward greater IL-2 transcription in adjuvant-treated mice. Importantly, mice immunized with nonadjuvanted VLPs did not differ in cytokine profile from previously infected mice. Thus, VLP vaccination induced a mostly balanced Th cytokine response, especially in mice without adjuvant.
FIG 6.

VLP immunization is associated with both Th1 and Th2 responses. Groups of mice were immunized with mock VLPs, M-VLP, M-VLP plus TMG, M-VLP plus αGC, F-VLP, F-VLP plus TMG, or F-VLP plus αGC on days 0 and 14 or infected with HMPV on day 0. All of the mice were then challenged with HMPV on day 28. Their lungs were collected and homogenized on day 5 postchallenge, and RNA was extracted and tested by real-time RT-PCR for multiple cytokines. All of the values were normalized to the housekeeping gene for HPRT and are reported as fold differences (determined by the ΔΔCT method) from mice that were mock VLP vaccinated. Comparisons of groups were made by one-way ANOVA with Dunnett's posttest (*, P < 0.01; **, P < 0.001; ***, P < 0.0001).
DISCUSSION
HMPV causes a significant disease burden in young infants. Furthermore, reinfection with HMPV occurs in healthy and immunocompromised humans, despite the presence of serum antibody (5, 61, 62). This may occur because of limited cross-protective immunity between different strains of HMPV or may indicate that antibody-mediated protection is not sufficient to prevent HMPV infection. However, studies with animal models indicate that passive transfer of anti-HMPV neutralizing antibodies can protect against HMPV replication and disease (45, 46, 63). Furthermore, in otherwise healthy adults, HMPV infection is typically limited to the upper respiratory tract, suggesting that neutralizing antibodies can ameliorate HMPV disease pathogenesis. Thus, an effective vaccine could prevent severe disease in children and adults.
T cell immunity is thought to be important for HMPV clearance and resolution of infection. For example, HMPV infections are more severe, and at times fatal, in HIV-infected or other immunocompromised patients (10–12, 64). The contribution of T cells to protection against HMPV remains poorly defined, although limited studies with mouse models suggest that T cells contribute to protective immunity (55, 65). Thus, an ideal HMPV vaccine would elicit both humoral and cellular immune responses (66).
VLPs are an attractive platform for an HMPV vaccine. VLPs are replication incompetent and nonpathogenic, with minimal concerns about adventitious agents. These particles can be administered via the parenteral or mucosal route with or without adjuvant. VLPs are amenable to large-scale production, and the use of biologics can be largely avoided, as in this study, where VLPs were produced in serum-free medium. The generation of VLPs in mammalian cells ensured proper glycosylation and folding of F, both of which are important for the induction of high-affinity neutralizing antibodies while avoiding enhanced respiratory disease (33). In addition, VLPs could be designed to incorporate all four F subtypes to ensure cross-protective immunity. While F-VLP particles are fusion competent (50), the length of the F protein measured by EM suggests that particles may contain both pre- and postfusion forms of F, as described for RSV (67). The fact that neutralizing-antibody titers were similar for HMPV and adjuvanted VLPs, while ELISA F-binding titers were significantly higher for F-VLP than for HMPV, suggests that the conformation of the F molecules on VLPs may not be uniform. This could affect immunogenicity, since for RSV, neutralizing epitopes are present in both forms of the F protein (68, 69). At least one HMPV F MAb epitope is conserved in both the pre- and postfusion forms (57).
HMPV VLPs induced CD8+ T cell responses, though the exact mechanism of this is not clear. Soluble antigens normally presented by major histocompatibility complex (MHC) class II can be cross-presented in the context of MHC class I, though at a low rate (70, 71). However, since HMPV VLPs are fusion competent, we speculate that fusion of the VLP envelope with the cell membrane might mediate the cytosolic delivery of HMPV F and M proteins, facilitating the loading of proteolytic peptides derived from F and M proteins onto MHC class I molecules. VLPs are capable of delivering proteins to the cytosol and mediating MHC class I presentation (72–76). The murine and human HMPV MHC class I epitopes described are generally conserved (55, 58, 77), and thus, CD8+ T cell memory would be expected to be broadly protective. Further studies are needed to answer this question, the answer to which could be applicable to the challenge of inducing effective T cell immunity with a nonreplicating vaccine.
A critical issue for any paramyxovirus vaccine, especially a nonreplicating vaccine, is the concern for enhanced disease such as that observed following FI RSV (29, 30). Enhanced disease associated with FI RSV is thought to be due in part to poor neutralizing-antibody induction and imbalanced Th2 responses (21, 31–34). Studies with rodents and primates show that FI HMPV is associated with a similar phenomenon, including Th2 bias (25, 26). We found that while αGC adjuvant was associated with increased IL-4 levels, VLPs alone were not, and VLPs with TMG were associated with increased IL-10 or IFN-γ levels. Vaccination with recombinant F protein or alphavirus-vectored F was not associated with enhanced disease in cotton rats (23, 47). It is important to note that rodent models do not always reflect human biology and that rodents are not faithful models of human respiratory disease. Studies with nonhuman primates before human clinical trials are important.
In summary, our results suggest that HMPV VLPs are promising HMPV vaccine candidates. Vaccination with VLPs induces a neutralizing-antibody response, stimulates an HMPV-specific CD8+ T cell response, and protects mice from HMPV infection of the lungs. Future directions of this work include mucosal delivery in the absence and presence of adjuvants, the effective cross-reactivity of antibody responses to other strains of HMPV, preclinical testing with other animal models, and determination of the duration of immunity.
ACKNOWLEDGMENTS
This work was financially supported by Public Health Service grants AI-085062 (J.V.W.), T32 AI-007611 (R.G.C.), T32 HL-069765 (J.C.B.), and GM-007347 (J.J.E.). Additional support was provided by a P.E.O. Scholar award from the Philanthropic Educational Organization (R.G.C.). The Vanderbilt Medical Center Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (NIH P30 CA-68485) and the Vanderbilt Digestive Disease Research Center (NIH DK-058404). The Islet Procurement and Analysis Core is supported by the Vanderbilt Diabetes Research and Training Center (NIH P60 DK-020593).
Footnotes
Published ahead of print 26 March 2014
REFERENCES
- 1.Peiris JS, Tang WH, Chan KH, Khong PL, Guan Y, Lau YL, Chiu SS. 2003. Children with respiratory disease associated with metapneumovirus in Hong Kong. Emerg. Infect. Dis. 9:628–633. 10.3201/eid0906.030009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ebihara T, Endo R, Kikuta H, Ishiguro N, Ishiko H, Hara M, Takahashi Y, Kobayashi K. 2004. Human metapneumovirus infection in Japanese children. J. Clin. Microbiol. 42:126–132. 10.1128/JCM.42.1.126-132.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, Wright PF, Crowe JE., Jr 2004. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med. 350:443–450. 10.1056/NEJMoa025472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sloots TP, Mackay IM, Bialasiewicz S, Jacob KC, McQueen E, Harnett GB, Siebert DJ, Masters BI, Young PR, Nissen MD. 2006. Human metapneumovirus, Australia, 2001-2004. Emerg. Infect. Dis. 12:1263–1266. 10.3201/eid1708.051239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams JV, Wang CK, Yang CF, Tollefson SJ, House FS, Heck JM, Chu M, Brown JB, Lintao LD, Quinto JD, Chu D, Spaete RR, Edwards KM, Wright PF, Crowe JE., Jr 2006. The role of human metapneumovirus in upper respiratory tract infections in children: a 20-year experience. J. Infect. Dis. 193:387–395. 10.1086/499274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh EE, Peterson DR, Falsey AR. 2008. Human metapneumovirus infections in adults: another piece of the puzzle. Arch. Intern. Med. 168:2489–2496. 10.1001/archinte.168.22.2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Widmer K, Zhu Y, Williams JV, Griffin MR, Edwards KM, Talbot HK. 2012. Rates of hospitalizations for respiratory syncytial virus, human metapneumovirus, and influenza virus in older adults. J. Infect. Dis. 206:56–62. 10.1093/infdis/jis309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esper F, Martinello RA, Boucher D, Weibel C, Ferguson D, Landry ML, Kahn JS. 2004. A 1-year experience with human metapneumovirus in children aged <5 years. J. Infect. Dis. 189:1388–1396. 10.1086/382482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papenburg J, Hamelin ME, Ouhoummane N, Carbonneau J, Ouakki M, Raymond F, Robitaille L, Corbeil J, Caouette G, Frenette L, De Serres G, Boivin G. 2012. Comparison of risk factors for human metapneumovirus and respiratory syncytial virus disease severity in young children. J. Infect. Dis. 206:178–189. 10.1093/infdis/jis333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams JV, Martino R, Rabella N, Otegui M, Parody R, Heck JM, Crowe JE., Jr 2005. A prospective study comparing human metapneumovirus with other respiratory viruses in adults with hematologic malignancies and respiratory tract infections. J. Infect. Dis. 192:1061–1065. 10.1086/432732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Englund JA, Boeckh M, Kuypers J, Nichols WG, Hackman RC, Morrow RA, Fredricks DN, Corey L. 2006. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann. Intern. Med. 144:344–349. 10.7326/0003-4819-144-5-200603070-00010 [DOI] [PubMed] [Google Scholar]
- 12.Shahda S, Carlos WG, Kiel PJ, Khan BA, Hage CA. 2011. The human metapneumovirus: a case series and review of the literature. Transpl. Infect. Dis. 13:324–328. 10.1111/j.1399-3062.2010.00575.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR, Collins PL, Buchholz UJ. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 78:12877–12887. 10.1128/JVI.78.23.12877-12887.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang RS, Mahmood K, Macphail M, Guzzetta JM, Haller AA, Liu H, Kaur J, Lawlor HA, Stillman EA, Schickli JH, Fouchier RA, Osterhaus AD, Spaete RR. 2005. A host-range restricted parainfluenza virus type 3 (PIV3) expressing the human metapneumovirus (hMPV) fusion protein elicits protective immunity in African green monkeys. Vaccine 23:1657–1667. 10.1016/j.vaccine.2004.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchholz UJ, Nagashima K, Murphy BR, Collins PL. 2006. Live vaccines for human metapneumovirus designed by reverse genetics. Expert Rev. Vaccines 5:695–706. 10.1586/14760584.5.5.695 [DOI] [PubMed] [Google Scholar]
- 16.Herfst S, de Graaf M, Schrauwen EJ, Sprong L, Hussain K, van den Hoogen BG, Osterhaus AD, Fouchier RA. 2008. Generation of temperature-sensitive human metapneumovirus strains that provide protective immunity in hamsters. J. Gen. Virol. 89:1553–1562. 10.1099/vir.0.2008/002022-0 [DOI] [PubMed] [Google Scholar]
- 17.Herfst S, Fouchier RA. 2008. Vaccination approaches to combat human metapneumovirus lower respiratory tract infections. J. Clin. Virol. 41:49–52. 10.1016/j.jcv.2007.10.022 [DOI] [PubMed] [Google Scholar]
- 18.Blanco JC, Boukhvalova MS, Shirey KA, Prince GA, Vogel SN. 2010. New insights for development of a safe and protective RSV vaccine. Hum. Vaccin. 6:482–492. 10.4161/hv.6.6.11562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. 162:80–99. 10.1016/j.virusres.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson LJ, Dormitzer PR, Nokes DJ, Rappuoli R, Roca A, Graham BS. 2013. Strategic priorities for respiratory syncytial virus (RSV) vaccine development. Vaccine 31(Suppl 2):B209–B215. 10.1016/j.vaccine.2012.11.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schickli JH, Dubovsky F, Tang RS. 2009. Challenges in developing a pediatric RSV vaccine. Hum. Vaccin. 5:582–591 [DOI] [PubMed] [Google Scholar]
- 22.Ramilo O. 2009. Evolution of prophylaxis: MoAb, siRNA, vaccine, and small molecules. Paediatr. Respir. Rev. 10(Suppl 1):23–25. 10.1016/S1526-0542(09)70011-9 [DOI] [PubMed] [Google Scholar]
- 23.Cseke G, Wright DW, Tollefson SJ, Johnson JE, Crowe JE, Jr, Williams JV. 2007. Human metapneumovirus fusion protein vaccines that are immunogenic and protective in cotton rats. J. Virol. 81:698–707. 10.1128/JVI.00844-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herfst S, de Graaf M, Schrauwen EJ, Ulbrandt ND, Barnes AS, Senthil K, Osterhaus AD, Fouchier RA, van den Hoogen BG. 2007. Immunization of Syrian golden hamsters with F subunit vaccine of human metapneumovirus induces protection against challenge with homologous or heterologous strains. J. Gen. Virol. 88:2702–2709. 10.1099/vir.0.83084-0 [DOI] [PubMed] [Google Scholar]
- 25.de Swart RL, van den Hoogen BG, Kuiken T, Herfst S, van Amerongen G, Yuksel S, Sprong L, Osterhaus AD. 2007. Immunization of macaques with formalin-inactivated human metapneumovirus induces hypersensitivity to hMPV infection. Vaccine 25:8518–8528. 10.1016/j.vaccine.2007.10.022 [DOI] [PubMed] [Google Scholar]
- 26.Yim KC, Cragin RP, Boukhvalova MS, Blanco JC, Hamlin ME, Boivin G, Porter DD, Prince GA. 2007. Human metapneumovirus: enhanced pulmonary disease in cotton rats immunized with formalin-inactivated virus vaccine and challenged. Vaccine 25:5034–5040. 10.1016/j.vaccine.2007.04.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chin J, Magoffin RL, Shearer LA, Schieble JH, Lennette EH. 1969. Field evaluation of a respiratory syncytial virus vaccine and a trivalent parainfluenza virus vaccine in a pediatric population. Am. J. Epidemiol. 89:449–463 [DOI] [PubMed] [Google Scholar]
- 28.Fulginiti VA, Eller JJ, Sieber OF, Joyner JW, Minamitani M, Meiklejohn G. 1969. Respiratory virus immunization. I. A field trial of two inactivated respiratory virus vaccines; an aqueous trivalent parainfluenza virus vaccine and an alum-precipitated respiratory syncytial virus vaccine. Am. J. Epidemiol. 89:435–448 [DOI] [PubMed] [Google Scholar]
- 29.Kapikian AZ, Mitchell RH, Chanock RM, Shvedoff RA, Stewart CE. 1969. An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am. J. Epidemiol. 89:405–421 [DOI] [PubMed] [Google Scholar]
- 30.Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89:422–434 [DOI] [PubMed] [Google Scholar]
- 31.Murphy BR, Prince GA, Walsh EE, Kim HW, Parrott RH, Hemming VG, Rodriguez WJ, Chanock RM. 1986. Dissociation between serum neutralizing and glycoprotein antibody responses of infants and children who received inactivated respiratory syncytial virus vaccine. J. Clin. Microbiol. 24:197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy BR, Walsh EE. 1988. Formalin-inactivated respiratory syncytial virus vaccine induces antibodies to the fusion glycoprotein that are deficient in fusion-inhibiting activity. J. Clin. Microbiol. 26:1595–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakurai H, Williamson RA, Crowe JE, Beeler JA, Poignard P, Bastidas RB, Chanock RM, Burton DR. 1999. Human antibody responses to mature and immature forms of viral envelope in respiratory syncytial virus infection: significance for subunit vaccines. J. Virol. 73:2956–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polack FP, Teng MN, Collins PL, Prince GA, Exner M, Regele H, Lirman DD, Rabold R, Hoffman SJ, Karp CL, Kleeberger SR, Wills-Karp M, Karron RA. 2002. A role for immune complexes in enhanced respiratory syncytial virus disease. J. Exp. Med. 196:859–865. 10.1084/jem.20020781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delgado MF, Polack FP. 2004. Involvement of antibody, complement and cellular immunity in the pathogenesis of enhanced respiratory syncytial virus disease. Expert Rev. Vaccines 3:693–700. 10.1586/14760584.3.6.693 [DOI] [PubMed] [Google Scholar]
- 36.Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP, Diaz L, Trento A, Chang HY, Mitzner W, Ravetch J, Melero JA, Irusta PM, Polack FP. 2009. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat. Med. 15:34–41. 10.1038/nm.1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider-Ohrum K, Ross TM. 2012. Virus-like particles for antigen delivery at mucosal surfaces. Curr. Top. Microbiol. Immunol. 354:53–73. 10.1007/82_2011_135 [DOI] [PubMed] [Google Scholar]
- 38.Garrone P, Fluckiger AC, Mangeot PE, Gauthier E, Dupeyrot-Lacas P, Mancip J, Cangialosi A, Du Chene I, LeGrand R, Mangeot I, Lavillette D, Bellier B, Cosset FL, Tangy F, Klatzmann D, Dalba C. 2011. A prime-boost strategy using virus-like particles pseudotyped for HCV proteins triggers broadly neutralizing antibodies in macaques. Sci. Transl. Med. 3:94ra71. 10.1126/scitranslmed.3002330 [DOI] [PubMed] [Google Scholar]
- 39.Haynes JR. 2009. Influenza virus-like particle vaccines. Expert Rev. Vaccines 8:435–445. 10.1586/erv.09.8 [DOI] [PubMed] [Google Scholar]
- 40.Kang SM, Pushko P, Bright RA, Smith G, Compans RW. 2009. Influenza virus-like particles as pandemic vaccines. Curr. Top. Microbiol. Immunol. 333:269–289. 10.1007/978-3-540-92165-3_14 [DOI] [PubMed] [Google Scholar]
- 41.Young KR, McBurney SP, Karkhanis LU, Ross TM. 2006. Virus-like particles: designing an effective AIDS vaccine. Methods 40:98–117. 10.1016/j.ymeth.2006.05.024 [DOI] [PubMed] [Google Scholar]
- 42.Hendrickx G, Vorsters A, Van Damme P. 2012. Advances in hepatitis immunization (A, B, E): public health policy and novel vaccine delivery. Curr. Opin. Infect. Dis. 25:578–583. 10.1097/QCO.0b013e328357e65c [DOI] [PubMed] [Google Scholar]
- 43.Schiller JT, Castellsague X, Villa LL, Hildesheim A. 2008. An update of prophylactic human papillomavirus L1 virus-like particle vaccine clinical trial results. Vaccine 26(Suppl 10):K53–K61. 10.1016/j.vaccine.2008.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 79:12608–12613. 10.1128/JVI.79.19.12608-12613.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams JV, Chen Z, Cseke G, Wright DW, Keefer CJ, Tollefson SJ, Hessell A, Podsiad A, Shepherd BE, Sanna PP, Burton DR, Crowe JE, Jr, Williamson RA. 2007. A recombinant human monoclonal antibody to human metapneumovirus fusion protein that neutralizes virus in vitro and is effective therapeutically in vivo. J. Virol. 81:8315–8324. 10.1128/JVI.00106-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulbrandt ND, Ji H, Patel NK, Riggs JM, Brewah YA, Ready S, Donacki NE, Folliot K, Barnes AS, Senthil K, Wilson S, Chen M, Clarke L, MacPhail M, Li J, Woods RM, Coelingh K, Reed JL, McCarthy MP, Pfarr DS, Osterhaus AD, Fouchier RA, Kiener PA, Suzich JA. 2006. Isolation and characterization of monoclonal antibodies which neutralize human metapneumovirus in vitro and in vivo. J. Virol. 80:7799–7806. 10.1128/JVI.00318-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mok H, Tollefson SJ, Podsiad AB, Shepherd BE, Polosukhin VV, Johnston RE, Williams JV, Crowe JE., Jr 2008. An alphavirus replicon-based human metapneumovirus vaccine is immunogenic and protective in mice and cotton rats. J. Virol. 82:11410–11418. 10.1128/JVI.01688-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skiadopoulos MH, Biacchesi S, Buchholz UJ, Amaro-Carambot E, Surman SR, Collins PL, Murphy BR. 2006. Individual contributions of the human metapneumovirus F, G, and SH surface glycoproteins to the induction of neutralizing antibodies and protective immunity. Virology. 345:492–501. 10.1016/j.virol.2005.10.016 [DOI] [PubMed] [Google Scholar]
- 49.Ryder AB, Tollefson SJ, Podsiad AB, Johnson JE, Williams JV. 2010. Soluble recombinant human metapneumovirus G protein is immunogenic but not protective. Vaccine 28:4145–4152. 10.1016/j.vaccine.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cox RG, Livesay SB, Johnson M, Ohi MD, Williams JV. 2012. The human metapneumovirus fusion protein mediates entry via an interaction with RGD-binding integrins. J. Virol. 86:12148–12160. 10.1128/JVI.01133-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams JV, Tollefson SJ, Johnson JE, Crowe JE., Jr 2005. The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J. Virol. 79:10944–10951. 10.1128/JVI.79.17.10944-10951.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohi M, Li Y, Cheng Y, Walz T. 2004. Negative staining and image classification—powerful tools in modern electron microscopy. Biol. Proced. Online 6:23–34. 10.1251/bpo70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cerundolo V, Silk JD, Masri SH, Salio M. 2009. Harnessing invariant NKT cells in vaccination strategies. Nat. Rev. Immunol. 9:28–38. 10.1038/nri2451 [DOI] [PubMed] [Google Scholar]
- 54.Gilchuk P, Spencer CT, Conant SB, Hill T, Gray JJ, Niu X, Zheng M, Erickson JJ, Boyd KL, McAfee KJ, Oseroff C, Hadrup SR, Bennink JR, Hildebrand W, Edwards KM, Crowe JE, Jr, Williams JV, Buus S, Sette A, Schumacher TN, Link AJ, Joyce S. 2013. Discovering naturally processed antigenic determinants that confer protective T cell immunity. J. Clin. Invest. 123:1976–1987. 10.1172/JCI67388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, Boyd KL, Johnson JE, Kim AS, Joyce S, Williams JV. 2012. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J. Clin. Invest. 122:2967–2982. 10.1172/JCI62860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harrison MS, Sakaguchi T, Schmitt AP. 2010. Paramyxovirus assembly and budding: building particles that transmit infections. Int. J. Biochem. Cell Biol. 42:1416–1429. 10.1016/j.biocel.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wen X, Krause JC, Leser GP, Cox RG, Lamb RA, Williams JV, Crowe JE, Jr, Jardetzky TS. 2012. Structure of the human metapneumovirus fusion protein with neutralizing antibody identifies a pneumovirus antigenic site. Nat. Struct. Mol. Biol. 19:461–463. 10.1038/nsmb.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herd KA, Mahalingam S, Mackay IM, Nissen M, Sloots TP, Tindle RW. 2006. Cytotoxic T-lymphocyte epitope vaccination protects against human metapneumovirus infection and disease in mice. J. Virol. 80:2034–2044. 10.1128/JVI.80.4.2034-2044.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee S, Stokes KL, Currier MG, Sakamoto K, Lukacs NW, Celis E, Moore ML. 2012. Vaccine-elicited CD8+ T cells protect against respiratory syncytial virus strain A2-line19F-induced pathogenesis in BALB/c mice. J. Virol. 86:13016–13024. 10.1128/JVI.01770-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Melendi GA, Zavala F, Buchholz UJ, Boivin G, Collins PL, Kleeberger SR, Polack FP. 2007. Mapping and characterization of the primary and anamnestic H-2(d)-restricted cytotoxic T-lymphocyte response in mice against human metapneumovirus. J. Virol. 81:11461–11467. 10.1128/JVI.02423-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okamoto M, Sugawara K, Takashita E, Muraki Y, Hongo S, Nishimura H, Matsuzaki Y. 2010. Longitudinal course of human metapneumovirus antibody titers and reinfection in healthy adults. J. Med. Virol. 82:2092–2096. 10.1002/jmv.21920 [DOI] [PubMed] [Google Scholar]
- 62.Falsey AR, Hennessey PA, Formica MA, Criddle MM, Biear JM, Walsh EE. 2010. Humoral immunity to human metapneumovirus infection in adults. Vaccine 28:1477–1480. 10.1016/j.vaccine.2009.11.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamelin ME, Couture C, Sackett M, Kiener P, Suzich J, Ulbrandt N, Boivin G. 2008. The prophylactic administration of a monoclonal antibody against human metapneumovirus attenuates viral disease and airways [sic] hyperresponsiveness in mice. Antivir. Ther. 13:39–46 [PubMed] [Google Scholar]
- 64.Madhi SA, Ludewick H, Kuwanda L, van Niekerk N, Cutland C, Klugman KP. 2007. Seasonality, incidence, and repeat human metapneumovirus lower respiratory tract infections in an area with a high prevalence of human immunodeficiency virus type-1 infection. Pediatr. Infect. Dis. J. 26:693–699. 10.1097/INF.0b013e3180621192 [DOI] [PubMed] [Google Scholar]
- 65.Kolli D, Bataki EL, Spetch L, Guerrero-Plata A, Jewell AM, Piedra PA, Milligan GN, Garofalo RP, Casola A. 2008. T lymphocytes contribute to antiviral immunity and pathogenesis in experimental human metapneumovirus infection. J. Virol. 82:8560–8569. 10.1128/JVI.00699-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feuillet F, Lina B, Rosa-Calatrava M, Boivin G. 2012. Ten years of human metapneumovirus research. J. Clin. Virol. 53:97–105. 10.1016/j.jcv.2011.10.002 [DOI] [PubMed] [Google Scholar]
- 67.Liljeroos L, Krzyzaniak MA, Helenius A, Butcher SJ. 2013. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc. Natl. Acad. Sci. U. S. A. 110:11133–11138. 10.1073/pnas.1309070110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, Zhou T, Graepel KW, Kumar A, Moin S, Boyington JC, Chuang GY, Soto C, Baxa U, Bakker AQ, Spits H, Beaumont T, Zheng Z, Xia N, Ko SY, Todd JP, Rao S, Graham BS, Kwong PD. 2013. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342:592–598. 10.1126/science.1243283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J. Virol. 85:7788–7796. 10.1128/JVI.00555-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delamarre L, Holcombe H, Mellman I. 2003. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J. Exp. Med. 198:111–122. 10.1084/jem.20021542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425:397–402. 10.1038/nature01911 [DOI] [PubMed] [Google Scholar]
- 72.Kaczmarczyk SJ, Sitaraman K, Young HA, Hughes SH, Chatterjee DK. 2011. Protein delivery using engineered virus-like particles. Proc. Natl. Acad. Sci. U. S. A. 108:16998–17003. 10.1073/pnas.1101874108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Win SJ, Ward VK, Dunbar PR, Young SL, Baird MA. 2011. Cross-presentation of epitopes on virus-like particles via the MHC I receptor recycling pathway. Immunol. Cell Biol. 89:681–688. 10.1038/icb.2010.161 [DOI] [PubMed] [Google Scholar]
- 74.Leclerc D, Beauseigle D, Denis J, Morin H, Pare C, Lamarre A, Lapointe R. 2007. Proteasome-independent major histocompatibility complex class I cross-presentation mediated by papaya mosaic virus-like particles leads to expansion of specific human T cells. J. Virol. 81:1319–1326. 10.1128/JVI.01720-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lacasse P, Denis J, Lapointe R, Leclerc D, Lamarre A. 2008. Novel plant virus-based vaccine induces protective cytotoxic T-lymphocyte-mediated antiviral immunity through dendritic cell maturation. J. Virol. 82:785–794. 10.1128/JVI.01811-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ruedl C, Storni T, Lechner F, Bachi T, Bachmann MF. 2002. Cross-presentation of virus-like particles by skin-derived CD8(-) dendritic cells: a dispensable role for TAP. Eur. J. Immunol. 32:818–825. [DOI] [PubMed] [Google Scholar]
- 77.Herd KA, Nissen MD, Hopkins PM, Sloots TP, Tindle RW. 2008. Major histocompatibility complex class I cytotoxic T lymphocyte immunity to human metapneumovirus (hMPV) in individuals with previous hMPV infection and respiratory disease. J. Infect. Dis. 197:584–592. 10.1086/526536 [DOI] [PubMed] [Google Scholar]