

ABSTRACT
Enteroviruses (EVs) are a genetically and antigenically diverse group of viruses infecting humans. A mostly distinct set of EV variants have additionally been documented to infect wild apes and several, primarily captive, Old World monkey (OWM) species. To investigate the prevalence and genetic characteristics of EVs infecting OWMs in the wild, fecal samples from mandrills (Mandrillus sphinx) and other species collected in remote regions of southern Cameroon were screened for EV RNA. Remarkably high rates of EV positivity were detected in M. sphinx (100 of 102 screened), Cercocebus torquatus (7/7), and Cercopithecus cephus (2/4), with high viral loads indicative of active infection. Genetic characterization in VP4/VP2 and VP1 regions allowed EV variants to be assigned to simian species H (EV-H) and EV-J (including one or more new types), while seven matched simian EV-B variants, SA5 and EV110 (chimpanzee). Sequences from the remaining 70 formed a new genetic group distinct in VP4/2 and VP1 region from all currently recognized human or simian EV species. Complete genome sequences were obtained from three to determine their species assignment. In common with EV-J and the EV-A A13 isolate, new group sequences were chimeric, being most closely related to EV-A in capsid genes and to EV-B in the nonstructural gene region. Further recombination events created different groupings in 5′ and 3′ untranslated regions. While clearly a distinct EV group, the hybrid nature of new variants prevented their unambiguous classification as either members of a new species or as divergent members of EV-A using current International Committee on Taxonomy of Viruses (ICTV) assignment criteria.
IMPORTANCE This study is the first large-scale investigation of the frequency of infection and diversity of enteroviruses (EVs) infecting monkeys (primarily mandrills) in the wild. Our findings demonstrate extremely high frequencies of active infection (95%) among mandrills and other Old World monkey species inhabiting remote regions of Cameroon without human contact. EV variants detected were distinct from those infecting human populations, comprising members of enterovirus species B, J, and H and a large novel group of viruses most closely related to species A in the P1 region. The viral sequences obtained contribute substantially to our growing understanding of the genetic diversity of EVs and the existence of interspecies chimerism that characterizes the novel variants in the current study, as well as in previously characterized species A and J viruses infecting monkeys. The latter findings will contribute to future development of consensus criteria for species assignments in enteroviruses and other picornavirus genera.
INTRODUCTION
Enteroviruses (EVs), members of the genus Enterovirus within the family Picornaviridae, are notable for their genetic and antigenic diversity with more than 300 types identified thus far. EVs are currently classified into nine species (labeled alphabetically as EV-A to EV-H and EV-J). This subdivision of the Enterovirus genus is based on degrees of sequence divergence, host range, similarities in replication, and a generally observed restriction of recombination between members of the same species (1). The enteroviruses infecting humans comprise EV-A to -D, while the remaining five contain viruses infecting primarily cattle (EV-E and EV-F; both termed bovine enteroviruses), pigs (EV-G; porcine enteroviruses) and monkeys (EV-H and EV-J; both simian enteroviruses) (1). However, EV variants classified as species A, B, and D have also been detected in several monkey and ape species (2–13).
The first simian EVs were discovered in the 1950s and 1960s in primate cell cultures or from primate tissue specimens used in biomedical research. These EVs were obtained from Old World monkey (OWM) species Macaca mulatta (rhesus macaque), Macaca fascicularis (cynomolgus monkey), Chlorocebus aethiops (African vervet monkey), and Papio cynocephalus (baboon) (2–7, 14). Subsequent genetic characterization indicated that some isolates were similar to human viruses (A13, SV19, SV43, and SV46 in species A; SA5 in EV-B), while others were genetically distinct (9–11) and now classified into two separate species (e.g., SV4 and SV28 in EV-H and SV6 in EV-J). More recent genetic characterization of EVs infecting captive primates (mostly rhesus macaques [83%]) in the Yerkes National Primate Center in the United States and at the Dhaka Zoo in Bangladesh identified four further simian EV types (EV92 in species A; EV103, EV112, and EV115 in species J) (13, 15), whereas no simian EVs were detected in synanthropic (e.g., pet) monkeys (12).
To investigate whether EVs also circulated in apes, we recently performed large-scale screening of fecal samples collected from wild populations of chimpanzees (Pan troglodytes) and gorillas (Gorilla gorilla) primarily in Cameroon. From these fecal samples, four EV types (EV76, EV89, and EV119 in species A; EV111 and EV120 in species D) and a novel species B variant (EV110) were identified (16, 17). However, as previously discussed (17), whether these represent endogenous infections in these species or have been acquired from contact with humans or monkeys remains unclear; remaining wild-living chimpanzee and gorilla populations are small and highly fragmented. The overall detection frequencies were however moderate (9 to 11%), and the populations sampled had minimal contact with human populations in sample collection areas.
To understand more about the natural circulation of EVs in wild primate populations, we analyzed a large collection of samples from mandrills (Mandrillus sphinx) and smaller numbers of other OWMs in isolated areas of Cameroon. Mandrills were selected because populations and groupings are large and more likely to support endogenous populations of EVs than apes. Genetic characterization of the extensive numbers of variants detected demonstrated the broad diversity of species and types circulating in these populations in the wild. A wider phylogenetic comparison of complete genome sequences of OWM, ape, and human EVs was performed to determine their species relationships to other simian and human EVs.
MATERIALS AND METHODS
Samples.
A total of 102 mandrill (Mandrillus sphinx), 7 red-capped mangabey (Cercocebus torquatus), and 4 moustached monkey (Cercopithecus cephus) stool samples were included in this study (Table 1; see Tables S1 and S2 in the supplemental material). Samples were collected from natural habitat areas of mandrills—remote and sparsely populated forested southern areas of Cameroon with minimal human contact (18). Samples were preserved in RNAlater (Ambion, Austin, TX), stored at room temperature at base camps for a maximum of 3 weeks, and subsequently transported to a central laboratory in Yaoundé, Cameroon, for storage at −20°C/−80°C. For all fecal samples, the species origin was also determined by mitochondrial DNA (mtDNA) analysis as described previously (19). Fecal DNA was extracted using the QIAamp stool DNA minikit (Qiagen, Valencia, CA).
TABLE 1.
Detection frequencies in study samples using primers from different genome regions
| Species | Detection frequency (no. of positive samples/total no. of samples tested) |
||
|---|---|---|---|
| 5′UTR | VP4/2 | VP1 | |
| Mandrillus sphinx | 100/102 | 88/100 | 39/88 |
| Cercocebus torquatus | 7/7 | 3/7 | 0/3 |
| Cercopithecus cephus | 2/4 | 2/2 | 0/2 |
Sample extraction and amplification.
RNA was extracted from fecal samples as previously described (20) and tested for enterovirus (EV) by one-step reverse transcriptase (RT)-PCR assay targeting the 5′ untranslated regions (5′UTRs) (see Table S3A in the supplemental material) (21). To estimate viral loads in EV-positive samples, samples were retested by real-time PCR calibrated using defined copy numbers of EV RNA transcripts (22). Additional primer sets were used to obtain sequence data from VP4 and VP1 region as described (Table S3B) (16, 17). The amplification of VP1 required two sets of primers, which amplify overlapping regions from each species, and additional primers specific for the species B variant SA5 (as indicated in the primer name). For each PCR, 6 μl extracted RNA was amplified by a combined RT and first-round PCR using Superscript III one-step RT-PCR system (Invitrogen, United Kingdom), and 1 μl of the first-round reaction was used for the nested PCR with second-round primers. PCR amplification included 30 cycles of denaturation (94°C, 20 s), annealing (50°C, 18 s), and elongation (72°C, 90 s) in a thermal cycler.
Whole-genome sequencing.
The whole genomes of EV variants representing three of the predominant types infecting mandrills were sequenced (samples GR2815, CPML8109, and CPML3961). Primers were designed using sequence data in the VP4 and VP1 regions available from the current study and sequences of viruses in species J. One microliter of cDNA synthesized by using random hexamer primers and Superscript III reverse transcriptase (Invitrogen, United Kingdom) was used as the template for nested PCR. The amplification conditions were the same as those of the second-round PCR mentioned above. Assemblies were created from sets of approximately 1.5-kb sequence fragments in which the regions of overlap were identical between fragments; this prevented generation of possible hybrid viruses from samples from monkeys that may be coinfected with two or more EV variants.
Direct sequencing of PCR products.
Positive second-round PCR products were sequenced in both directions using the inner sense and inner antisense primers used in the second round of amplification, using BigDye Terminator v3.1 (Applied Biosystems). Nucleotide sequences were assembled, annotated, and aligned using the SSE sequence editor version 1.1 (23).
Genetic analysis of EV sequences.
Sequence comparisons were made between the Old World monkey-derived sequences obtained in the current study with all available sequences from VP1, VP4, 3Dpol, 5′UTR, and 3′UTR sequences of variants previously reported for other nonhuman primates. All data sets additionally included a single representative sequence derived from complete genome sequences of each human EV type. Calculation of pairwise distances between sequences and divergence scans were performed using SSE. Phylogenetic trees constructed using maximum likelihood methods as implemented in the MEGA 6 software package (24). The optimum maximum likelihood model (lowest Bayesian information criterion score and typically greatest maximum likelihood value) for each sequence data set was first determined and used for phylogenetic reconstruction. For each genome region (5′UTR, VP4/VP2, VP1, and 3CD), this was general time reversible (GTR) with a gamma (γ) distribution (5 rates) and invariant sites (I) (GTR plus γ plus I). Phylogenetic analysis of each data set used 100 bootstrap resamplings to determine the robustness of grouping.
Nucleotide sequence accession numbers.
Sequences obtained in the current study have been submitted to GenBank and have been assigned the accession numbers KJ420625 to KJ420749.
RESULTS
EV RNA detection frequencies and viral loads.
RNA was extracted from 102 mandrill, 7 red-capped mangabey, and 4 moustached monkey fecal samples and screened for EV RNA sequences using primers targeting a region of the 5′untranslated region (5′UTR). From these fecal samples, 100 of 102 mandrill samples (98%), all red-capped mangabey samples, and two moustached monkey samples were positive. Sampling sites were located throughout southern Cameroon (Fig. 1; see Tables S1 and S2 in the supplemental material). The mean viral loads of EV in mandrill fecal samples were ≈10-fold higher than those from a representative set of human samples tested in the same assay (25) (Fig. 2), with mean threshold cycle (CT) values of 27.5 compared to 29.8 in human samples (P = 0.001 by the Kruskal-Wallis test).
FIG 1.
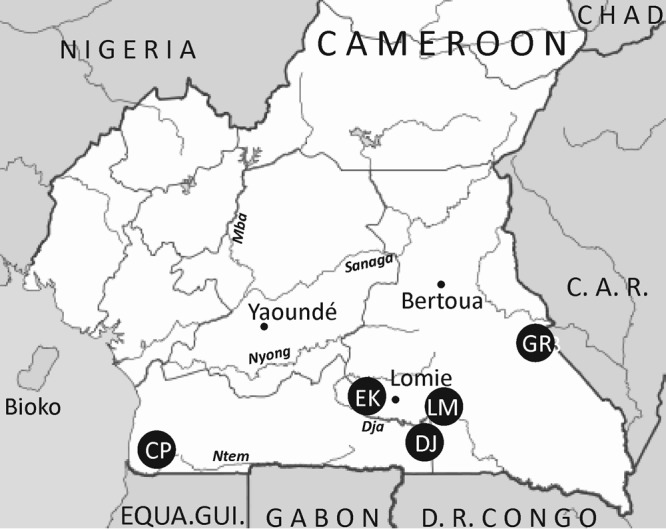
Collection sites in Cameroon of samples used in the current study. The collection sites in Cameroon were abbreviated as follows: CP, Campo, locations Oveng, Melen, and Grothes (CPOV, CPLM, and CPGR, respectively); DJ, Djoum; EK, Ekom; LM, Lomie; GR, Gribi. C. A. R., Central African Republic, EQUA.Gui., Equatorial Guinea, D. R. Congo, Democratic Republic of the Congo.
FIG 2.
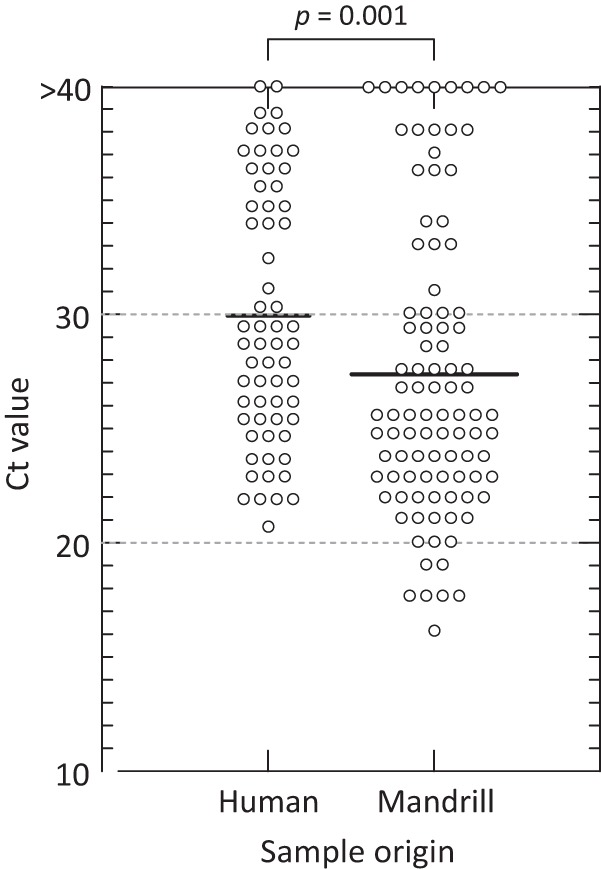
Comparison of CT values of EV RNA detected by real-time PCR in fecal samples collected from mandrills with those in human samples. Each symbol represents the value for one fecal sample. Bars show mean values. Statistical significance was performed using Kruskal-Wallis nonparametric test (above graph).
EV type assignments.
To genetically characterize EV variants infecting these OWMs, sequences were amplified from the VP4/2 region. A total of 88 of 100 samples from mandrills were positive, from which 84 were successfully sequenced, as were 3 of 7 mangabey samples and two Cercopithecus (moustached monkey) samples (Fig. 3A). For the purposes of further type identification and new type assignments, sequences were amplified in the VP1 region from 39 mandrill samples (Fig. 3B).
FIG 3.

Maximum likelihood analysis (GTR plus γ plus I model) of VP4/partial VP2 region (A) and whole VP1 (positions 743 to 1168 and 2477 to 3376, respectively, numbered using the PV3 reference sequence) (B). Sequences amplified from study samples are shown as white and solid black symbols, while those previously assigned within EV species A to D, J, and H are color coded according to the key (human EV-B and EV-C sequences were monophyletic and have been collapsed to clarify presentation of simian sequences). Bootstrap resampling of maximum likelihood (ML) trees was performed to indicate robustness of grouping (values of >70% shown).
The viruses identified fell into three previously identified enterovirus species. Four could be assigned to species H that includes the simian viruses A-2 plaque virus and SEV-A (isolated from the OWM species Macaca mulatta). VP1 sequences showed 14.5 to 15.5% nucleotide sequence divergence from previously characterized simian variants in this species, suggesting that they should be classified as EV-H1. However, the four samples containing species H viruses were collected from the same site on the same day; due to the nature of sampling from the jungle floor and their high sequence identity, it was likely that they originated from the same group or possibly even the same mandrill.
Other variants grouped with the simian enteroviruses SA5 and EV-B110 within species B. Five of these variants could be assigned as EV110 (20.6% to 20.8% divergence in VP1), a recently described EV type detected in a chimpanzee from Cameroon (17), while a further two could be assigned as SA5 (15.5% divergence in VP1) that was originally isolated from a vervet monkey (Cercopithecus aethiops [4]).
VP4/2 sequences of a further 10 variants clustered in species J (7 derived from mandrills and 3 derived from mangabeys), a species into which the simian viruses SV6 (M. mulatta), EV103 (Macaca nemestrina), and EV108 (Papio cynocephalus) are currently assigned. From these, a VP1 sequence was obtained from sample CPOV3336, which was >25% divergent from assigned types within this species. It is likely, however, that several other variants correspond to further EV-J types based on their sequence divergence in VP4/2.
The remainder of viruses form a monophyletic group in both VP4/2 (67 mandrill sequences and 2 Cercopithecus sequences) and VP1 (30 mandrill sequences). In the VP1 region, sequences showed substantial nucleotide sequence divergence from each other and supported their assignment into 5 types (Fig. 3B) based on >25% pairwise nucleotide distances. How these should be named is dependent on their final species assignment, and this remains uncertain based on phylogenetic analysis of VP1 and VP4/2 region sequences.
Species assignment.
To clarify the species assignment of the new, predominantly mandrill, group and investigate its relationships with other simian and human EVs, complete genome sequences were obtained from variants representing 3 of the 5 putative types (samples GR2815, CPML8109, and CPML3961) within the group. This allowed current molecular demarcation criteria of amino acid sequence identity in the polyprotein, P1, and combined nonstructural gene region 2C plus 3CD (1) to be used for species assignments (Table 2).
TABLE 2.
P1, 3CD, and whole-genome amino acid sequence distances between mandrill variants and other species
| Species and subgroup | Amino acid sequence distancea |
||
|---|---|---|---|
| Whole genome | P1 | 2C + 3CD | |
| EV-J | 0.264 | 0.384 | 0.1682 |
| EV-C | 0.403 | 0.514 | 0.2934 |
| EV-A | |||
| All | 0.351 | 0.3714 | 0.3334 |
| A1 | 0.3595 | 0.379 | 0.341 |
| A2 | 0.3235 | 0.346 | 0.3074 |
| EV-B | |||
| All | 0.2981 | 0.5107 | 0.158 |
| Human | 0.2996 | 0.511 | 0.1596 |
| SA5 | 0.2489 | 0.493 | 0.0613 |
| EV-C | 0.4034 | 0.514 | 0.2934 |
| EV-D | 0.375 | 0.463 | 0.2853 |
| EV-H | 0.3905 | 0.465 | 0.3085 |
Amino acid sequence distances below species assignment thresholds are shown in boldface type (complete coding region, 30%; P1, 40%; 2C plus 3CD, 30%).
As suspected from the differences in phylogenetic groupings between the new variants and other EV species (Fig. 3B), amino acid sequence distances between this group and species J and A viruses in P1 were close to but consistently below the 40% previously proposed species assignment threshold (38.4% and 37.9%, respectively; Table 2). Furthermore, if species A viruses were divided into the two subclades apparent in VP1 (one comprising exclusively human serotypes, termed A1), and the other simian and more recently discovered human-derived types EV76, -89, -90, and -91 (termed A2) (26), then further problematic relationships arise. The new mandrill variants showed only 34.5% divergence from the predominantly simian A2 group. A2 variants themselves show only 38.9% divergence from species J.
Sequence divergence in the combined 2C plus 3CD region did not resolve the species status of the mandrill sequences and indeed revealed further inconsistent sequence relationships with other variants. With a proposed 30% amino acid divergence threshold, new variants showed remarkably low divergence from both EV-J (16.8%) and EV-B (15.8%). Furthermore, mandrill variants and the simian species B isolate SA5 showed only 6.1% divergence. These interrelationships are mirrored by the greater than expected sequence similarity of EV-J to EV-B (21.8% divergence). These inconsistencies in sequence relationships in different genome regions were not resolved by comparison of whole polyprotein sequences, where mandrill variants showed below-species divergence thresholds (set at 30%) of 26.4% with EV-J and 29.8% with EV-B.
Information for the mandrill variants that contribute to the other species assignment criteria (1) was largely unresolved or ambiguous, including host range (infects at least two different OWM species), host cell receptors (unknown), and compatibility in proteolytic processing, replication, encapsidation, and genetic recombination (unknown). However, the three variants show a narrowly constrained range of G+C compositions (44.3% to 44.6%; threshold range of 2.5%).
Chimerism in EV genomes.
It is possible that the difficulty associated with a species designation of the new variants using conventional criteria had originated from their possession of chimeric genomes comprising sequences originating from different EV species. To investigate this, relationships between inter- and intraspecies distances in P1 and 3CD were compared (Fig. 4). For the presumed nonchimeric human species A, B, and C sequences, sequence distances in P1 were approximately proportional to those in 3CD (Fig. 4A). Comparison of variants assigned to different species invariably showed distances of >40% in P1 and >25% in 3CD (including EV-D; not shown). The main exception to the linear relationship between distances was the frequently suppressed variability of 3CD sequences between different types within a species. As documented elsewhere, this originates through promiscuous intertype recombination in the nonstructural gene regions, where different types within a species frequently share a pool of less-divergent NS gene region sequences (27–30). Species A and C, however, contain subgroups with more-divergent NS gene region sequences (15% to 23%; Fig. 4A). In species A, these represent distances between subgroups corresponding to the previously described EV-A1 (all human) and -A2 groups (EV76, -89, -90, and -91) (26). In species C, they originate from the previously identified more-divergent variants CAV1, CAV19, and CAV22 (31) and EV104 (32).
FIG 4.
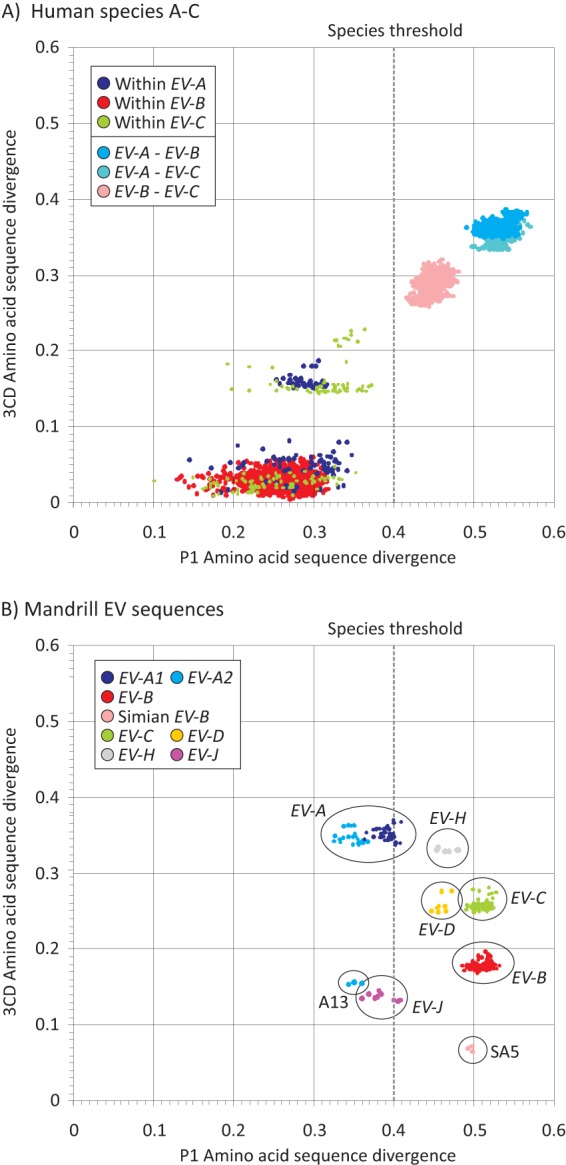
(A) Distributions of pairwise amino acid distances in P1 (positions 743 to 3376) and 3CD regions (positions 5429 to 7360) between representative human-derived sequences within EV-A, EV-B, and EV-C. Color coding has been used to identify within- and between-species sequences distances for each. The 40% amino acid distance threshold for species assignments using P1 is shown as vertical gray broken line. (B) Pairwise distances between three representative mandrill sequences with EV species A to D, J, and H.
An equivalent comparison of sequences from the mandrill group with other species showed combinations of P1 and 3CD regions that were markedly inconsistent with relationships between human-derived viruses (Fig. 4B). Only comparisons with EV-C, EV-D, and EV-H produced pairwise distances consistently above 40% (P1) and 25% (3CD) indicative of unambiguous separate species status. In contrast, most sequence comparisons of mandrill viruses with species B and J variants were in the intraspecies range in 3CD, with divergence values of 15% to 23% that were comparable to those between subgroups of EV-A and EV-C (Fig. 4A). Furthermore, mandrill sequences differed by only 6.8 to 7.3% from SA5, which are typical of within-recombination pool distances in human EV-A–EV-C (Fig. 4A). Mandrill viruses showed the opposite relationship with EV-A with P1 distances of <40% (intraspecies range) but >25% in 3CD (interspecies).
Comparison of sequence distances in P1 and 3CD sequences therefore provided evidence for chimerism (from previous interspecies recombination) in the mandrill sequences between these regions. To identify sites where sequence exchanges occurred, divergence plots were constructed between mandrill sequences and the nonrecombinant human species A to D (Fig. 5A) and with simian viruses A13, SA5, and species H and J (Fig. 5B). As expected from their consistent sequence relationships in VP1 and 3CD, divergence between mandrill viruses and human species A, C, and D and simian species H were comparable across the EV coding region (Fig. 5A and B). Comparison with species B, however, identified highly divergent capsid sequences, and an abrupt decline into interspecies divergence levels at the P1/P2 (VP1/2A) boundary. A different breakpoint was observed with the simian EV-B sequence SA5 (Fig. 5B), situated approximately 400 bases downstream from P1/P2 within 2A.
FIG 5.
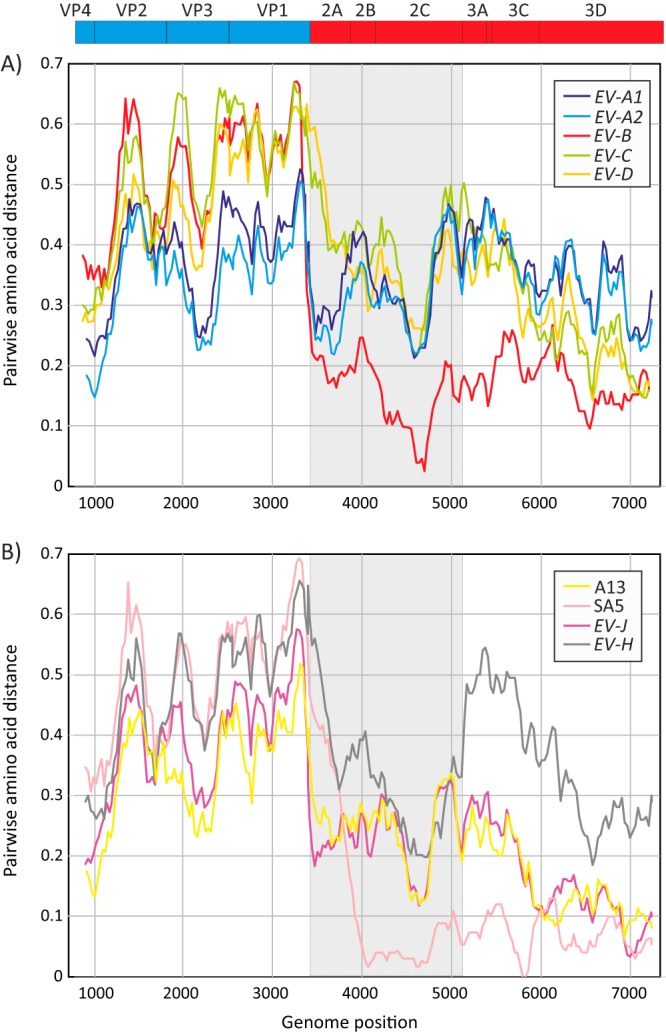
Amino acid sequence divergence between mandrill sequences and A1 and A2 subgroups in EV-A, human variants in species B to D (A) and simian viruses in species A, B, J, and H (B). The values on the y axes show mean pairwise distances between 300-base windows across the coding region, in increments of 30 bases between fragments. To show gene boundaries, the P2 region has been shaded gray, and a representation of gene positions in the EV polyprotein (drawn to scale) is shown above the graphs.
Comparison of mandrill viruses with simian variants EV-J and A13 provided evidence for further chimerism, with sequences from the P2 region (shaded area in Fig. 5B) close to the within-species range (20% to 35%) and comparable to distances from EV-H. However, in P3, distances were substantially lower in the within-species range. These observations provide evidence for further genetic exchanges in the evolution of mandrill viruses, one or more of which are likely ancestral to their split from EV-J.
Genetic exchange of 5′- and 3′UTR sequences.
To investigate the genetic relatedness of noncoding regions at the ends of the EV genomes, phylogenies were constructed from whole 5′UTR and 3′UTR and compared to those of 3CD (Fig. 6) and previously constructed trees from structural gene regions VP4/2 and VP1 (Fig. 3). Sequence relationships in both noncoding regions were distinct from those of coding regions and from each other, with consistent separate groupings of simian-derived viruses from human variants and in the case of the 5′UTR, the existence of individual clades containing human variants of more than one species, EV-A and -B in one and EV-C, -D, and -A in another (33). In the 3′UTR, mandrill viruses, EV-J and SA5 grouped together as observed in 3CD, but this group excluded A13 and human species B. The other striking phylogeny difference was the complete separation of human A2 viruses from A1 that were in turn distinct from simian A2 viruses. Although the 3′UTR was short and tree construction was intrinsically less robust than for other genome regions, these observations provide evidence for genetic exchanges, additional to those between nonstructural gene regions and distinct from those occurring in the 5′UTR.
FIG 6.

Phylogenetic analysis of parts of 5′UTR (A), 3CD (B), and 3′UTR (C). (A) 5′UTR, positions 1 to 742; (B) 3CD, positions 5429 to 7366; (C) 3′UTR, positions 7377 to 7431. Bootstrapped ML trees were constructed as described for in the legend to Fig. 3 with the exception of the 3′UTR where its short sequence length (55 bases) precluded meaningful model fitting. The tree is therefore shown as a phenogram by neighbor joining of uncorrected nucleotide p-distances.
DISCUSSION
This study is the first large-scale investigation of infection frequencies and genetic characterization of enteroviruses infecting Old World monkeys in the wild. The extraordinarily high prevalence of active infection and genetic diversity of EVs found provides evidence for the extensive circulation of these viruses in wild primate populations. The sampling strategy that concentrated collection on remote areas with no or minimal human contact and the reclusive nature of the species (34) helped rule out human sources of infection in monkeys, as did the finding of virus variants that were consistently genetically distinct from human enteroviruses. Primate sampling of feces has been commonly used to investigate infection frequencies of simian immunodeficiency virus (18), and collection methods used for the current study were specifically designed to avoid environmental contamination. Furthermore, samples showed generally high viral loads (mean CT values of 27.5), indicating substantially higher viral loads than typically found in human samples (Fig. 2) and demonstrating active infections in the sampled populations.
The nature of the sampling, however, prevented more-focused investigations of EV persistence and disease associations in mandrills or other monkey species. This limitation additionally prevented clear attribution of samples to individual (different) monkeys. This sampling bias is exemplified by the four species H variants that originated from samples collected on the same day from the same site. It is likely that they originated from members of the same group (and thus are epidemiologically linked infections) or conceivably from the same mandrill. The development of genetic fingerprinting for mandrills and other monkey species, as used in ape samples, would be needed to investigate this further. Nevertheless, the vast majority of EV variants characterized in the study were genetically distinct from each other and originated from widespread sampling (throughout southern Cameroon) over a prolonged period (2008 to 2012). In the main, therefore, they represent independent infections.
Infection frequencies of greater than 95% in mandrills were substantially higher than the 10% detected in apes (chimpanzees and gorillas) from similar sampling areas (16). Although little is known about possible differences in susceptibility or persistence of EV infections in OWMs, apes, and humans, a major factor likely contributing to the circulation of EVs in mandrills is the large and highly interactive populations of mandrills in the wild, which typically live in hordes of between 500 and 1,000 (34). This population size and their interactions with other groups of mandrills and other OWMs may create the necessary conditions to permit ongoing transmission of acute virus infections over long periods. Indeed, even under captive conditions, infections with EV variants found previously in monkeys were frequently detected even in small isolated OWM populations in a primate center and a zoo (13, 15), while only human serotypes were detected among nonhuman primates living in human environments (12). Our findings support the broader hypothesis that EV infections are prevalent in at least one OWM species and continue to circulate in the absence of human contact or intervention.
The EV types detected in the current study fell into species H and J and a large, new group of currently undefined taxonomic status within the Enterovirus genus. No variants that corresponded to types or species found in humans were detected, confirming the absence of human sources of infection in mandrills and other sampled OWM species. Among the 89 EV variants characterized by VP4/2 sequencing, several could be classified as members of species H type 1 variants previously detected in a simian organ cultures from an Asian ape, M. mulatta (SV2, SV4, and SV28) (6) but also from human subjects with hepatitis in the early 1960s (35, 36). Others grouped in species J, a group now listed as containing six assigned types (but with sequences available from only three [SV6, EV103, and EV108]), to which the sequence of CPOV_3336 may contribute an additional type based on its divergent VP1 sequence. Although most EV-J variants have been identified in African OWMs, the originally described SV6 isolate originated in an Asian macaque (6). Finally, several mandrill-derived variants could be classified as members of the simian subgroup of species B, one corresponding to SA5 and the other to EV110, previously identified in chimpanzees from the same geographical area as the mandrill samples (Lomie [LM] in Fig. 1) (17).
Genetic characterization of the remaining, predominantly mandrill-derived EVs was performed by extensive sequence analysis of VP4/2 and complete VP1 sequences. From the latter, the existence of at least 5 types differing by >25% in nucleotide sequence could be inferred, although their definitive assignment awaits their species designation. Determining this, however, may prove problematic because they reproduced the previously identified hybrid nature of many simian enterovirus genomes (9, 10); variants such as A13 were identified as showing closer sequence relatedness to one species in the capsid-encoding region (EV-A) and to another in the NS region (EV-B). The subsequent characterization of further simian viruses, the identification of subgroups with species A (A1 and A2 [26]) and B (human viruses and SA5) and the new complete genome sequence data from mandrill-derived viruses provided the opportunity for a reevaluation of the occurrence of recombination in the genomes of simian (and human) viruses, from which various species classification possibilities might be proposed.
Genome configurations.
Despite the seeming complexity of sequence relationships between simian and human viruses in different genome regions (Fig. 4 to 6 and Fig. 6 in reference 9), structural and nonstructural gene regions of all OWM monkey-derived viruses ultimately show only three genome configurations.
(i) Group 1.
Group 1 comprises SV19, SV43, SV46, and EV92 which form part of the A2 subgroup in species A, along with the human viruses EV76 and EV89 to -91 (that may indeed be zoonotically acquired) (26).
(ii) Group 2.
Group 2 comprises A13 (in species A2 in P1), EV-J, and the mandrill viruses. All three possess P1 sequences classified as or closely related to EV-A with pairwise distances below (A13 and mandrill viruses) or around the species assignment threshold (EV-J) of 40% for this region (Table 2; Fig. 4B). However, all of these viruses possess species B-like NS gene regions, differing from each other and from EV-B by 6% to 21% in the 2C plus 3CD region and a well-defined breakpoint at the P1/P2 boundary (Fig. 4B; see Fig. S1 in the supplemental material). This commonality is further manifested by similar distributions of pairwise distances to other EV species (Fig. 4B and Fig. S2).
(iii) Group 3.
Group 3 is represented by SA5 that is most closely related to human EV-B sequences throughout its genome. Several further examples of this group potentially exist among samples obtained in the current study from mandrills. Group 3 viruses likely represent the source of the species B-like NS gene regions of group B viruses, through recombination, accounting for the remarkably similar 3CD sequences of SA5 and mandrill viruses (6% divergence).
As previously discussed (9, 33, 37), there is further recombination between coding regions and 5′UTRs and 3′UTRs within human and simian enteroviruses. Sequence relationships in the 5′UTR and 3′UTR are distinct, although both regions show different propensities for species mergers and splits. For example, human species A1 and B variants are phylogenetically interspersed in the 5′UTR, as are species C and D along with some A2 viruses, while EV-J, simian EV-B (SA5) and mandrill viruses group closely together in the 3′UTR. A common feature, however, is the universally distinct grouping of simian- and human-derived viruses from each other in both regions. This observation is consistent with the existence of host range determinants in these genome regions and their potentially limiting effect on the occurrence of zoonotic infections. Further genetic characterization of simian viruses and in vitro studies are required to formally examine this hypothesis.
Enterovirus classification.
The final classification of mandrill and other OWM viruses obtained in the current study may necessitate an update to the current species assignment criteria (1). By definition, viruses such as those obtained in the current study, A13 and EV-J which show evidence for interspecies chimerism between capsid and NS gene regions cannot simultaneously fulfill criteria 2 (<40% divergence in P1) and 3 (<30% divergence in 2C plus 3CD) for any one species. This phenomenon similarly limits the use of divergence thresholds for complete coding region sequences (criterion 1). As the capsid likely possesses elements that primarily determine EV host and cellular tropisms and other biological properties, it may perhaps be appropriate to regard P1 divergence as the primary criterion for species assignment, although this will require further discussion with the International Committee for the Taxonomy Study Group.
Findings in the current study additionally highlight the need to reexamine the species assignment threshold for P1, currently set at 40%. Pairwise distances between EV-A (particularly A2) types, A13, mandrill viruses, and EV-J are mostly below this threshold (Fig. 4B), and simian viruses in these groups all possess a common (EV-B-like) NS gene region (and mostly group together in the 3′UTR). A slight elevation of the 40% threshold would allow their assignment as species A and would substantially simplify the current classification of simian EVs (i.e., all members of groups 1 and 2 described above would be classifiable as EV-A). Although this would create a species containing both nonrecombinant and recombinant viruses, this is not different from EV-C, which also contains variants with evidence for chimerism in NS gene regions and in the 5′UTR (31, 32).
The alternative would be to lower the P1 threshold 40% to 32% so that EV-J and the mandrill viruses could be classified as separate species. The immediate problem with this is that this reduction would have the undesirable effect of splitting species C and A. As described above, the other problem is that structural gene sequences of EV-A, EV-J, and mandrill viruses fail to form clearly resolvable monophyletic clusters when included in the same data set (Fig. 2A and B), and bootstrap support for these groupings requires the whole P1 region (see Fig. S3 in the supplemental material). The diversity of simian viruses is likely considerably undersampled, and it is likely that further viruses showing loose sequence affiliations to these variants will be characterized in the future and create further complications with EV-J and mandrill EV species assignments.
Overall, this study provides substantial new information on the genetic diversity and prevalence of naturally occurring enteroviruses in wild primate populations. It is increasingly clear that relationships between variants infecting humans and nonhuman primates are complex, notwithstanding the growing evidence for zoonotic infections in both directions (12, 38) and continuing uncertainty about the timescales for the original divergence of types and species and appearance of interspecies chimeras. Understanding which genome regions ultimately determine host range and disease outcomes are essential in assessment of the risk of simian enteroviruses as potential zoonotic and pathogenic infections in humans.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff and the SIV team from PRESICA for logistical support in Cameroon and the Cameroonian Ministries of Health, Environment and Forestry, and Research for permission to collect samples in Cameroon.
This work was supported in part by grants from the National Institutes of Health (RO1 AI 50529), the Agence Nationale de Recherches sur le SIDA (ANRS 12125/12182/12255), and the Wellcome Trust (VIZIONS, WT/093724).
Footnotes
Published ahead of print 12 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00088-14.
REFERENCES
- 1.Knowles NJ, Hovi T, Hyypia T, King AMQ, Lindberg AM, Pallansch MA, Palmenberg AC, Simmonds P, Skern T, Stanway G, Yamashita T, Zell R. 2012. Picornaviridae, p 855–880 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA [Google Scholar]
- 2.Heberling RL, Cheever FS. 1965. Some characteristics of the simian enteroviruses. Am. J. Epidemiol. 81:106–123 [DOI] [PubMed] [Google Scholar]
- 3.Fuentes-Marins R, Rodriguez AR, Kalter SS, Hellman A, Crandell RA. 1963. Isolation of enteroviruses from the “normal” baboon (Papio doguera). J. Bacteriol. 85:1045–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malherbe H, Harwin R. 1963. The cytopathic effects of vervet monkey viruses. S. Afr. Med. J. 37:407–411 [PubMed] [Google Scholar]
- 5.Hull RN, Minner JR, Mascoli CC. 1958. New viral agents recovered from tissue cultures of monkey kidney cells. III. Recovery of additional agents both from cultures of monkey tissues and directly from tissues and excreta. Am. J. Hyg. 68:31–44 [DOI] [PubMed] [Google Scholar]
- 6.Hull RN, Minner JR, Smith JW. 1956. New viral agents recovered from tissue cultures of monkey kidney cells. I. Origin and properties of cytopathogenic agents S.V.1, S.V.2, S.V.4, S.V.5, S.V.6, S.V.11, S.V.12 and S.V.15. Am. J. Hyg. 63:204–215 [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez AR, Kalter SS, Heberling RL, Helmke RJ, Guajardo JE. 1977. Viral infections of the captive Kenya baboon (Papio cynocephalus): a five-year epidemiologic study of an outdoor colony. Lab. Anim. Sci. 27:356–371 [PubMed] [Google Scholar]
- 8.Oberste MS, Jiang X, Maher K, Nix WA, Jiang B. 2008. The complete genome sequences for three simian enteroviruses isolated from captive primates. Arch. Virol. 153:2117–2122. 10.1007/s00705-008-0225-4 [DOI] [PubMed] [Google Scholar]
- 9.Oberste MS, Maher K, Pallansch MA. 2007. Complete genome sequences for nine simian enteroviruses. J. Gen. Virol. 88:3360–3372. 10.1099/vir.0.83124-0 [DOI] [PubMed] [Google Scholar]
- 10.Oberste MS, Maher K, Pallansch MA. 2002. Molecular phylogeny and proposed classification of the simian picornaviruses. J. Virol. 76:1244–1251. 10.1128/JVI.76.3.1244-1251.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poyry T, Kinnunen L, Hovi T, Hyypia T. 1999. Relationships between simian and human enteroviruses. J. Gen. Virol. 80:635–638 [DOI] [PubMed] [Google Scholar]
- 12.Oberste MS, Feeroz MM, Maher K, Nix WA, Engel GA, Hasan KM, Begum S, Oh G, Chowdhury AH, Pallansch MA, Jones-Engel L. 2013. Characterizing the picornavirus landscape among synanthropic nonhuman primates in Bangladesh, 2007 to 2008. J. Virol. 87:558–571. 10.1128/JVI.00837-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oberste MS, Feeroz MM, Maher K, Nix WA, Engel GA, Begum S, Hasan KM, Oh G, Pallansch MA, Jones-Engel L. 2013. Naturally acquired picornavirus infections in primates at the Dhaka zoo. J. Virol. 87:572–580. 10.1128/JVI.00838-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffert WR, Bates ME, Cheever FS. 1958. Study of enteric viruses of simian origin. Am. J. Hyg. 68:15–30 [DOI] [PubMed] [Google Scholar]
- 15.Nix WA, Jiang B, Maher K, Strobert E, Oberste MS. 2008. Identification of enteroviruses in naturally infected captive primates. J. Clin. Microbiol. 46:2874–2878. 10.1128/JCM.00074-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harvala H, Van Nguyen D, McIntyre C, Ahuka-Mundeke S, Mpoudi NE, Delaporte E, Peeters M, Simmonds P. 4 November 2013. Co-circulation of enteroviruses between apes and humans. J. Gen. Virol. 10.1099/vir.0.059048-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvala H, Sharp CP, Ngole EM, Delaporte E, Peeters M, Simmonds P. 2011. Detection and genetic characterisation of enteroviruses circulating among wild populations of chimpanzees in Cameroon: relationship with human and simian enteroviruses. J. Virol. 85:4480–4486. 10.1128/JVI.02285-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH. 2006. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 313:523–526. 10.1126/science.1126531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neel C, Etienne L, Li Y, Takehisa J, Rudicell RS, Bass IN, Moudindo J, Mebenga A, Esteban A, Van Heuverswyn F, Liegeois F, Kranzusch PJ, Walsh PD, Sanz CM, Morgan DB, Ndjango JB, Plantier JC, Locatelli S, Gonder MK, Leendertz FH, Boesch C, Todd A, Delaporte E, Mpoudi-Ngole E, Hahn BH, Peeters M. 2010. Molecular epidemiology of simian immunodeficiency virus infection in wild-living gorillas. J. Virol. 84:1464–1476. 10.1128/JVI.02129-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharp CP, Lebreton M, Kantola K, Nana A, Diffo JL, Djoko CF, Tamoufe U, Kiyang JA, Babila TG, Ngole EM, Pybus OG, Delwart E, Delaporte E, Peeters M, Soderlund-Venermo M, Hedman K, Wolfe ND, Simmonds P. 2010. Widespread infection of chimpanzees and gorillas with homologues of human parvoviruses B19, PARV4 and human bocavirus in the wild. J. Virol. 84:10289–10296. 10.1128/JVI.01304-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett S, Harvala H, Witteveldt J, McWilliam Leitch EC, McLeish N, Templeton K, Gunson R, Carman WF, Simmonds P. 2011. Rapid simultaneous detection of enterovirus and parechovirus RNAs in clinical samples by one-step real-time reverse transcription-PCR assay. J. Clin. Microbiol. 49:2620–2624. 10.1128/JCM.02445-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLeish NJ, Witteveldt J, Clasper L, McIntyre C, McWilliam Leitch EC, Hardie A, Bennett S, Gunson R, Carman WF, Feeney SA, Coyle PV, Vipond B, Muir P, Benschop K, Wolthers K, Waris M, Osterback R, Johannessen I, Templeton K, Harvala H, Simmonds P. 2012. Development and assay of RNA transcripts of enterovirus species A to D, rhinovirus species A to C, and human parechovirus: assessment of assay sensitivity and specificity of real-time screening and typing methods. J. Clin. Microbiol. 50:2910–2917. 10.1128/JCM.01172-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmonds P. 2012. SSE: a nucleotide and amino acid sequence analysis platform. BMC Res. Notes 5:50. 10.1186/1756-0500-5-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30:2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harvala H, McIntyre CL, McLeish NJ, Kondracka J, Palmer J, Molyneaux P, Gunson R, Bennett S, Templeton K, Simmonds P. 2012. High detection frequency and viral loads of human rhinovirus species A to C in fecal samples: diagnostic and clinical implications. J. Med. Virol. 84:536–542. 10.1002/jmv.23203 [DOI] [PubMed] [Google Scholar]
- 26.Oberste MS, Maher K, Michele SM, Belliot G, Uddin M, Pallansch MA. 2005. Enteroviruses 76, 89, 90 and 91 represent a novel group within the species human enterovirus A. J. Gen. Virol. 86:445–451. 10.1099/vir.0.80475-0 [DOI] [PubMed] [Google Scholar]
- 27.Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J. 2005. Recombination in circulating human enterovirus B: independent evolution of structural and non-structural genome regions. J. Gen. Virol. 86:3281–3290. 10.1099/vir.0.81264-0 [DOI] [PubMed] [Google Scholar]
- 28.McWilliam Leitch EC, Bendig J, Cabrerizo M, Cardosa J, Hyypia T, Ivanova OE, Kelly A, Kroes AC, Lukashev A, Macadam A, McMinn P, Roivainen M, Trallero G, Evans DJ, Simmonds P. 2009. Transmission networks and population turnover of echovirus 30. J. Virol. 83:2109–2118. 10.1128/JVI.02109-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukashev AN. 2005. Role of recombination in evolution of enteroviruses. Rev. Med. Virol. 15:157–167. 10.1002/rmv.457 [DOI] [PubMed] [Google Scholar]
- 30.Simmonds P. 2006. Recombination and selection in the evolution of picornaviruses and other mammalian positive-stranded RNA viruses. J. Virol. 80:11124–11140. 10.1128/JVI.01076-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown B, Oberste MS, Maher K, Pallansch MA. 2003. Complete genomic sequencing shows that polioviruses and members of human enterovirus species C are closely related in the noncapsid coding region. J. Virol. 77:8973–8984. 10.1128/JVI.77.16.8973-8984.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tapparel C, Junier T, Gerlach D, Van Belle S, Turin L, Cordey S, Muhlemann K, Regamey N, Aubert JD, Soccal PM, Eigenmann P, Zdobnov E, Kaiser L. 2009. New respiratory enterovirus and recombinant rhinoviruses among circulating picornaviruses. Emerg. Infect. Dis. 15:719–726. 10.3201/eid1505.081286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santti J, Hyypia T, Kinnunen L, Salminen M. 1999. Evidence of recombination among enteroviruses. J. Virol. 73:8741–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrison MJS. 1988. The mandrill in Gabon's rain forest—ecology, distribution and status. Oryx 22:218–228. 10.1017/S0030605300022365 [DOI] [Google Scholar]
- 35.Liu Z, Donahue RE, Young NS, Brown KE. 2000. Sequencing and characterization of A-2 plaque virus: a new member of the Picornaviridae family. Virology 272:168–176. 10.1006/viro.2000.0355 [DOI] [PubMed] [Google Scholar]
- 36.Shaw ED, McKee AP, Rancourt M, Hollenbeck L. 1973. Induction of hepatitis B antibody in experimental animals by immunization with A-2 plaque virus. J. Virol. 12:1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smura T, Blomqvist S, Paananen A, Vuorinen T, Sobotova Z, Bubovica V, Ivanova O, Hovi T, Roivainen M. 2007. Enterovirus surveillance reveals proposed new serotypes and provides new insight into enterovirus 5′-untranslated region evolution. J. Gen. Virol. 88:2520–2526. 10.1099/vir.0.82866-0 [DOI] [PubMed] [Google Scholar]
- 38.Harvala H, McIntyre CL, Imai N, Clasper L, Djoko CF, Lebreton M, Vermeulen M, Saville A, Mutapi F, Tamoufe U, Kiyang J, Biblia TG, Midzi N, Mduluza T, Pepin J, Njouom R, Smura T, Fair JN, Wolfe ND, Roivainen M, Simmonds P. 2012. High seroprevalence of enterovirus infections in apes and Old World monkeys. Emerg. Infect. Dis. 18:283–286. 10.3201/eid1802.111363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.