

ABSTRACT
Members of the family Picornaviridae consist of small positive-sense single-stranded RNA (+ssRNA) viruses capable of infecting various vertebrate species, including birds. One of the recently identified avian picornaviruses, with a remarkably long (>9,040-nucleotide) but still incompletely sequenced genome, is turkey hepatitis virus 1 (THV-1; species Melegrivirus A, genus Megrivirus), a virus associated with liver necrosis and enteritis in commercial turkeys (Meleagris gallopavo). This report presents the results of the genetic analysis of three complete genomes of megriviruses from fecal samples of chickens (chicken/B21-CHV/2012/HUN, GenBank accession no. KF961186, and chicken/CHK-IV-CHV/2013/HUN, GenBank accession no. KF961187) (Gallus gallus domesticus) and turkey (turkey/B407-THV/2011/HUN, GenBank accession no. KF961188) (Meleagris gallopavo) with the largest picornavirus genome (up to 9,739 nucleotides) so far described. The close phylogenetic relationship to THV-1 in the nonstructural protein-coding genome region and possession of the same internal ribosomal entry site type (IVB-like) suggest that the study strains belong to the genus Megrivirus. However, the genome comparisons revealed numerous unique variations (e.g., different numbers of potential 2A peptides, unusually long 3′ genome parts with various lengths of a potential second open reading frame, and multiple repeating sequence motifs in the 3′ untranslated region) and heterogeneous sequence relationships between the structural and nonstructural genome regions. These differences suggest the classification of chicken megrivirus-like viruses into a candidate novel species in the genus Megrivirus. Based on the different phylogenetic positions of chicken megrivirus-like viruses at the structural and nonstructural genome regions, the recombinant nature of these viruses is plausible.
IMPORTANCE The comparative genome analysis of turkey and novel chicken megriviruses revealed numerous unique genome features, e.g., up to four potential 2A peptides, unusually long 3′ genome parts with various lengths containing a potential second open reading frame, multiple repeating sequence motifs, and heterogeneous sequence relationships (possibly due to a recombination event) between the structural and nonstructural genome regions. Our results could help us to better understand the evolution and diversity (in terms of sequence and genome layout) of picornaviruses.
INTRODUCTION
Picornaviruses (family Picornaviridae), with small, nonenveloped viruses with single-stranded, positive-sense genomic RNA, are one of at least five virus families forming the order Picornavirales (1; http://ictvonline.org/Virustaxonomy.asp). The picornaviruses are currently classified into 17 official genera (Aphthovirus, Aquamavirus, Avihepatovirus, Cardiovirus, Cosavirus, Dicipivirus, Enterovirus, Erbovirus, Hepatovirus, Kobuvirus, Megrivirus, Parechovirus, Salivirus, Sapelovirus, Senecavirus, Teschovirus, and Tremovirus) (1, 2). Nine new genera (“Avisivirus,” “Gallivirus,” “Hunnivirus,” “Mischivirus,” “Mosavirus,” “Oscivirus,” “Pasivirus,” “Passerivirus,” and “Rosavirus”) are awaiting approval by the International Committee on Taxonomy of Viruses (http://talk.ictvonline.org/), and approximately 18 candidate species await classification (http://www.picornaviridae.com/).
In general, the polyadenylated picornaviral genome consists of a single polyprotein coding region flanked by highly structured 5′ and 3′ untranslated regions (UTRs), although the discovery of canine picodicistroviruses (genus Dicipivirus) with dicistronic genome organization and an additional intergenic internal ribosome entry site (IRES) at the junction of P1 and P2 (P1/P2) makes it the first picornavirus with two long open reading frames (ORFs) (3, 4). Furthermore, the presence of a putative second ORF in partial nucleotide sequences of chicken and turkey picornaviruses has been identified in silico (5).
Most picornaviruses have only one mature 2A peptide/protein, although the number of picornaviruses with multiple potential 2As with mainly unknown function, e.g., duck hepatitis A virus (DHAV), turkey hepatitis virus (THV), swine pasivirus, bluegill picornavirus, turkey avisivirus, and pigeon mesivirus (6–11), is increasing.
The size of a picornavirus genome ranges from 7.1 to over 9.1 kb. In general, the size variations among the picornavirus genomes are derived from differences in the genome organization (e.g., presence or absence of L proteins, single or multiple 2A or 3B peptides) (12) and from the different lengths of the untranslated regions. The largest (still incomplete) picornavirus genome is THV, a virus associated with liver necrosis and enteritis in turkeys, which belongs to the species Melegrivirus A (genus Megrivirus) and consists of >9,040 nucleotides (nt), although neither the full length of the 5′ UTR nor the complete 3′ UTR were determined (7). Additionally, two partial 3D/3′ UTR sequence fragments (1,327 nt), closely related to THV, have been identified in turkeys and chickens (5). Recently, two novel picornaviruses (mesivirus 1 and 2) most closely related to megriviruses were identified from wild pigeons. These viruses may either be a novel species within the genus Megrivirus or the prototypes of a novel picornavirus genus (11).
In this paper, we report three complete genomes of megrivirus-like picornaviruses from fecal samples of commercial turkeys (Meleagris gallopavo) (turkey/B407-THV/2011/HUN) and for the first time from those of chickens (Gallus gallus domesticus) (chicken/B21-CHV/2012/HUN and chicken/CHK-IV-CHV/2013/HUN). The high sequence identities and close phylogenetic relationship at the nonstructural region suggest that megrivirus-like picornaviruses in chickens and turkeys belong to one genus, Megrivirus. However, the genome comparisons of these megriviruses revealed numerous unique genome variations (e.g., uneven numbers of multiple 2As, the presence of a potential ORF2 with a different size at the 3′ part of the genome, different numbers of sequence repeats in the 3′ UTR) and a heterogeneous sequence relationship between the structural and nonstructural genome regions. These differences suggest the classification of chicken megrivirus-like viruses into a novel species in the genus Megrivirus.
MATERIALS AND METHODS
454 pyrosequencing and metagenomic analysis.
A fecal sample (B-21) collected in January 2012 from a 21-day-old broiler chicken (Gallus gallus domesticus) showing malabsorption syndrome in a poultry farm located in northwest Hungary was selected for viral metagenomic analysis. Phosphate-buffered saline (PBS)-diluted specimens were passed through a 0.45-μm-pore-size sterile filter and centrifuged at 6,000 × g for 5 min. The filtrate was digested with a mixture of nucleases to enrich for particle-protected nucleic acids (13). Nucleic acids were extracted using a QIAamp viral RNA minikit (QIAgen, Hilden, Germany) according to the manufacturer's instructions. Viral cDNA libraries derived from RNA and DNA molecules were constructed by sequence-independent random reverse transcription-PCR (RT-PCR) amplification as previously described (13). Pyrosequencing using 454 GS FLX technology was then performed as previously described (13, 14). The assembled sequence contigs from the pyrosequencing reads (singletons) were compared to the GenBank nucleotide and protein databases using BLASTn and BLASTx, respectively.
Genome acquisition of turkey and chicken picornaviruses.
RNA extracts from 150-μl suspensions (35% to 40% [vol/vol] in 0.1 M phosphate-buffered saline) of fecal samples from two chickens (B-21 and CHK-IV) collected in January 2012 and April 2013 and from a turkey (B-407) collected in November 2011 from three different poultry farms located in northwest Hungary were prepared using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Specific and generic primer sets were designed based on the available sequence contigs from the pyrosequencing analysis and the available THV sequences (GenBank accession no. HQ189775 and HM751199) to confirm the available pyrosequencing contigs of chicken/B-21-CHV/2012/HUN and to determine the complete nucleotide sequences of the picornaviruses chicken/B-21-CHV/2012/HUN, chicken/CHK-IV-CHV/2013/HUN, and turkey/B407-THV/2011/HUN using reverse transcription-PCR (RT-PCR) and long-range RT-PCR methods as described previously (15–17). The primer sequences are available on request. The steps of the genome acquisition of chicken and turkey picornaviruses are shown in Fig. 1. The 5′ and the 3′ ends of the genome were determined using a 5′/3′ rapid amplification of cDNA ends (RACE) PCR kit (Roche, Mannheim, Germany) as previously described (18). PCR products were sequenced directly with a BigDye Terminator cycle sequencing ready reaction kit (PE Applied Biosystems, Warrington, United Kingdom) using the primer-walking method and were run on an automated sequencer (ABI PRISM 310 Genetic Analyzer; Applied Biosystems).
FIG 1.

(A) Genome organization with conserved picornaviral motifs and predicted cleavage sites of chicken/B21-CHV/2012/HUN (B21-chicken, GenBank accession no. KF961186) and turkey/B407-THV/2011/HUN (B407-turkey, GenBank accession no. KF961188) megriviruses. Nucleotide (upper numbers) and amino acid (lower numbers) lengths are indicated in each gene box. The pairwise nucleotide (and amino acid) sequence identity values of each genome regions can be found between the two genome maps. The positions of the conserved picornaviral amino acid motifs of chicken/B21-CHV/2012/HUN are indicated with the first amino acid positions of the motif. The locations of the two consensus sequences of chicken/B21-CHV/2012/HUN acquired from pyrosequencing are indicated with gray bars. The steps (reaction no. R1 to R8) of the genome amplification and the locations of the primers are depicted as horizontal lines. (B) Amino acid sequence alignment of VP1-2B genome region of chicken/B21-CHV/2012/HUN (B21) and turkey/B407-THV/2011/HUN (B407). Predicted cleavage sites of the study strains (black arrows) together with the identified Hbox-NC motifs (dotted boxes) are indicated. The locations of the predicted cleavage sites (FQ ↓ AP) are illustrated with a gray background. The presumed cleavage site of genome region “2A0” of turkey/B407-THV/2011/HUN is indicated with a black box. Letters with a black background represent the identical amino acids of the alignment.
Sequence and phylogenetic analysis.
Nucleotide and deduced amino acid sequences of picornaviruses closely related to the study strains were obtained from the GenBank database and aligned using Clustal X software (version 2.0.3) (19), and pairwise identity calculations were performed using BioEdit software (version 7.1.3.0) (20). Phylogenetic trees of the amino acid alignments were created using the neighbor-joining method based on the Jones-Taylor-Thornton matrix-based model of MEGA software (version 5.2) (21). Bootstrap values (based on 1,000 replicates) for each node are given if >50%. The secondary structure of the 5′ UTR was predicted using the Mfold program (22) and compared to the predicted IRES structures of DHAV and a quail picornavirus (23, 24); the two-dimensional (2D) model was drawn using Corel Draw Graphics Suite (version 12). The nucleotide distance plot analysis was performed with RDP software version 4.16 (25) using the Jukes-Cantor model with a window size of 200 nt and a step size of 20 nt. The complete genome sequences of chicken/B21-CHV/2012/HUN, chicken/CHK-IV-CHV/2013/HUN, and turkey/B407-THV/2011/HUN were submitted to GenBank.
Cultivation of turkey and chicken picornaviruses in embryonated chicken eggs.
The fecal samples of turkey (B407) and chickens (B21 and CHK-IV) used for the complete genome characterization of turkey/B407-THV/2011/HUN, chicken/B21-CHV/2012/HUN, and chicken/CHK-IV-CHV/2013/HUN were used for viral cultivation in embryonated chicken eggs. The protocol was based on the study of Snoeyenbos and coworkers (1959) (26). In brief, the fecal suspensions were subjected to membrane filtration using Ultrafree-MC membrane filters with a 0.45-μm pore size (Millipore, Billerica, MA). Filtered suspensions (100 to 100 μl) containing gentamicin at 0.15 μg/ml, amphotericin B at 5 μg/ml, and vancomycin at 0.25 μg/ml were inoculated into the yolk sacs of 6-day-old embryonated chicken eggs. The yolk fluids were extracted and the physical conditions of the embryos were visually examined every 2 days until 8 days postinoculation (p.i.). Phosphate-buffered saline (0.1 M) with antibiotics was used for the control inoculations. The virus growth was monitored using the SYBR green RT-quantitative PCR (RT-qPCR) method and Maxima SYBR green/ROX qPCR master mix (Thermo Scientific) with type IV megrivirus IRES-specific primer pairs (Megri-IRES-R [5′ GAC ATA GTG GCA AAT CAA CTG 3′] and Apical8-5UTR-F [5′ TGG TGC TGA AAT ATY GCA AG 3′]) in the yolk fluid samples. To verify the presence of the study strains, the RT-PCR method, using a generic reverse primer targeting the conserved 3′ end of 2A2 (Megri-2A2-gen-R [5′ AAY TCA GCA TCA ATY TCA TCA TC 3′]) and two strain-specific forward primers designed to correspond to the divergent 3′ end of VP1 (Chicken megrivirus-VP1-F [5′ CAG ATG ATC CAG GYA AYA TGG G 3′] and Turkey megrivirus-VP1-F [5′ GGT GAT ATG GGT ACY ATY TTT GT 3′]), was used on the RNA extracts of yolk fluid samples. The size of the RT-PCR amplicons was either 1,400 or 1,215 bp, depending on the presence (turkey megriviruses) or absence (chicken megriviruses) of the presumed 2A0 region. The PCR amplicons were sequenced in both directions to verify the existence of the study strains.
Nucleotide sequence accession numbers.
The complete genome sequences of chicken/B21-CHV/2012/HUN, chicken/CHK-IV-CHV/2013/HUN, and turkey/B407-THV/2011/HUN were submitted to GenBank and assigned accession numbers KF961186 to KF961188.
RESULTS
Genome acquisition and analysis of megrivirus-like picornaviruses in chickens and turkeys.
From chicken fecal sample B-21, 70 sequence contigs (ranging between 106 and 1,215 nt) that yielded two continuous consensus sequences were acquired using viral metagenomics covering 56% of a picornavirus genome (Fig. 1). The two consensus sequences were 442 and 4,760 nucleotide (nt) long and showed 42% nt (36% amino acid) and 85% nt (95% amino acid) sequence identity to the VP0/VP3 junction and 2B-3D of THV-1 strain 2993D (GenBank accession no. HM751199) of the genus Megrivirus (the closest match in GenBank). The picornaviral genome of strain chicken/B21-CHV/2012/HUN (GenBank accession no. KF961186) was determined using the primer walking method (Fig. 1). For the verification of the genome features of chicken/B21-CHV/2012/HUN, another complete genome of this picornavirus (chicken/CHK-IV-CHV/2013/HUN; GenBank accession no. KF961187) was determined from chicken feces collected a year later (2013) from a different farm using the same primer sets and RT-PCR methods.
Overlapping sequences representing a complete picornavirus genome related to chicken/B-21-CHV/2012/HUN were amplified from turkey fecal sample B-407 using generic primer sets (Fig. 1) designed based on the available THV-1 sequences (GenBank accession no. HQ189775 and HM751199) and virus strain chicken/B21-CHV/2012/HUN.
The complete genomes of megrivirus-like picornaviruses of chickens, including chicken/B21-CHV/2012/HUN (GenBank accession no. KF961186) and chicken/CHK-IV-CHV/2013/HUN, (GenBank accession no. KF961187), and of turkey (turkey/B407-THV/2011/HUN; GenBank accession no. KF961188) consisted of 9,560 and 9,566 nt and 9,739 nt, respectively [excluding the poly(A) tail], the largest picornavirus genomes so far described. The G+C contents of the genomes were in the same range, 44.8% and 45.6%, respectively. The search for potential ORFs of both viruses revealed the presence of a long ORF encoding the viral polyproteins of 2,750 and 2,745 amino acids (aa) and 2,816 aa, with similar picornaviral genome organizations—P1 (VP0-3-1) and P2 (2A-B-C)-P3 (3A-B-C-D)—preceded by 639- and 636-nt and 642-nt 5′ UTRs and followed by remarkably long (668- and 692-nt and 646-nt) 3′ regions and a poly(A) tail containing a potential ORF2 (see below) (Fig. 1). For practical reasons, for the detailed sequence analyses, the chicken/B21-CHV/2012/HUN (GenBank accession no. KF961186) and turkey/B407-THV/2011/HUN (GenBank accession no. KF961188) sequences were used; the supporting data from the chicken/CHK-IV-CHV/2013/HUN (GenBank accession no. KF961187) sequence were included if necessary.
The complete P1 (2,436 and 2,403 nt; 812 and 801 aa), P2 (3,261 and 3,492 nt; 1,087 and 1,164 aa), and P3 (2,556 and 2,556 nt; 851 and 851 aa) genome regions of chicken/B21-CHV/2012/HUN and turkey/B407-THV/2011/HUN showed 37% and 93%, 97% and 94%, and 95% and 98% amino acid identity to THV-1 strain 2993D (GenBank accession no. HM751199), which was the closet relative. The position of the STOP codon in the nucleotide and amino acid alignments was the same in all of the study strains and the THVs (GenBank accession no. HQ189775 and HM751199). The high sequence identities at the nonstructural genome regions (P2–P3) between the study sequences and THV-1 strongly suggest the classification of these picornaviruses in the genus Megrivirus.
Analysis of the 5′ UTR.
Until now, neither the complete 5′ UTR nor the predicted secondary RNA structure of the IRES of turkey megrivirus was known. The predicted initiation codons of chicken/B21-CHV/2012/HUN and turkey/B407-THV/2011/HUN were found in an optimal Kozak context (GCCACTA640/643TGT, optimal nucleotides being underlined) in both viruses. The 639- and 642-nt-long 5′ UTRs of the study sequences showed 87% and 88% nucleotide identity to the available 461-nt-long partial 5′ UTR of THVs (GenBank accession no. HQ189775 and HM751199) and 88% nucleotide identity to each other. The 5′ UTR of the study sequences contained the highly conserved 20-nt-long motif forming an apical “8”-like structure which is also present in DHAV (7), quail picornavirus (QPV; GenBank accession no. JN674502), and several other picornaviruses, mostly of avian origin (24) (Fig. 2). The predicted secondary RNA structures of IRESs of both study viruses were similar to those of domains II and III (III1, III2, III3, III4, IIIa, IIIc, IIId, IIIe, and IIIf) of the hepacivirus/pestivirus-like (HP) type IVB IRES (Fig. 2). The predicted lengths of the pseudoknot stem 1, stem 2, linker, and spacer (23) are 11, 8, 2, and 10 nt, respectively. However, important structural differences were also observed: in similarity to the IRES of QPV, the apical part of domain III did not end with the short IIIb hairpin but had a long (>80-nt) secondary structure with the “8”-like motif at the apical position (Fig. 2).
FIG 2.
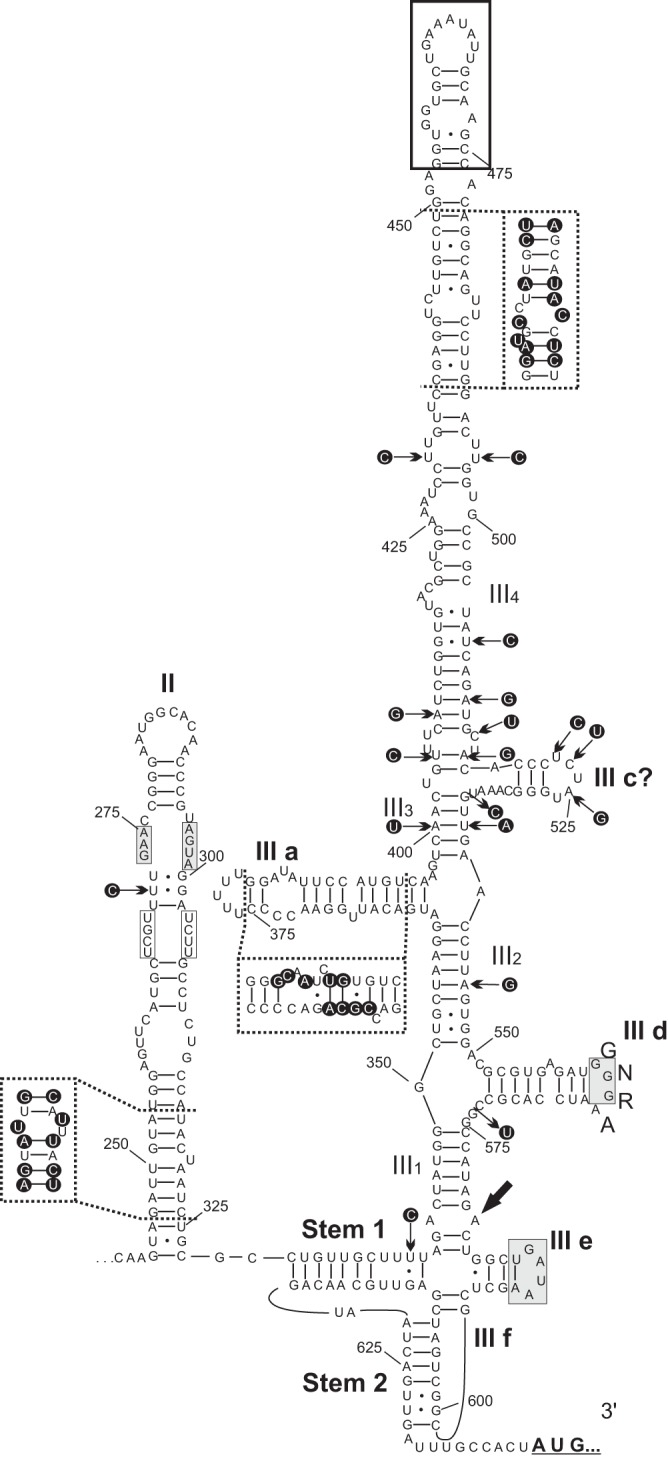
Predicted two-dimensional secondary RNA structure of the 5′ UTR IRES of chicken megrivirus (GenBank accession no. KF961186) with the positions of different nucleotides (in dark circles) indicated by black arrows and within frames of dashed lines compared to the IRES of turkey/B407-THV/2011/HUN (GenBank accession no. KF961188). The main domains (II and III), helical segments (III 1 to 4), and hairpins (III a; III c to f) and stems (Stem 1 and Stem 2) similar to those of the similarly labeled structures of hepacivirus/pestivirus-like type IVB IRES are depicted. Gray boxes indicate conserved motifs of IVB IRESes (23). White boxes indicate the conserved unpaired base pairs with respect to DHAVs within the middle loop of domain II. The sequence and location of the apical “8” structure also identified in other avian picornaviruses are indicated with a black frame box (24).
Analysis of the 3′ genome region.
The 668- and 646-nt-long 3′ apparent UTRs (sequences after the STOP codon of the viral polyprotein-coding regions) of chicken/B21-CHV/2012/HUN and turkey/B407-THV/2011/HUN show 88% and 91% nucleotide identity to partial sequences of chicken/CHK148/USA/2010 (GenBank accession no. JF424831) and turkey/TRK22/USA/2010 (GenBank accession no. JF424832) picornaviruses which probably also belong to the genus Megrivirus (5). Similarly to CHK148 and TRK22, the study sequences could also contain a second, downstream ORF (5) encoding putative short (114- and 98-aa-long) proteins of unknown function, although the predicted initiation codons were in a weak Kozak context (Fig. 1) (3). The presence of this putative ORF2 among the THVs is uncertain due to incompleteness of the 3′ UTR of the prototype strains (0091.1 and 2993D). The analysis of the 324- and 349 nt-long 3′ UTRs (sequences after the second ORF) of study strains revealed multiple conservative sequence repeats (provisionally named “unit A” and “unit B”) and 77- to 101-nt-long AUG-rich regions (where the cytosine content is lower than 4%), which could also be identified in a second strain of chicken picornavirus (chicken/CHK-IV-CHV/2013/HUN) and among the 3′ UTRs of the recently identified pigeon mesiviruses (PiMEVs) (GenBank accession no. KC876003 and KC811837) (11). Interestingly, the genome organization of the 3′ UTRs of PiMEVs and those of the study sequences and THVs were essentially the same, with the exception of the potential ORF2, which is missing in the PiMEVs (Fig. 3). It is interesting that the localization of unit A is always adjacent to a STOP codon (Fig. 3). The nucleotide and amino acid sequence identities and relative positions of the putative ORF2 and of the conservative unit As and unit Bs are shown in Fig. 3.
FIG 3.

(A) The organization of the 3′ parts of chicken/B21-CHV/2012/HUN (B21, GenBank accession no. KF961186), chicken/CHK-IV-CHV/2013/HUN (CHK-IV, GenBank accession no. KF961187), turkey/B407-THV/2011/HUN (B407, GenBank accession no. KF961188), and pigeon mesiviruses (PiMEVs: GenBank accession no. KC876003 and KC811837). The presumed initiation codons in a weak Kozak context (optimal nucleotides are underlined) of ORF2 are shown in italics after the STOP codon (black star) of the end of the viral polyprotein (3D). Nucleotide (upper numbers) and amino acid (lower numbers) lengths of the presumed ORF2 are indicated in each gene box. Dotted lines corresponding to PiMEVs indicate the deletion of ORF2 region. The identified repeating elements of “unit A” and “unit B” (“B”) together with the location of AUG-rich genome parts are depicted within the gray and white arrows. The pairwise nucleotide (and amino acid) sequence identities between the different regions (indicated by double arrows) of 3′ genome parts are also illustrated. (B) Alignments of the “unit A” and “unit B” sequences of the study strains (B21; CHK-IV and B407), pigeon mesiviruses (PiMEV-1 and -2), and the members of genus Megrivirus (0091.1, 2993D, CHK148, and TRK22). Similar nucleotides are shaded. Asterisks indicate identical nucleotides.
Analysis of the viral polyprotein.
The possible cleavage sites of the viral polyprotein of turkey/B407-THV/2011/HUN and chicken/B21-CHV/2012/HUN were mapped based on the amino acid alignments of the available THVs (GenBank accession no. HQ189775 and HM751199). All of the previously predicted cleavage sites of THVs (7) were identifiable in the study sequences, although the genome alignments revealed previously unknown sequence stretches and novel potential cleavage sites in the N-terminal part of P2 (Fig. 1).
Similarly to the THVs, neither the presence of a potential L-protein nor the cleavage of VP0 into VP4 and VP2 were recognizable in the study sequences. The VP0 (384 and 392 aa), VP3 (181 and 166 aa), and VP1 (247 and 243 aa) genomic regions of chicken/B21-CHV/2012/HUN and turkey/B407-THV/2011/HUN were similar in length to those of THV-1 (388 aa, 166 aa, and 244 aa). The presence of Rhv-like capsid domains was also identifiable in both study sequences using the conserved domain database (CDD) search of NCBI (27) (Fig. 1).
The analysis of aligned sequences revealed a 246-nt (82-aa)-long insertion after the predicted C-terminal cleavage site of VP1 in the turkey/B407-THV/2011/HUN sequence (and in those of both known THV strains) compared to the two chicken picornavirus sequences (chicken/B21-CHV/2012/HUN and chicken/CHK-IV-CHV/2013/HUN), which insertion was terminated with a previously supposed cleavage site (FQ883/SP) (7) (Fig. 1). Due to the absence of this peptide among the chicken origin picornaviruses, this genome region was referred to as 2A0 (Fig. 1). According to Honkavuori et al. (7), the viral polypeptide region between VP1 and 2B was cleaved at least at the position of FQ1199/AP (in THV strains 0091.1 and 2993D), causing the release of a 2A peptide which belongs to the group of Hbox-NC (noncoding) proteins (7, 28). The sequence and location of this motif (FQ/AP)—as the C-terminal cleavage site of an Hbox-NC-containing 2A—were the same in the study sequences; furthermore, this sequence motif was also present 104 aa upstream, suggesting an additional possible cleavage site and, therefore, the release of a 104-aa-long 2A peptide during the maturation process of the study strains (Fig. 1). In the amino acid alignments, at the location of the N-terminal cleavage site of the 104-aa-long 2A, an identical, previously unrecognized cleavage motif (FQ1095/AP) was also present in the THVs (strains 0091.1 and 2993D). Therefore, the presumed mature 2A peptides numbered four (due to the presence of 2A0) in the case of THVs and turkey/B407-THV/2011/HUN and numbered three in the case of chicken/B21-CHV/2012/HUN and chicken/CHK-IV-CHV/2013/HUN (Fig. 1). The function of the suspected multiple 2As is unknown. In the nonstructural protein region (P2–P3), all the previously identified conserved amino acid motifs of the 2C helicase, 3C protease, and 3D RNA-dependent RNA polymerase of THVs (7) were identified in the study sequences (Fig. 1).
Sequence comparison and phylogenetic analysis.
The amino acid identities, based on the pairwise comparisons of different genome regions of chicken/B21-CHV/2012/HUN and turkey/B407-THV/2011/HUN, show significant differences with respect to the P1 and P2–P3 genome regions (Fig. 1 and Table 1). The pairwise amino acid sequence identities of genome regions of P1 (VP0-VP3-VP1) and 2A1 were significantly lower (<46%) than the identity values (>90%) of genome regions after 2A2 (2A3-2B-2C-3A-3B-3C-3D) (Fig. 1 and Table 1). The results of the pairwise nucleotide sequence similarity comparisons show that the transition between the heterogeneous and homogenous genome regions at the 2A area is not sharp (Fig. 4).
TABLE 1.
Amino acid sequence identity values of different genome regions of chicken/B21-CHV/2012/HUN and turkey/B407-THV/2011/HUN compared to the members of genus Megrivirus and to the second strain of chicken megrivirus-like picornavirus chicken/CHK-IV-CHV/2013/HUNa
| Genome region | % amino acid sequence identity |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 0091.1 |
2993D |
CHK148 |
TRK22 |
CHK-IV |
||||||
| B21 | B407 | B21 | B407 | B21 | B407 | B21 | B407 | B21 | B407 | |
| VP0 | 34.4 | 91.6 | 35.0 | 91.6 | N.D. | N.D. | N.D. | N.D. | 93.5 | 35.4 |
| VP3 | 39.2 | 96.4 | 39.2 | 96.4 | N.D. | N.D. | N.D. | N.D. | 91.2 | 39.8 |
| VP1 | 35.5 | 89.3 | 34.9 | 93.0 | N.D. | N.D. | N.D. | N.D. | 87.0 | 34.5 |
| 2A0 | N/A | 87.8 | N/A | 89.0 | N.D. | N.D. | N.D. | N.D. | N/A | N/A |
| 2A1 | 46.4 | 92.6 | 46.0 | 93.5 | N.D. | N.D. | N.D. | N.D. | 92.3 | 43.7 |
| 2A2 | 74.0 | 100.0 | 75.0 | 99.0 | N.D. | N.D. | N.D. | N.D. | 97.1 | 72.1 |
| 2A3 | 87.9 | 92.0 | 88.4 | 92.0 | N.D. | N.D. | N.D. | N.D. | 96.0 | 87.9 |
| 2B | 92.2 | 95.3 | 92.2 | 94.3 | N.D. | N.D. | N.D. | N.D. | 95.8 | 89.1 |
| 2C | 94.4 | 98.1 | 94.1 | 98.4 | N.D. | N.D. | N.D. | N.D. | 98.9 | 95.2 |
| 3A | 94.0 | 99.3 | 94.0. | 99.3 | N.D. | N.D. | N.D. | N.D. | 97.3 | 93.3 |
| 3B | 100.0 | 100.0 | 100.0 | 100.0 | N.D. | N.D. | N.D. | N.D. | 100.0 | 100.0 |
| 3C | 96.0 | 96.0 | 96.5 | 96.5 | N.D. | N.D. | N.D. | N.D. | 98.0 | 94.6 |
| 3D | 98.3 | 99.2 | 97.9 | 98.7 | 99.6b | 97.3b | 97.8b | 100.0b | 99.8 | 98.1 |
B21, chicken/B21-CHV/2012/HUN (GenBank accession no. KF961186); B407, turkey/B407-THV/2011/HUN (GenBank accession no. KF961188); 0091.1, 2993D, CHK148, and TRK22, members of genus Megrivirus; CHK-IV, chicken/CHK-IV-CHV/2013/HUN (GenBank accession no. KF961187); N.D., not determined (no sequences were available for the comparisons); N/A, not applicable (the genome part is missing from the sequence).
Only the available 223-aa-long C-terminal parts of the 3Dpol genome regions were used for comparisons.
FIG 4.

Distance plot analysis based on the comparison of the complete genome of chicken/B21-CHV/2012/HUN (GenBank accession no. KF961186) to those of turkey/B407-THV/2011/HUN (B407-THV; GenBank accession no. KF961188) and pigeon mesivirus 1 (GenBank accession no. KC876003). The genome regions of chicken megrivirus-like picornavirus are drawn to scale. Dashed lines show the borders of 5′ UTR/P1, P1/2A, 2A/2B, and 3D/ORF2.
The sequence heterogeneity between structural and nonstructural genome regions was conspicuous in the phylogenetic trees generated from the amino acid alignments of different genome regions of megriviruses and the closest relatives (Fig. 4). Interestingly, turkey/B407-THV/2011/HUN grouped together with the THVs in all trees, while the chicken megrivirus-like viruses showed significant divergence from the megriviruses in the P1 and VP1 genome regions and parallel close relationships with the same group at the 2C and 3CD phylogenetic trees (Fig. 5).
FIG 5.

Phylogenetic positions of the study strains (indicated in bold and by black arrows) among the closest relatives of picornaviruses based on amino acid sequences of the different picornavirus coding regions: P1 (A), VP1 (B), 2C (C), and 3CD (D). Bars indicate 0.2 amino acid substitutions per site. The names of virus species in the different picornavirus genera are underlined.
Cultivation of turkey and chicken picornaviruses.
Two of the three embryos (inoculated with B21 and B407) were found dead at day 8 p.i., and the third (inoculated with CHK-IV) showed signs of growth depression. The megrivirus RNA was detectable using megrivirus IRES-specific primer pairs in every infected yolk fluid sample, with the exception of the day 8 p.i. yolk samples of the dead embryos. Increasing amounts of viral RNA were detected from day 2 to 6 p.i., with the highest level of RNA in the yolk samples collected on day 6 p.i. (data not shown). Furthermore, the study strains were identified by strain-specific VP1/2A2 primer pairs in every megrivirus RNA-containing yolk fluid sample. The 2A0-containing turkey megrivirus RNA was not present in the yolk fluids from eggs inoculated with chicken samples B21 and CHK-IV; similarly, the 2A0-less chicken megrivirus-like viral RNA was also not detectable in the yolk fluids from eggs inoculated with a turkey sample (B407).
DISCUSSION
From this study, using metagenomic and RT-PCR approaches, we report the complete genome characterization and analysis of picornaviruses related to megrivirus from fecal samples of chickens and turkeys collected from different poultry farms in Hungary. Based on the phylogenetic analysis and the high sequence identities in the nonstructural protein-coding region, the available chicken and turkey megrivirus-like picornaviruses analyzed most likely belong to the genus Megrivirus. The specific taxonomic position of these strains within the genus Megrivirus is clear in the case of strain turkey/B407-THV/2011/HUN, which is phylogenetically indistinguishable from THV-1 and therefore belongs to the species Melegrivirus A. Considerable phylogenetic and sequence divergence of chicken megrivirus-like picornaviruses from the strains of Melegrivirus A was found in the P1 capsid polypeptide precursor; the P1 amino acid identities were less than 40% (representing one of the criteria of demarcation between members of different picornavirus genera) (1). Furthermore, the absence of the presumed 2A0 region from the chicken megrivirus-like picornaviruses, parallel with the close sequence relationship in the P2–P3 region, makes the classification of chicken megrivirus-like picornaviruses complicated. Moreover, if the chicken megrivirus-like viruses were members of the genus Megrivirus, then, based on the phylogenetic position of the currently unclassified pigeon mesiviruses (located between the Melegrivirus A and the chicken megrivirus-like viruses in the P1 genome region), the PiMEVs might also belong to the genus Megrivirus.
The sequence heterogeneity between structural and nonstructural genome regions in the chicken megriviruses and the results of the similarity plot analysis raised the possibility that these strains had emerged from an intra- or interspecies recombination event, although, due to the lack of one of the potential parent sequences, the evidence for recombination is incomplete at present (data not shown). The gradual transition between the heterogeneous P1 region and the homogenous P2–P3 genome regions could suggest that if the chicken megrivirus-like viruses are indeed recombinant viruses, then the recombination event took place some time ago. The continuous mutations affecting the 2A1 to 2A2 regions (maybe due to the adaptation process of a novel host) masked the potential recombination breakpoint.
Until now, only the two prototype genome sequences in the species Melegrivirus A (THV strains 0091.1 and 2993D) with incomplete 5′ UTR and 3′ UTR sequences were available. Additionally, two partial 3D-3′ UTR sequences of chicken/CHK148/USA/2010 (1,312 nt; GenBank accession no. JF424831) and turkey/TRK22/USA/2010 (1,327 nt; GenBank accession no. JF424832) from the United States (5) have been assumed to be THV based on their close sequence relationships; however, our findings suggest that species assignment of these viruses will require sequences representing the capsid-coding region. The 5′ UTR sequence length of megriviruses was extended by 180 nt compared to the available THVs, probably reaching the actual 5′ end of the genome. Results of the secondary RNA structure analysis of the 5′ UTR IRESs suggest that the chicken and turkey megriviruses have a hepacivirus/pestivirus-like type IVB IRES based on the sequence and secondary structural similarities to the IRESes of DHAVs and hepatitis C virus (23), although the considerable structural differences, e.g., the presence of an extended hairpin and the conservative apical “8” structure—which is probably functioning as a mobile genetic element similar to the stem-loop-2-like motif (s2m) found as a highly conserved fragment near the 3′ end of many positive-stranded RNA viruses (29)—at the top of domain III, in analogy to other picornaviruses of avian origin (24), may suggest the possibility that this is a variant of the type IV IRES.
The 3′ genome regions (sequences after the STOP codon of the viral polyprotein-coding regions) of the chicken and turkey megriviruses were the longest after those of the newly described rosavirus (30) and, as identified in the megrivirus strains of CHK148 (GenBank accession no. JF424831) and TRK22 (GenBank accession no. JF424832) (5), could contain a minor ORF. The translation and the possible function of the putative polypeptides encoded by the minor ORF are currently unknown, although the relatively low sequence identity and considerable length difference suggest different roles (if any). Further experimental evidence is necessary to verify the presence of a second ORF in these picornavirus genomes, although the recently identified canine cadiciviruses with a dicistronic genome organization (4) and the previously identified functional small ORFs in the genomes of two cardioviruses (30, 31, 35) are good examples that deviate from the picornavirus single-ORF model.
The similar organizations of the 3′ UTRs of megriviruses and mesiviruses and the presence of conserved repeating sequence motifs (“unit A” and “unit B”) served as more evidence for the closer relationship and a common ancestor of the members of these virus groups, which may be of avian origin. The role of these various sequence repeats in the buildup of the 3′ UTR is currently unknown. A similarly short and conserved nucleotide motif, called a “barbell-like structure,” was identified in the 3′ UTR of other avian picornaviruses of the genera Avihepatovirus and “Gallivirus” (16). The presence of these repeating units of megri- and mesiviruses among other viral sequences is worthy of further investigation.
Another interesting genome feature of megri- and mesiviruses is the relatively long 3′ UTR region with significantly low cytosine content, called an AUG-rich element, in which the one or more “unit B” motifs were located. The presence of such AUG-rich elements is not novel among picornaviruses; similar regions with short oligonucleotide repeats were identified in 3′ UTRs of some entero- and rhinoviruses (32). The role of these regions is currently unknown, although similar AUG-rich elements were identifiable in the so-called late mRNAs of certain human papillomaviruses, where these elements act as mRNA instability elements in papillomavirus late gene expression (33).
The predictions of possible proteolytic cleavage sites, based mainly on sequence alignments, were difficult and did not always result in the successful mapping of all of the cleavage sites. In this study, we confirmed all the cleavage sites predicted by Honkavuori et al. (7) of megriviruses and mapped a previously unknown additional 2A cleavage site (7). In the chicken megriviruses, three putative mature 2A peptides were predicted, in similarity to the avian picornaviruses of the genus Avihepatovirus and the recently identified “Avisivirus” (6, 10) in turkeys. The presence of a 82-aa-long stretch located at the junction of VP1/2A terminating with a potential cleavage site in the THVs and turkey/B407-THV/2011/HUN would result in the release of a fourth mature 2A, which makes the turkey megriviruses unique among the known picornaviruses, and members of genus Megrivirus may therefore represent an ancient group of picornaviruses showing multiple 2A genome features (6, 34).
The unique genome features, such as the heterogenic sequence relationship of the new chicken megriviruses to the THV in the P1 and P2–P3 regions, should be interpreted as another reminder, as in the case of the interspecies recombinant ovine enteroviruses (17), that without the knowledge of the complete genome sequence, the sequence-based taxonomic classification of a picornavirus (regardless the size or the location of the particular sequence) could be misleading.
Until now, there had been no direct evidence of the propagation of megriviruses in embryonated chicken eggs. In this study, we demonstrated that the study strains of both 2A0-containing turkey megrivirus and the 2A0-less chicken megrivirus-like viruses were cultivable in yolk sac. Death of the embryos was observed at the 8th day p.i. Similarly, embryonic deaths with the same p.i. time were observed when embryos were infected with an undetermined picornavirus (possibly a megrivirus) historically isolated from fecal and liver samples of turkeys with signs of viral hepatitis (26). However, the idea of a direct link between the observed deaths and the megrivirus infection should be treated with caution due to the potential presence of other coinfective agents, including other picornaviruses, in the fecal samples.
Avian-origin picornaviruses, including megriviruses and mesiviruses, have a unique genome and unusual sequence features among picornaviruses. It is highly possible that there are further avian picornaviruses with potentially interesting genome features. Analysis of the evolutional connections of this group of viruses could help us to better understand the evolution of picornaviruses.
ACKNOWLEDGMENTS
This work was supported by grants from the Hungarian Scientific Research Fund (OTKA, K83013, and K111615) and the European Union and the state of Hungary, cofinanced by the European Social Fund in the framework of the TÁMOP-4.2.4.A/2-11/1-2012-0001 “National Excellence Program.” G.R. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and E.D. by NHLBI grant R01HL083254. N.J.K. was supported by core funding provided to the Pirbright Institute by the Biotechnology and Biological Sciences Research Council, United Kingdom.
We gratefully acknowledge the help of Istvan Jankovics for the viral cultivation experiments.
Footnotes
Published ahead of print 26 March 2014
REFERENCES
- 1.Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, Palmenberg AC, Simmonds P, Skern T, Stanway G, Yamashita T, Zell R. 2011. Picornaviridae, p 855–880 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 2.Adams MJ, King AMQ, Carstens EB. 2013. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch. Virol. 158:2023–2030. 10.1007/s00705-013-1688-5 [DOI] [PubMed] [Google Scholar]
- 3.Racaniello V. 2007. Picornaviridae: the viruses and their replication, p 795–838 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Woo PC, Lau SK, Choi GK, Huang Y, Teng JL, Tsoi HW, Tse H, Yeung ML, Chan KH, Jin DY, Yuen KY. 2012. Natural occurrence and characterization of two internal ribosome entry site elements in a novel virus, canine picodicistrovirus, in the picornavirus-like superfamily. J. Virol. 86:2797–2808. 10.1128/JVI.05481-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farkas T, Fey B, Hargitt E, Parcells M, Ladman B, Murgia YS. 2012. Molecular detection of novel picornaviruses in chickens and turkeys. Virus Genes 44:262–272. 10.1007/s11262-011-0695-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tseng CH, Knowles NJ, Tsai HJ. 2007. Molecular analysis of duck hepatitis virus type 1 indicates that it should be assigned to a new genus. Virus Res. 123:190–203. 10.1016/j.virusres.2006.09.007 [DOI] [PubMed] [Google Scholar]
- 7.Honkavuori KS, Shivaprasad HL, Briese T, Street C, Hirschberg DL, Hutchison SK, Lipkin WI. 2011. Novel picornavirus in turkey poults with hepatitis, California, USA. Emerg. Infect. Dis. 17:480–487. 10.3201/eid1703.101410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauvage V, Ar Gouilh M, Cheval J, Muth E, Pariente K, Burguiere A, Caro V, Manuguerra JC, Eloit M. 2012. A member of a new Picornaviridae genus is shed in pig feces. J. Virol. 86:10036–10046. 10.1128/JVI.00046-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbknecht M, Sepsenwol S, Leis E, Tuttle-Lao M, Gaikowski M, Knowles NJ, Lasee B, Hoffman MA. 11 December 2014. Characterization of a new picornavirus isolated from the freshwater fish Lepomis macrochirus. J. Gen. Virol. 10.1099/vir.0.061960-0 [DOI] [PubMed] [Google Scholar]
- 10.Boros A, Nemes C, Pankovics P, Kapusinszky B, Delwart E, Reuter G. 2013. Genetic characterization of a novel picornavirus in turkeys (Meleagris gallopavo) distinct from turkey galliviruses and megriviruses and distantly related to the members of the genus Avihepatovirus. J. Gen. Virol. 94:1496–1509. 10.1099/vir.0.051797-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phan TG, Vo NP, Boros Á, Pankovics P, Reuter G, Li OT, Wang C, Deng X, Poon LL, Delwart E. 2013. The viruses of wild pigeon droppings. PLoS One 8:e72787. 10.1371/journal.pone.0072787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmenberg A, Neubauer D, Skern T. 2010. Genome organization and encoded proteins, p 3–17 In Ehrenfeld E, Domingo E, Roos RP. (ed), The picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 13.Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. 2009. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 83:4642–4651. 10.1128/JVI.02301-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapoor A, Victoria J, Simmonds P, Slikas E, Chieochansin T, Naeem A, Shaukat S, Sharif S, Alam MM, Angez M, Wang C, Shafer RW, Zaidi S, Delwart E. 2008. A highly prevalent and genetically diversified Picornaviridae genus in South Asian children. Proc. Natl. Acad. Sci. U. S. A. 105:20482–20487. 10.1073/pnas.0807979105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reuter G, Farkas T, Berke T, Jiang X, Matson DO, Szücs G. 2002. Molecular epidemiology of human calicivirus gastroenteritis outbreaks in Hungary, 1998 to 2000. J. Med. Virol. 68:390–398. 10.1002/jmv.10216 [DOI] [PubMed] [Google Scholar]
- 16.Boros A, Nemes C, Pankovics P, Kapusinszky B, Delwart E, Reuter G. 2012. Identification and complete genome characterization of a novel picornavirus in turkey (Meleagris gallopavo). J. Gen. Virol. 93:2171–2182. 10.1099/vir.0.043224-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boros A, Pankovics P, Knowles NJ, Reuter G. 2012. Natural interspecies recombinant bovine/porcine enterovirus in sheep. J. Gen. Virol. 93:1941–1951. 10.1099/vir.0.041335-0 [DOI] [PubMed] [Google Scholar]
- 18.Boros A, Pankovics P, Simmonds P, Reuter G. 2011. Novel positive-sense, single-stranded RNA (+ssRNA) virus with di-cistronic genome from intestinal content of freshwater carp (Cyprinus carpio). PLoS One 6:e29145. 10.1371/journal.pone.0029145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 http://jwbrown.mbio.ncsu.edu/JWB/papers/1999Hall1.pdf [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hellen CU, de Breyne S. 2007. A distinct group of hepacivirus/pestivirus-like internal ribosomal entry sites in members of diverse picornavirus genera: evidence for modular exchange of functional noncoding RNA elements by recombination. J. Virol. 81:5850–5863. 10.1128/JVI.02403-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pankovics P, Boros A, Reuter G. 2012. Novel picornavirus in domesticated common quail (Coturnix coturnix) in Hungary. Arch. Virol. 157:525–530. 10.1007/s00705-011-1192-8 [DOI] [PubMed] [Google Scholar]
- 25.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snoeyenbos GH, Bosch HI, Sevoian M. 1959. An infectious agent producing hepatitis in turkeys. Avian Dis. 3:377–388. 10.2307/1587577 [DOI] [Google Scholar]
- 27.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229. 10.1093/nar/gkq1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes PJ, Stanway G. 2000. The 2A proteins of three diverse picornaviruses are related to each other and to the H-rev107 family of proteins involved in the control of cell proliferation. J. Gen. Virol. 81:201–207 [DOI] [PubMed] [Google Scholar]
- 29.Kofstad T, Jonassen CM. 2011. Screening of feral and wood pigeons for viruses harbouring a conserved mobile viral element: characterization of novel astroviruses and picornaviruses. PLoS One 6:e25964. 10.1371/journal.pone.0025964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phan TG, Kapusinszky B, Wang C, Rose RK, Lipton HL, Delwart E. 2011. The fecal viral flora of wild rodents. PLoS Pathog. 7:e1002218. 10.1371/journal.ppat.1002218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loughran G, Firth AE, Atkins JF. 2011. Ribosomal frameshifting into an overlapping gene in the 2B-encoding region of the cardiovirus genome. Proc. Natl. Acad. Sci. U. S. A. 108:E1111–E1119. 10.1073/pnas.1102932108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pilipenko EV, Maslova SV, Sinyakov AN, Agol VI. 1992. Towards identification of cis-acting elements involved in the replication of enterovirus and rhinovirus RNAs: a proposal for the existence of tRNA-like terminal structures. Nucleic Acids Res. 20:1739–1745. 10.1093/nar/20.7.1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy IM, Haddow JK, Clements JB. 1991. A negative regulatory element in the human papillomavirus type 16 genome acts at the level of late mRNA stability. J. Virol. 65:2093–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johansson S, Niklasson B, Maizel J, Gorbalenya AE, Lindberg AM. 2002. Molecular analysis of three Ljungan virus isolates reveals a new, close-to-root lineage of the Picornaviridae with a cluster of two unrelated 2A proteins. J. Virol. 76:8920–8930. 10.1128/JVI.76.17.8920-8930.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong WP, Roos RP. 1991. Alternative translation initiation site in the DA strain of Theiler's murine encephalomyelitis virus. J. Virol. 65:3395–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]