

ABSTRACT
Herpesvirus capsid morphogenesis occurs in the nucleus, while final maturation takes place in the cytosol, requiring translocation of capsids through the nuclear envelope. The nuclear egress complex, consisting of homologs of herpes simplex virus pUL31 and pUL34, is required for efficient nuclear egress via primary envelopment and de-envelopment. Recently, we described an alternative mode of nuclear escape by fragmentation of the nuclear envelope induced by replication-competent pUL31 and pUL34 deletion mutants of the alphaherpesvirus pseudorabies virus (PrV), which had been selected by serial passaging in cell culture. Both passaged viruses carry congruent mutations in seven genes, including UL46, which encodes one of the major tegument proteins. Herpesvirus pUL46 homologs have recently been shown to activate the PI3K-Akt and ERK1/2 signaling pathways, which are involved in regulation of mitosis and apoptosis. Since in uninfected cells fragmentation of the nuclear envelope occurs during mitosis and apoptosis, we analyzed whether pUL46 of PrV is involved in signaling events impairing the integrity of the nuclear envelope. We show here that PrV pUL46 is able to induce phosphorylation of ERK1/2 and, thus, expression of ERK1/2 target genes but fails to activate the PI3K-Akt pathway. Deletion of UL46 from PrV-ΔUL34Pass and PrV-ΔUL31Pass or replacement by wild-type UL46 resulted in enhanced nuclear envelope breakdown, indicating that the mutations in pUL46 may limit the extent of NEBD. Thus, although pUL46 induces ERK1/2 phosphorylation, controlling the integrity of the nuclear envelope is independent of the ERK1/2 signaling pathway.
IMPORTANCE Herpesvirus nucleocapsids can leave the nucleus by regulated, vesicle-mediated transport through the nuclear envelope, designated nuclear egress, or by inducing nuclear envelope breakdown (NEBD). The viral proteins involved in NEBD are unknown. We show here that the pseudorabies virus tegument protein pUL46 induces the ERK1/2 signaling pathway and modulates NEBD. However, these two processes are independent and ERK1/2 signaling induced by pUL46 is not involved in herpesvirus-induced NEBD.
INTRODUCTION
Nuclear egress of nucleocapsids to the cytosol is an important step in the herpesvirus replication cycle. For translocation through the nuclear envelope, nucleocapsids bud at the inner nuclear membrane to form a primary enveloped virion located in the perinuclear cleft. The subsequent fusion with the outer nuclear membrane then releases the nucleocapsid into the cytosol, where final maturation takes place (reviewed in references 1, 2, and 3). This process is mediated by the nuclear egress complex (NEC), consisting of homologs of the herpes simplex virus 1 (HSV-1) proteins pUL31 and pUL34. The type II membrane protein pUL34 is targeted to the nuclear envelope and binds the nucleoplasmic pUL31, thereby linking it to the inner nuclear membrane (2, 3). The pUL34/pUL31 complex recruits viral and cellular kinases to the budding sites, where they phosphorylate lamins, resulting in local dissolution of the nuclear lamina (4–7).
Deletion of either UL31 or UL34 from the pseudorabies virus (PrV) genome results in mutants that are unable to leave the nucleus efficiently and that replicate to drastically reduced titers (8, 9). However, their residual infectivity was used for serial passaging, resulting in the isolation of mutant viruses PrV-ΔUL31Pass and PrV-ΔUL34Pass, which replicated to titers comparable to that of the wild type in the absence of the NEC. Ultrastructural analysis showed that unlike wild-type PrV, both passaged mutants induced fragmentation of the nuclear envelope (nuclear envelope breakdown [NEBD]) to escape from the nuclear compartment for subsequent cytoplasmic final maturation (10, 11).
To elucidate the molecular basis for PrV-ΔUL31Pass- and PrV-ΔUL34Pass-induced NEBD, different inhibitors of key enzymes were used to probe for involvement of cellular pathways. We showed that roscovitine, an inhibitor of cyclin-dependent kinase 1 (cdk 1), cdk 2, cdk 5, and, when used in high concentrations, extracellular signal-regulated kinase ERK1/2, impaired replication of both passaged viruses, whereas wild-type PrV was significantly less affected. U0126, an inhibitor of MEK1/2, the kinase upstream of ERK1/2, was able to inhibit replication of PrV-ΔUL31Pass and PrV-ΔUL34Pass without affecting wild-type PrV (10). In parallel, whole genomes of PrV-ΔUL34Pass, parental PrV-ΔUL34, and wild-type PrV strain Kaplan (PrV-Ka) were sequenced. Genes mutated in PrV-ΔUL34Pass were subsequently analyzed in PrV-ΔUL31Pass. Sequence comparison identified seven genes mutated in both passaged viruses, including UL46 (10). The alterations in the UL46 sequences of PrV-ΔUL31Pass and PrV-ΔUL34Pass resulted in a 29-amino-acid (aa) duplication of aa 514 to 542 for PrV-ΔUL34Pass pUL46 and a duplication of 37 bp, creating a frameshift after codon 662, for PrV-ΔUL31Pass (10).
pUL46 is an abundant, yet nonessential tegument protein (12–14). By yeast two-hybrid analyses, it was shown to interact with different capsid proteins and with tegument proteins such as pUL37, pUL48, and pUL49 (15). It has been suggested that pUL46 enhances trans-activation of immediate early genes induced by the α-transinducing factor pUL48 (13, 16, 17). Although a direct interaction between pUL48 and pUL46 could be identified, the possible mechanism of modulation is still unclear (18). For HSV-1 pUL46, an association with cellular membranes and the viral capsid has been shown (12, 16).
Recently, data indicating that HSV-1 pUL46 (also designated VP11/12) and its homologs are able to manipulate cellular signaling pathways and might mimic an activated growth factor receptor have accumulated (19). HSV-1 pUL46 activates a member of the Src family of tyrosine kinases, the lymphocyte-specific protein tyrosine kinase Lck, and is in turn phosphorylated by Lck (20). This led to the suggestion that pUL46 participates in an Lck-mediated signaling pathway controlling downstream signaling events. The phosphoinositide 3-kinase (PI3K)-Akt pathway, located downstream of Lck, is activated through direct interaction of PI3K with phosphorylated pUL46 (21, 22). Currently, it is still unclear which Akt target is affected by pUL46. Although the Akt target mTORC1 is activated during HSV-1 infection, its activation is not induced by pUL46, suggesting that other viral proteins activate Akt signaling as well (21). The varicella-zoster virus (VZV) homolog of pUL46, ORF12, also induces the PI3K-Akt pathway by binding to the PI3K p85 subunit. It was shown that cells infected with an ORF12-deficient VZV mutant had a reduced cell population in the M phase compared to wild-type-virus-infected cells, indicating that VZV influences cell cycle progression by manipulating PI3K-Akt signaling via ORF12 (23). In addition, VZV ORF12 triggers phosphorylation of the mitogen-activated protein kinases (MAPKs) p38 and ERK1/2, resulting in the activation of the downstream factor AP-1. The ERK1/2 signaling pathway regulates apoptosis induction (24), and in line with this observation, ORF12-deficient VZV proved to be more susceptible to treatment with the apoptosis inducer staurosporine than did wild-type virus (25).
The ERK1/2 and PI3K-Akt signaling cascades are two of the central pathways within the cell. PI3K-Akt is mainly involved in the prevention of apoptosis, but also in the regulation of cell cycle progression or cell survival (reviewed in references 26 and 27), whereas the ERK1/2 pathway mediates cellular processes including proliferation, differentiation, and cell survival, but also induction of apoptosis or stress responses (reviewed in references 28 and 29). ERK1/2 has multiple targets, e.g., transcription factors, cytoskeletal proteins, and MAPK-activated protein kinases, explaining the different functions within the ERK1/2 cascade (reviewed in reference 29). To activate nuclear targets, like the transcription factor Elk-1, phosphorylated ERK1/2 translocates into the nucleus (30). Elk-1 is rapidly phosphorylated by ERK1/2 and regulates the expression of various genes (31–33).
We were interested to analyze whether PrV pUL46 or the mutated isoforms present in the passaged virus mutants may influence cellular signaling pathways. We show here that PrV pUL46 induces the phosphorylation of ERK1/2 and activates Elk-1, a nuclear target of ERK1/2. Deletion of UL46 from either wild-type PrV or PrV-ΔUL34Pass and PrV-ΔUL31Pass genomes reduced Elk-1 activation, whereas deletion of UL46 from the passaged mutants as well as replacement by wild-type UL46 enhanced NEBD, indicating that the mutated pUL46 limits NEBD in a fashion independent of induction of ERK1/2 target genes.
MATERIALS AND METHODS
Cells and viruses.
RK13-SREluc cells, which stably express an Elk-1 reporter inducing firefly luciferase, were established after transfection of rabbit kidney cells (RK13) with pGL4.33[luc2P/SRE/Hygro] (Promega, Mannheim, Germany) and a plasmid expressing Renilla luciferase under a murine cytomegalovirus immediate early promoter (a gift from Günther Keil, Friedrich-Loeffler-Institut) followed by hygromycin B (200 μg/ml; Sigma-Aldrich, Hamburg, Germany) selection. PrV strain Kaplan (PrV-Ka [34]) was used as the parental virus. Passaging of UL31- and UL34-deficient mutants has been described (10, 11), as has generation of deletion mutants PrV-ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46gfp (10), and PrV-ΔUL46w (14). Replacement of PrV-ΔUL31Pass UL46 by green fluorescent protein (GFP) was performed in analogy to PrV-ΔUL34Pass/ΔUL46gfp using pUC-UL27/47gfp (14). To exclude any effects caused by GFP insertion, UL46 deletion mutants without marker gene insertion were constructed. To this end, genomic DNA of PrV-ΔUL31Pass/ΔUL46gfp and PrV-ΔUL34Pass/ΔUL46gfp was cotransfected with pUC19-SphI/UL47 (14), followed by selection of nonfluorescing plaques. For generation of passaged mutants expressing wild-type pUL46, genomic DNA of PrV-ΔUL34Pass/ΔUL46gfp and PrV-ΔUL31Pass/ΔUL46gfp were cotransfected with a 4.3-kbp PstI subfragment of wild-type BamHI fragment 1 containing the complete UL46 open reading frame (ORF) cloned into pUC19, followed by selection of nonfluorescing plaques. Correct deletion of GFP sequences and insertion of wild-type UL46 were verified by restriction enzyme digestion and sequencing after PCR amplification of the corresponding genomic region (data not shown).
Plasmids.
Generation of pcDNA-PrV-UL46 has been described (14). pcDNA-PrV-UL46(ΔUL34Pass) was generated similarly using PrV-ΔUL34Pass genomic DNA as the template and primers UL46For and UL46Rev for amplification. The PCR product was cloned after cleavage with EcoRI and NotI for which restriction sites had been added with the primers and subsequent insertion into appropriately digested mammalian expression vector pcDNA3 (Invitrogen). Generation of pcDNA-HSV-UL46 and pcDNA-VZV-ORF12 has been reported (25).
Plasmid assay.
HEK293T cells, seeded in a 6-well plate, were transfected with 2 μg pcDNA-PrV-UL46, pcDNA-PrV-UL46(ΔUL34Pass), pcDNA-HSV-UL46, or pcDNA-VZV-ORF12 per well using Lipofectamine 2000. After 48 h, the cells were harvested, lysed, and processed for Western blotting. Membranes were incubated with polyclonal rabbit antibodies against p-ERK1/2, ERK1/2, p-Akt, or Akt (Cell Signaling Technology). Bound antibody was detected with peroxidase-conjugated goat anti-rabbit secondary antibodies and subsequent incubation with chemiluminescent substrate and analyzed using an imager (VersaDoc; Bio-Rad).
Western blotting.
For detection of ERK1/2 phosphorylation by immunoblotting, serum-starved RK13 cells were used. To this end, 1 day after seeding the cells to subconfluency, the monolayer was washed three times with phosphate-buffered saline (PBS) and further incubated with serum-free medium for 3 days. To avoid signaling induced by the medium added with the virus stocks, the supernatant of infected cells, after removal of cellular debris, was centrifuged at 20,000 rpm for 1 h, and the virus pellet was washed and dissolved in PBS. Cells were infected by adding purified wild-type PrV-Ka, PrV-ΔUL46, PrV-ΔUL34Pass, or PrV-ΔUL31Pass suspension to the starvation medium at a multiplicity of infection (MOI) of 5. At 0, 0.5, 1, 2, 4, and 6 h postinfection, cells were scraped into the supernatant, pelleted by centrifugation, washed twice with PBS, and lysed in sodium dodecyl sulfate (SDS) sample buffer. Cell lysates were separated on SDS-12% polyacrylamide gels and blotted onto nitrocellulose. Membranes were blocked with 6% skim milk in Tris-buffered saline-Tween 20 (TBS-T) and subsequently incubated with polyclonal rabbit antibody against p-ERK1/2 (Cell Signaling Technology, Frankfurt, Germany). Bound antibody was detected with peroxidase-conjugated goat anti-rabbit secondary antibodies (Dianova, Hamburg, Germany) or, after incubation with anti-α-tubulin antibodies (Sigma-Aldrich, Hamburg, Germany), with secondary peroxidase-conjugated goat anti-mouse antibodies (Dianova, Hamburg, Germany). After incubation with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific), the blots were analyzed using an imager (VersaDoc; Bio-Rad).
For characterization of viral protein profiles, RK13 cells were infected with wild-type PrV-Ka, PrV-ΔUL46gfp, PrV-ΔUL46w, PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, and PrV-ΔUL34Pass/UL46wt at an MOI of 3 or left uninfected. After 18 h, the cells were scraped into the supernatant, and cell lysates were separated on SDS-10 or -12% polyacrylamide gels. After Western blotting, membranes were incubated with monospecific rabbit sera against pUL31 (8), pUL34 (9), pUL38 (unpublished), pUL46 (14), pUL47 (14), pUL48 (35), or pUL49 (36) and processed as described above.
Luciferase assay.
RK13-SREluc cells were seeded to subconfluency and serum starved starting the following day for 72 h as described above. The cells were either mock infected or infected by adding wild-type PrV-Ka, PrV-ΔUL46w, PrV-ΔUL46gfp, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/UL46wt, PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, or PrV-ΔUL31Pass/UL46wt virus suspension to the starvation medium at an MOI of 3. At 7 h postinfection (p.i.), the cells were washed once with PBS and then lysed for 15 min with lysing buffer (125 mM Tris-HCl [pH 7.8], 10 mM EDTA, 10 mM dithiothreitol [DTT], 50% glycerol, and 5% Triton X-100). The supernatant was diluted, when necessary, and firefly luciferase (a measure of Elk-1 activation) and Renilla luciferase activities were measured using the Dual Luciferase Reporter system (Promega, Mannheim, Germany). The Renilla luciferase activity in infected cells was normalized to the value of mock-infected cells to allow calculation of the corresponding firefly luciferase activity. Mean values of 3 to 6 independent duplicate experiments and the corresponding standard deviations are given. For statistical analysis, the repeated-measures analysis of variance (ANOVA) with a Tukey posttest was used (*, P < 0.05; **, P < 0.01; and ***, P < 0.001).
One-step replication kinetics.
RK13 cells were infected with wild-type PrV-Ka, PrV-ΔUL46gfp, PrV-ΔUL46w, PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, and PrV-ΔUL34Pass/UL46wt at an MOI of 3 and harvested at different times after a low-pH treatment (37). Cells were scraped into the supernatant and lysed by freezing at −80°C and thawing. Cellular debris was removed by centrifugation, and infectivity titers were determined on RK13 cells. The mean values of three independent experiments were calculated and plotted. The corresponding standard deviations are given.
Electron microscopy.
RK13 cells were infected with PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, and PrV-ΔUL34Pass/UL46wt at an MOI of 1. After 1 h, the inoculum was replaced by fresh medium. At 14 to 18 h p.i., the cells were fixed and processed for electron microscopy as described before (9). For quantitation of NEBD, approximately 70 nuclei per virus infection were counted and differentiated into ruptured or intact nuclei. Quantitation was performed in two independent experiments. For statistical analysis, ANOVA with a Tukey posttest was used (*, P < 0.05; **, P < 0.01; and ***, P < 0.001).
RESULTS
ERK1/2 is activated by wild-type and PrV-ΔUL34Pass pUL46.
The genes encoding the tegument protein pUL46 isoforms in PrV-ΔUL31Pass and PrV-ΔUL34Pass both comprise sequence duplications. While PrV-ΔUL34Pass expresses a pUL46 with a duplication of aa 514 to 542, the sequence duplication in PrV-ΔUL31Pass resulted in a frameshift after codon 662 with a stop after codon 725 (10). Recently, it was shown that overexpression of the VZV pUL46 homolog ORF12 induces ERK1/2 phosphorylation (25). To elucidate if the expression of PrV-Ka pUL46 and the PrV-ΔUL34Pass pUL46 isoform also results in phosphorylation of ERK1/2, plasmids expressing wild-type PrV-Ka and PrV-ΔUL34Pass UL46, VZV ORF12, HSV-1 UL46, or the empty vector pcDNA3.1 were transfected into cells. After 48 h, the cell lysates were harvested, blotted onto nitrocellulose, and analyzed with an antibody specific for p-ERK1/2. Transfection of plasmids expressing wild-type or PrV-ΔUL34Pass pUL46 resulted in a similar signal of p-ERK1/2 (Fig. 1, lanes 2 and 3), although this was weaker than in VZV ORF12-transfected cells (lane 5). HSV-1 pUL46-transfected cells showed only a very faint signal for p-ERK1/2 (lane 4), while no p-ERK1/2 reactivity was detected after transfection of the control vector pcDNA3.1 (lane 1). Antibody against ERK1/2 was used as a loading control.
FIG 1.
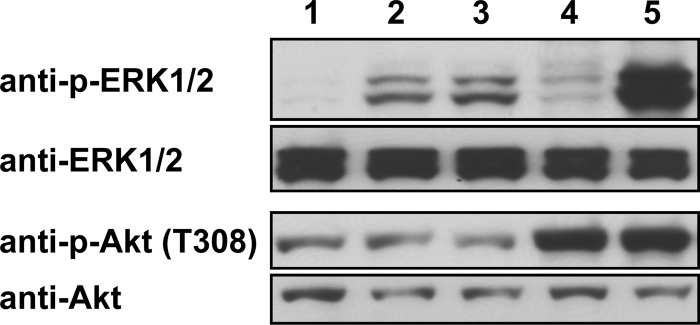
Activation of ERK1/2 and Akt by transient expression of pUL46 homologs. HEK293T cells were transfected with plasmids expressing wild-type PrV-Ka UL46 (lane 2), PrV-ΔUL34Pass UL46 (lane 3), HSV-1 UL46 (lane 4), VZV ORF12 (lane 5), or the empty pcDNA3.1 vector (lane 1) using Lipofectamine 2000. At 48 h posttransfection, cells were harvested, lysed for Western blotting, and analyzed with antisera as indicated on the left.
The VZV ORF12 protein, as well as the homologous HSV-1 pUL46, also activates Akt, which functions in various signaling pathways (21, 23). To test if PrV pUL46 could also induce phosphorylation of Akt, the same samples were incubated with a p-Akt-specific antibody. In contrast to HSV-1 pUL46 (Fig. 1, lane 4) and VZV ORF12 (lane 5), which both induced Akt phosphorylation, pUL46 of wild-type PrV-Ka (lane 2) and PrV-ΔUL34Pass (lane 3) did not result in enhanced phosphorylation of Akt compared to the negative control (lane 1). As a loading control, antibody against Akt was used.
Infection with PrV mutants lacking pUL46 results in reduced ERK1/2 phosphorylation.
To determine the effect of PrV pUL46 on ERK1/2 activation during virus infection, we infected serum-starved RK13 cells with virus-enriched suspensions in PBS of wild-type PrV-Ka and PrV-ΔUL46w at an MOI of 5. Cell lysates were harvested after the times indicated, blotted, and analyzed with an antibody against p-ERK1/2. As a loading control, α-tubulin was used since the corresponding ERK1/2-specific antibody did not react well with RK13 cell lysates (data not shown). ERK1/2 activation during wild-type PrV-Ka infection has already been described (38). Consistent with these data, we found that ERK1/2 phosphorylation increased with a first peak at 30 min after infection and a second peak approximately 4 h after infection. The phosphorylation of ERK1/2 after infection of starved RK13 cells with PrV-ΔUL46w virions was significantly weaker, with no peak after 30 min, while signal intensity increased 4 to 6 h p.i. (Fig. 2A). This finding supports the observation in the transfection assay that PrV pUL46 is able to induce ERK1/2 phosphorylation.
FIG 2.

Activation of the ERK1/2 signaling cascade. (A) RK13 cells were infected with wild-type PrV-Ka (WT) or PrV-ΔUL46w preparations in PBS at an MOI of 5 and harvested at times indicated. Cell lysates were separated on SDS-polyacrylamide gels (12%), blotted onto nitrocellulose membranes, and incubated with antisera as indicated on the right. Molecular masses of marker proteins (in kDa) are shown on the left. (B) RK13-SREluc cells were infected at an MOI of 3 for 7 h with wild-type PrV-Ka (WT), PrV-ΔUL46w, and PrV-ΔUL46gfp or mock infected. Cells were lysed, and firefly luciferase (as readout for Elk-1 activation) and Renilla luciferase activity was measured. Mean values and standard deviations of five independent experiments performed in duplicates are given. For statistical analysis, the repeated-measures ANOVA with a Tukey posttest was used (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (C and D) RK13 cells were infected with wild-type PrV-Ka (C) and PrV-ΔUL31Pass (D) or with wild-type PrV-Ka (WT) and PrV-ΔUL34Pass at an MOI of 5. At times indicated, cells were harvested; cell lysates were separated on SDS-polyacrylamide gels (12%), blotted, and incubated with antisera as indicated. (E) RK13-SREluc cells were mock infected or infected with wild-type PrV, PrV-ΔUL31Pass, or PrV-ΔUL34Pass at an MOI of 3. At 7 h p.i., cells were lysed, and firefly luciferase activity (as readout for Elk-1 activation) and Renilla luciferase activity were measured. Mean values of six independent experiments performed in duplicates and the corresponding standard deviations are shown. For statistical analysis, the repeated-measures ANOVA and a Tukey posttest were used (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
To analyze transcription of ERK1/2 target genes, we generated a cell line, RK13-SREluc, stably expressing an Elk-1 reporter inducing firefly luciferase. As an internal standard, the cell line also expresses Renilla luciferase. The cells were starved for 72 h with serum-free medium as described above and infected by adding the virus suspensions to the serum-free medium, or they were mock infected. Maximum firefly luciferase activity was detected 7 h postinfection, when cells were lysed and the activity of firefly and Renilla luciferases was measured. In HSV-1, a US3 deletion mutation was shown to significantly induce ERK1/2 phosphorylation (39). For control of our assay, a PrV-ΔUS3 deletion mutant was included, and an approximate 2.5-fold luciferase induction was observed (data not shown). When the cells were infected with wild-type PrV-Ka, PrV-ΔUL46gfp, or PrV-ΔUL46w at an MOI of 3, the expression of ERK1/2 target genes, reflected by luciferase activation, was induced and compared to that in mock-infected cells. While PrV-Ka showed approximately 2-fold-higher values, activation of luciferase was significantly reduced after infection with PrV-ΔUL46w and PrV-ΔUL46gfp (Fig. 2B), again indicating that pUL46 supports ERK1/2-activation.
Infection with PrV-ΔUL34Pass, but not with PrV-ΔUL31Pass, reduces ERK1/2 phosphorylation.
Inhibitors roscovitine and U0126, which impair the ERK1/2 signaling pathway, affected replication of the two passaged viruses to a similar extent, while they had less effect on wild-type PrV-Ka (10). PrV-ΔUL31Pass and PrV-ΔUL34Pass carry mutations in the UL46 gene, suggesting a connection between the activation of the ERK1/2 signaling pathway and NEBD. To elucidate how infection with the passaged viruses influences the activation of ERK1/2, serum-starved RK13 cells were infected at an MOI of 5 with wild-type PrV-Ka, PrV-ΔUL31Pass, or PrV-ΔUL34Pass virus preparations in PBS. Cell lysates were harvested at the indicated times and analyzed using antibodies against p-ERK1/2 and α-tubulin. While infection with PrV-ΔUL31Pass resulted in a signal pattern for p-ERK1/2 similar to that of wild-type PrV-Ka, p-ERK1/2 was reduced after infection with PrV-ΔUL34Pass with no discernible peaks early or late after infection (Fig. 2C and D). To confirm the results obtained by Western blotting and analyze the transcription of ERK1/2 target genes, the Elk-1 reporter cell line was starved for 72 h and infected with wild-type PrV-Ka, PrV-ΔUL31Pass, or PrV-ΔUL34Pass at an MOI of 3 or mock infected. At 7 h postinfection, the cells were lysed, and firefly and Renilla luciferase activities were measured. PrV-Ka, as well as PrV-ΔUL31Pass and PrV-ΔUL34Pass, significantly induced luciferase activity compared to the mock-infected control. While values for PrV-ΔUL31Pass were rather similar to those of wild-type PrV-Ka, infection with PrV-ΔUL34Pass resulted in reduced luciferase levels (Fig. 2E).
PrV-ΔUL34Pass and PrV-ΔUL31Pass lacking UL46 show reduced activation of ERK1/2 target genes.
To test the influence of mutated pUL46 in the passaged viruses on induction of ERK1/2 target genes, UL46 was either deleted from the genomes of PrV-ΔUL31Pass and PrV-ΔUL34Pass or replaced by wild-type UL46.
Protein expression of the newly generated virus mutants was examined by Western blotting. PrV-ΔUL31Pass and the mutants derived thereof (Fig. 3, lanes 5 to 8) do not express pUL31, as expected. Similarly, the PrV-ΔUL34Pass mutants (lanes 9 to 12) lack pUL34. pUL46 is absent from PrV-ΔUL46gfp (lane 3), PrV-ΔUL46w (lane 4), PrV-ΔUL31Pass/ΔUL46gfp (lane 6), PrV-ΔUL31Pass/ΔUL46w (lane 7), PrV-ΔUL34Pass/ΔUL46gfp (lane 10), and PrV-ΔUL34Pass/ΔUL46w (lane 11). The duplication in PrV-ΔUL34Pass pUL46 results in a shift in molecular mass compared to wild-type pUL46 (lane 9). Expression of pUL47 and pUL48 is reduced in the GFP-expressing UL46-negative mutants (lanes 3, 6, and 10) but not in the markerless deletion mutants. pUL38 served as a loading control, and pUL49 was used to differentiate the passaged viruses and the mutants derived thereof. pUL49 of PrV-ΔUL31Pass contains a mutation resulting in a smaller expression product (lanes 5 to 8), and UL49 is deleted in PrV-ΔUL34Pass (lanes 9 to 12).
FIG 3.

Protein expression of PrV-ΔUL31Pass and PrV-ΔUL34Pass mutants. RK13 cells were infected with wild-type PrV-Ka (lane 2), PrV-ΔUL46gfp (lane 3), PrV-ΔUL46w (lane 4), PrV-ΔUL31Pass (lane 5), PrV-ΔUL31Pass/ΔUL46gfp (lane 6), PrV-ΔUL31Pass/ΔUL46w (lane 7), PrV-ΔUL31Pass/UL46wt (lane 8), PrV-ΔUL34Pass (lane 9), PrV-ΔUL34Pass/ΔUL46gfp (lane 10), PrV-ΔUL34Pass/ΔUL46w (lane 11), and PrV-ΔUL34Pass/UL46wt (lane 12) at an MOI of 3 or were left uninfected (lane 1). At 18 h postinfection, cell lysates were harvested, separated on SDS-polyacryamide gels (10 or 12%), blotted, and incubated with antisera as indicated on the right. Molecular masses (in kDa) of marker proteins are indicated on the left.
The mutant viruses were further tested for in vitro replication. To this end, RK13 cells were infected at an MOI of 3 and scraped into the supernatant at various times after infection. Titration on RK13 cells showed that wild-type PrV-Ka and PrV-ΔUL46w replicated to comparable titers, whereas maximum titers of PrV-ΔUL46gfp are reduced approximately 10-fold (Fig. 4A). Similar results were obtained for mutants derived from PrV-ΔUL31Pass (Fig. 4B) and PrV-ΔUL34Pass (Fig. 4C). Whereas PrV-ΔUL31Pass/ΔUL46w and PrV-ΔUL31Pass/UL46wt as well as PrV-ΔUL34Pass/ΔUL46w and PrV-ΔUL34Pass/UL46wt reached similar titers, titers of PrV-ΔUL31Pass/ΔUL46gfp and PrV-ΔUL34Pass/ΔUL46gfp were reduced about 10-fold, most likely due to the effect of the GFP insertion on UL47 and UL48 expression.
FIG 4.

Replication kinetics of UL46-mutants. RK13 cells were infected with wild-type PrV-Ka, PrV-ΔUL46gfp, and PrV-ΔUL46w (A), PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, and PrV-ΔUL31Pass/UL46wt (B) or PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, and PrV-ΔUL34Pass/UL46wt (C) at an MOI of 3. At the time points indicated, cells were scraped into the supernatant, and virus progeny titers were determined on RK13 cells. Mean values of three independent assays and the corresponding standard deviations are indicated.
To analyze the induction of ERK1/2 target genes by these mutants, the starved Elk-1 reporter cells were either mock infected or infected with wild-type PrV-Ka, PrV-ΔUL46gfp, PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, or PrV-ΔUL34Pass/UL46wt at an MOI of 3. At 7 h postinfection, the cells were lysed, and firefly and Renilla luciferase activity was measured. As previously shown for PrV-ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46gfp, and PrV-ΔUL34Pass/ΔUL46gfp, infected cells exhibited a reduced luciferase activity in comparison to infection with the corresponding parental viruses, indicating that pUL46, independent of the mutations present in the passaged mutants, is able to induce ERK1/2 activation (Fig. 5). No difference could be observed in the luciferase activity induced by PrV-ΔUL31Pass/UL46wt compared to PrV-ΔUL31Pass, or PrV-ΔUL34Pass/UL46wt compared to PrV-ΔUL34Pass, demonstrating that the mutated pUL46 isoforms and wild-type pUL46 activate ERK1/2 equally well (Fig. 5). Infection with PrV-ΔUL46gfp or PrV-ΔUL46w reduced luciferase levels to the same extent (Fig. 2B), excluding an effect of GFP insertion on luciferase levels.
FIG 5.

ERK1/2 activation by PrV-ΔUL31Pass or PrV-ΔUL34Pass UL46-mutants. RK13-SREluc cells were infected at an MOI of 3 with wild-type PrV-Ka, PrV-ΔUL46gfp, PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, or PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, or PrV-ΔUL34Pass/UL46wt or were mock infected. At 7 h p.i., cells were lysed, and firefly luciferase (as readout for Elk-1 activation) and Renilla luciferase activity was measured. Mean values and corresponding standard deviations of three independent experiments performed in duplicate are shown. For statistical analysis the repeated-measures ANOVA with a Tukey posttest was used (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Deletion of UL46 from PrV-ΔUL31Pass and PrV-ΔUL34Pass enhances NEBD.
To elucidate if pUL46 regulates NEBD, RK13 cells were infected with PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, or PrV-ΔUL34Pass/UL46wt at an MOI of 1 for 14 to 18 h. Ultrastructural analysis demonstrated NEBD in cells infected with PrV-ΔUL31Pass (Fig. 6A) and PrV-ΔUL31Pass/ΔUL46gfp (Fig. 6B), as well as PrV-ΔUL34Pass (Fig. 6C) and PrV-ΔUL34Pass/ΔUL46gfp (Fig. 6D). No morphological difference in NEBD between the UL46 deletion mutants expressing GFP, the markerless UL46 deletion mutants, or wild-type UL46-expressing mutants was observed (data not shown). Subsequent quantitation demonstrated that in cells infected with PrV-ΔUL31Pass and PrV-ΔUL34Pass, as published before (10, 11), approximately one-half of the nuclei showed NEBD, whereas after deletion of UL46 or substitution with wild-type UL46 the percentage of cells showing NEBD increased to approximately 60% in PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, and PrV-ΔUL31Pass/UL46wt and to about 75% in PrV-ΔUL34Pass-derived viruses PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, and PrV-ΔUL34Pass/UL46wt (Fig. 6E).
FIG 6.

Electron microscopic analysis of cells infected with UL46-deficient PrV-ΔUL31Pass and PrV-ΔUL34Pass mutants. RK13 cells were infected with PrV-ΔUL31Pass (A), PrV-ΔUL31Pass/ΔUL46gfp (B), PrV-ΔUL34Pass (C), and PrV-ΔUL34Pass/ΔUL46gfp (D) at an MOI of 1 for 14 to 18 h and subsequently processed for electron microscopy. Bars represent 4 μm, and arrows indicate NEBD. N, nucleus. (E) For quantitation of NEBD in cells infected with PrV-ΔUL31Pass, PrV-ΔUL31Pass/ΔUL46gfp, PrV-ΔUL31Pass/ΔUL46w, PrV-ΔUL31Pass/UL46wt, PrV-ΔUL34Pass, PrV-ΔUL34Pass/ΔUL46gfp, PrV-ΔUL34Pass/ΔUL46w, and PrV-ΔUL34Pass/UL46wt, approximately 70 nuclei per virus infection were assessed for ruptured or intact nuclear envelopes. Quantitation was performed twice in independent experiments, and subsequently ANOVA with a Tukey posttest was used for statistical analysis (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
DISCUSSION
Herpesviruses leave the nucleus via an envelopment/de-envelopment pathway that requires the presence of the nuclear egress complex (NEC), consisting of viral proteins pUL31 and pUL34. Although PrV mutants lacking either protein reach only very low titers (8, 9), reversion analysis by serial passaging in cell culture yielded virus mutants with the ability to replicate efficiently in the absence of the NEC by inducing NEBD (10, 11).
NEBD is of great importance in uninfected cells, e.g., during mitosis or apoptosis. Inhibitor studies demonstrated that the ERK1/2 signaling cascade could be involved in the induction of NEBD by PrV (10). Parallel whole-genome sequencing revealed seven surprisingly congruent mutations in PrV-ΔUL34Pass and PrV-ΔUL31Pass, including alterations in UL46 encoding a major tegument protein (10). VZV ORF12, which is homologous to PrV and HSV-1 pUL46, activates the ERK1/2 signaling cascade, as well as the PI3K-Akt pathway (23, 25), whereas HSV-1 pUL46 activates the PI3K-Akt cascade (21, 22).
In contrast to VZV ORF12 and HSV-1 pUL46, neither wild-type PrV-Ka nor PrV-ΔUL34Pass pUL46 induced the phosphorylation of Akt, whereas phosphorylation of ERK1/2, as well as the expression of ERK1/2 target genes as measured with the luciferase reporter assay, was induced by PrV pUL46 independent of the mutations present in the pUL46 isoforms of the passaged, NEBD-inducing viruses. However, activation of the ERK1/2 signaling cascade via pUL46 had no profound effect on PrV replication, since PrV-ΔUL46w replicated to wild-type levels with no obvious defects in nuclear egress, virion morphogenesis, or virus release (14). Similar results were also obtained for the passaged viruses. Deletion of UL46 or substitution of UL46 from the passaged viruses by wild-type UL46 did not have a measurable effect on virus replication. Nevertheless, mutants lacking UL46 or expressing wild-type UL46 showed significantly enhanced NEBD in comparison to parental PrV-ΔUL31Pass and PrV-ΔUL34Pass, although the extent of increase differed between the two viral lineages. This finding indicates that the mutations in PrV-ΔUL31Pass and PrV-ΔUL34Pass pUL46 influence NEBD and may limit NEBD to restrict a complete breakdown of cellular compartmentalization. Whether wild-type pUL46 has an effect on the integrity of the nuclear envelope in viral backgrounds other than the passaged viruses remains to be analyzed.
Similar to what was seen in wild-type PrV, deletion of UL46 from the passaged viruses resulted in reduced ERK1/2 activation. In contrast, deletion of UL46 as well as expression of wild-type pUL46 significantly enhanced NEBD, demonstrating that the observed increase in NEBD occurs independently of ERK1/2 signaling.
We also observed a difference between PrV-ΔUL31Pass and PrV-ΔUL34Pass regarding ERK1/2 activation. Whereas PrV-ΔUL31Pass induced ERK1/2 phosphorylation to a level comparable to that of wild-type PrV-Ka, infection with PrV-ΔUL34Pass led to a considerably lower activation. This cannot be explained by mutations in the UL46 genes of the passaged mutants, because replacement of the mutated UL46 from either passaged virus with wild-type UL46 did not alter the ability to activate Elk-1 beyond the level observed in the parental viruses.
It is likely that one of the other genes mutated in PrV-ΔUL34Pass (UL50, UL49, UL46, UL27, UL29, UL41, UL26, UL21, US1, US7, US9, or US2 genes) is responsible for the impaired ERK1/2 activation. To date, only the tegument protein pUS2 has been shown to manipulate ERK1/2. The US2 gene of PrV-ΔUL34Pass contains a frameshift after codon 180, while US2 is deleted in PrV-ΔUL31Pass. Whereas HSV-1 pUS2 does not interact with ERK1/2 (40), PrV pUS2 binds ERK1/2 in the common docking domain and maintains it at the plasma membrane, thus inhibiting the activation of ERK1/2 target genes (38, 41). However, in cells infected with wild-type PrV Becker or a US2 null mutant, ERK1/2 is activated in a similar fashion immediately after infection, indicating that ERK1/2 activation is independent of pUS2 (38).
Deletion of ORF12 from VZV increases sensitivity to apoptosis induction (25). However, neither treatment with a pancaspase inhibitor nor treatment with an apoptosis inducer affected replication of PrV-ΔUL31Pass or PrV-ΔUL34Pass to a greater extent than PrV-Ka (10). In contrast, the cell cycle inhibitor roscovitine and the MEK inhibitor U0126 impaired the replication of both passaged viruses, whereas wild-type PrV-Ka was less sensitive (10). It is therefore much more likely that the passaged viruses manipulate mitosis to induce NEBD. More specifically, PrV-ΔUL31Pass and PrV-ΔUL34Pass possibly influence the spindle assembly checkpoint during mitosis. While the nuclear envelope is fragmented, the cell cycle stops to ensure that the previous phase has been completed correctly and that conditions are optimal to continue. Many viruses have been shown to manipulate this checkpoint to optimize viral replication (42), and herpesvirus pUL46 may also manipulate this checkpoint to regulate NEBD.
In summary, the data presented here demonstrate that PrV pUL46 has two important functions. First, it is able to induce phosphorylation of the extracellular signal-regulated kinase ERK1/2, and second, it regulates maintenance of the integrity of the nuclear envelope. The mutations in pUL46 of the passaged mutants restrict the extent of NEBD induced during infection with PrV-ΔUL31Pass and PrV-ΔUL34Pass. This seemingly contradictory function may be necessary to curb excessive NEBD, resulting in the destruction of cellular compartmentalization, which is detrimental for virus replication but, at the same time, allows nuclear escape in the absence of the NEC.
ACKNOWLEDGMENTS
We thank C. Meinke, M. Sell, and P. Meyer for expert technical assistance, M. Jörn for photographic help, and M. Ziller for help with statistical analysis.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG Me 854/12-1) and the intramural research program of the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print 12 March 2014
REFERENCES
- 1.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell. Microbiol. 15:170–178. 10.1111/cmi.12044 [DOI] [PubMed] [Google Scholar]
- 2.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537–1547. 10.1128/JVI.76.4.1537-1547.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. 10.1038/nrmicro2559 [DOI] [PubMed] [Google Scholar]
- 4.Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5:e1000275. 10.1371/journal.ppat.1000275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470. 10.1128/JVI.00380-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. 10.1126/science.1071506 [DOI] [PubMed] [Google Scholar]
- 7.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 80:494–504. 10.1128/JVI.80.1.494-504.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378. 10.1128/JVI.76.1.364-378.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063–10073. 10.1128/JVI.74.21.10063-10073.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grimm KS, Klupp BG, Granzow H, Muller FM, Fuchs W, Mettenleiter TC. 2012. Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J. Virol. 86:6512–6521. 10.1128/JVI.00068-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klupp BG, Granzow H, Mettenleiter TC. 2011. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 85:8285–8292. 10.1128/JVI.00741-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy MA, Bucks MA, O'Regan KJ, Courtney RJ. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376:279–289. 10.1016/j.virol.2008.03.018 [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, McKnight JL. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kopp M, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. 2002. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 76:8820–8833. 10.1128/JVI.76.17.8820-8833.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. 10.1016/j.virol.2008.05.035 [DOI] [PubMed] [Google Scholar]
- 16.Kato K, Daikoku T, Goshima F, Kume H, Yamaki K, Nishiyama Y. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149–2162. 10.1007/s007050070045 [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571. 10.1128/JVI.79.15.9566-9571.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strunk U, Saffran HA, Wu FW, Smiley JR. 2013. Role of herpes simplex virus VP11/12 tyrosine-based motifs in binding and activation of the Src family kinase Lck and recruitment of p85, Grb2, and Shc. J. Virol. 87:11276–11286. 10.1128/JVI.01702-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zahariadis G, Wagner MJ, Doepker RC, Maciejko JM, Crider CM, Jerome KR, Smiley JR. 2008. Cell-type-specific tyrosine phosphorylation of the herpes simplex virus tegument protein VP11/12 encoded by gene UL46. J. Virol. 82:6098–6108. 10.1128/JVI.02121-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner MJ, Smiley JR. 2011. Herpes simplex virus requires VP11/12 to activate Src family kinase-phosphoinositide 3-kinase-Akt signaling. J. Virol. 85:2803–2812. 10.1128/JVI.01877-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner MJ, Smiley JR. 2009. Herpes simplex virus requires VP11/12 to induce phosphorylation of the activation loop tyrosine (Y394) of the Src family kinase Lck in T lymphocytes. J. Virol. 83:12452–12461. 10.1128/JVI.01364-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Cohen JI. 2013. Varicella-zoster virus ORF12 protein activates the phosphatidylinositol 3-kinase/Akt pathway to regulate cell cycle progression. J. Virol. 87:1842–1848. 10.1128/JVI.02395-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331. 10.1126/science.270.5240.1326 [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Li Q, Dowdell K, Fischer ER, Cohen JI. 2012. Varicella-zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J. Virol. 86:3143–3151. 10.1128/JVI.06923-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. 2003. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia 17:590–603. 10.1038/sj.leu.2402824 [DOI] [PubMed] [Google Scholar]
- 27.Hemmings BA, Restuccia DF. 2012. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 4:a011189. 10.1101/cshperspect.a011189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wortzel I, Seger R. 2011. The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2:195–209. 10.1177/1947601911407328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roskoski R., Jr 2012. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66:105–143. 10.1016/j.phrs.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 30.Chen RH, Sarnecki C, Blenis J. 1992. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol. 12:915–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boros J, Donaldson IJ, O'Donnell A, Odrowaz ZA, Zeef L, Lupien M, Meyer CA, Liu XS, Brown M, Sharrocks AD. 2009. Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res. 19:1963–1973. 10.1101/gr.093047.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cruzalegui FH, Cano E, Treisman R. 1999. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 18:7948–7957 [DOI] [PubMed] [Google Scholar]
- 33.Marais R, Wynne J, Treisman R. 1993. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 73:381–393. 10.1016/0092-8674(93)90237-K [DOI] [PubMed] [Google Scholar]
- 34.Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407. 10.1016/0042-6822(59)90068-6 [DOI] [PubMed] [Google Scholar]
- 35.Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 76:6729–6742. 10.1128/JVI.76.13.6729-6742.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brack AR, Klupp BG, Granzow H, Tirabassi R, Enquist LW, Mettenleiter TC. 2000. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 74:4004–4016. 10.1128/JVI.74.9.4004-4016.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625. 10.1016/0042-6822(89)90635-1 [DOI] [PubMed] [Google Scholar]
- 38.Lyman MG, Randall JA, Calton CM, Banfield BW. 2006. Localization of ERK/MAP kinase is regulated by the alphaherpesvirus tegument protein Us2. J. Virol. 80:7159–7168. 10.1128/JVI.00592-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chuluunbaatar U, Roller R, Mohr I. 2012. Suppression of extracellular signal-regulated kinase activity in herpes simplex virus 1-infected cells by the Us3 protein kinase. J. Virol. 86:7771–7776. 10.1128/JVI.00622-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang MH, Roy BB, Finnen RL, Le Sage V, Johnston SM, Zhang H, Banfield BW. 2013. The Us2 gene product of herpes simplex virus 2 is a membrane associated ubiquitin interacting protein. J. Virol. 87:9590–9603. 10.1128/JVI.00994-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang MH, Banfield BW. 2010. Pseudorabies virus tegument protein Us2 recruits the mitogen-activated protein kinase extracellular-regulated kinase (ERK) to membranes through interaction with the ERK common docking domain. J. Virol. 84:8398–8408. 10.1128/JVI.00794-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaurushiya MS, Weitzman MD. 2009. Viral manipulation of DNA repair and cell cycle checkpoints. DNA Repair (Amst.) 8:1166–1176. 10.1016/j.dnarep.2009.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]