

ABSTRACT
Viruses with positive-strand RNA genomes amplify their genomes in replication complexes associated with cellular membranes. Little is known about the mechanism of replication complex formation in cells infected with Nodamura virus. This virus is unique in its ability to lethally infect both mammals and insects. In mice and in larvae of the greater wax moth (Galleria mellonella), Nodamura virus-infected muscle cells exhibit mitochondrial aggregation and membrane rearrangement, leading to disorganization of the muscle fibrils on the tissue level and ultimately in hind limb/segment paralysis. However, the molecular basis for this pathogenesis and the role of mitochondria in Nodamura virus infection remains unclear. Here, we tested the hypothesis that Nodamura virus establishes RNA replication complexes that associate with mitochondria in mammalian cells. Our results showed that Nodamura virus replication complexes are targeted to mitochondria, as evidenced in biochemical, molecular, and confocal microscopy studies. More specifically, we show that the Nodamura virus RNA-dependent RNA polymerase interacts with the outer mitochondrial membranes as an integral membrane protein and ultimately becomes associated with functional replication complexes. These studies will help us to understand the mechanism of replication complex formation and the pathogenesis of Nodamura virus for mammals.
IMPORTANCE This study will further our understanding of Nodamura virus (NoV) genome replication and its pathogenesis for mice. NoV is unique among the Nodaviridae in its ability to infect mammals. Here we show that NoV establishes RNA replication complexes (RCs) in association with mitochondria in mammalian cells. These RCs contain newly synthesized viral RNA and feature a physical interaction between mitochondrial membranes and the viral RNA-dependent RNA polymerase (RdRp), which is mediated by two membrane-associated regions. While the nature of the interaction needs to be explored further, it appears to occur by a mode distinct from that described for the insect nodavirus Flock House virus (FHV). The interaction of the NoV RdRp with mitochondrial membranes is essential for clustering of mitochondria into networks that resemble those described for infected mouse muscle and that are associated with fatal hind limb paralysis. This work therefore provides the first link between NoV RNA replication complex formation and the pathogenesis of this virus for mice.
INTRODUCTION
Nodamura virus (NoV) is the type species of the Alphanodavirus genus of the virus family Nodaviridae. Originally isolated from the mosquito Culex tritaeniorhynchus, NoV lethally infects insects (mosquitoes, wax moth larvae, and honey bees) and mammals (suckling mice and suckling hamsters), causing hind segment or hind limb paralysis, respectively (1–7). The family Nodaviridae consists of two genera: members of the Alphanodavirus genus, which includes NoV, Flock House virus (FHV), and Black beetle virus (BBV), primarily infect insects, while the betanodaviruses, including Greasy grouper nervous necrosis virus (GGNNV) and Atlantic halibut nodavirus (AHNV), have been isolated only from fish (8, 9). NoV provides an excellent model for deciphering the basic mechanism of viral RNA replication, due to its genetic simplicity, tremendous levels of RNA amplification, and ability to replicate in a wide range of host cells. While NoV remains the only member of the family Nodaviridae that is naturally able to infect mammals, the genomic RNAs of both NoV and FHV can replicate in cultured mammalian cells.
Nodaviruses contain bipartite single-stranded positive-sense RNA genomes (8, 9). The larger genome segment, RNA1, encodes the 110-kDa viral contribution of the RNA-dependent RNA polymerase (RdRp), which replicates both genomic and subgenomic RNA segments via negative-strand RNA replication intermediates. RNA2 encodes the precursor to the viral capsid protein and is dispensable for RNA1 replication (10–12). During RNA1 replication, subgenomic RNA3 is synthesized. RNA3 is 3′ coterminal with RNA1 and encodes proteins B1 and B2 from overlapping open reading frames (ORFs). The function of B1 is unknown, while the B2 proteins of FHV and NoV suppress the cellular RNA interference (RNAi) response in insect, plant, and mammalian cells (13–16).
Positive-strand RNA viruses replicate their genomes in replication complexes (RCs) associated with intracellular membranes, often via interactions between the membranes and one or more viral nonstructural proteins (17–19). The source of the membranes varies with the virus: many viral RCs are associated with modified membranes derived from the endoplasmic reticulum (ER), including members of the arteri-, bromo-, corona-, flavi-, and picornavirus families (20–29). However, the RCs of other virus families localize to membranes derived from other organelles, including chloroplasts and peroxisomes (tombusviruses), endosomes and lysosomes (togaviruses), and mitochondria (tombus- and nodaviruses) (30–34).
Two other members of the Nodaviridae replicate their genomes in association with mitochondrial membranes, namely, FHV (in yeast and cultured Drosophila melanogaster cells) and the betanodavirus GGNNV (in cultured sea bass cells) (32, 35, 36). The FHV RdRp and RCs colocalize to spherules within the outer mitochondrial membrane (32, 36, 37). The RdRp of a second betanodavirus, AHNV, localizes to mitochondria in infected fish cells and in transfected African green monkey kidney COS-7 cells when expressed from a plasmid in the absence of viral RNA replication, but the significance of this localization for the life cycle of the virus is unknown (38).
It is difficult to simply extrapolate the localization of the NoV RdRp from that of FHV, since the two proteins are markedly different. Although both contain canonical RdRp domains, they share only 44% sequence identity at the amino acid level (39, 40). Their template specificities are also unique, with each viral RdRp preferring its own RNA templates (41, 42). Finally, the NoV RdRp enzymatic activity is more thermostable than that of FHV, in that it retains enzymatic activity at temperatures up to 37°C, while the FHV RdRp is inactive at temperatures above 31°C (11). Nevertheless, the literature does suggest a role for mitochondria in the NoV life cycle. Transmission electron micrographs of muscle tissue from NoV-infected mice show localized increases in the number of mitochondria, rearrangement of mitochondrial membranes, and aggregation of mitochondria. Similar rearrangements in muscle tissue are also visible in NoV-infected larvae of the greater wax moth, Galleria mellonella, which exhibit hind segment paralysis upon infection with NoV (1, 43, 44). However, the role of mitochondria in NoV infection has not been addressed previously.
In the present study, we tested the hypothesis that NoV establishes RNA replication complexes that associate with mitochondria in mammalian cells. Here, we show that the NoV RdRp contains two predicted membrane-associated regions (MARs). Our results demonstrate that the NoV RdRp localized to mitochondria and caused them to cluster in cultured mammalian cells at 37°C. Deletion of the predicted MARs from the RdRp resulted in loss of mitochondrial localization, abolition of mitochondrial clustering, and reductions in levels of RdRp protein and of RNA1 replication products. The results of selective membrane permeabilization and confocal microscopy experiments showed that the NoV RdRp is targeted to the outer mitochondrial membrane, where it remains exposed to the cytoplasm. We also demonstrate via biochemical assays that the NoV RdRp is closely associated with intracellular membranes. These results firmly establish the role of mitochondria and mitochondrial membranes in NoV RNA replication in mammalian cells and form the basis for further studies on the mechanism of NoV RC formation.
MATERIALS AND METHODS
Cells, virus stocks, and tissue culture.
All recombinant plasmids were amplified in Escherichia coli strain NEB10β (New England BioLabs) grown in 2× YT broth or on 2× YT-agar plates supplemented with ampicillin (45). BSR-T7/5 cells, which are baby hamster kidney BHK21 cells that constitutively express cytoplasmic T7 RNA polymerase (46), were grown at 37°C in Glasgow minimal essential medium or Dulbecco's modified Eagle's medium (Gibco) supplemented with 5% concentrations each of newborn calf serum and fetal bovine serum and 1 mg/ml G-418 (Life Technologies). The full-length RNA1 cDNA clone of Nodamura virus (family Nodaviridae, genus Alphanodavirus, species Nodamura virus) was derived from the Mag-115 strain, as described previously (47). Its GenBank ID is AF174533.
BSR-T7/5 cells were seeded in 6-well plates (10-cm2 wells) at a density of 5 × 105 cells/well; for immunofluorescence microscopy, the cells were plated over sterilized glass coverslips. Cells were transfected with 2 μg of the appropriate pNoV1(0,0)-based plasmid by using Lipofectamine 2000 and serum-free Opti-MEM (Invitrogen, Carlsbad, CA), as described previously (47). Cells were incubated with DNA-liposome complexes for 4 h at 37°C and supplemented with complete growth medium, and the incubation was continued for 20 h prior to harvest. For the proteasome inhibition assays, transfected cells were treated with the cell-permeable proteasome inhibitor MG132 (48) at a concentration of 5 μM for the 8 h prior to harvest, beginning at 16 h posttransfection (hpt).
In silico predictions and sequence alignments.
Computer protein topology predictions were performed on the primary sequence of the NoV RdRp ORF by using the following prediction programs: TopPred II (49), PSIPRED (50, 51), and SOSUI (52). Hydrophobicity was calculated by using TopPredII and the method of Kyte and Doolittle (53) with a core window size of 11 amino acids (aa) and a full window size of 21 aa. We computed the identity between the NoV and GGNNV RdRp primary sequences by using the ClustalW2 multiple sequence alignment program (54) (data not shown).
Plasmid constructions.
Expression of NoV RNA1 and the viral RdRp in mammalian cells was directed by plasmid pNoV1(0,0), in which the full-length RNA1 cDNA (accession number NC_002690.1) is under transcriptional control of a bacteriophage T7 promoter (47). The predicted MARs were deleted from the RdRp ORF in pNoV1(0,0), either singly (Δ12-34 and Δ42-64) or together (Δ12-64) by circular PCR-based mutagenesis with the overlapping oligonucleotide primers shown in Table 1, followed by DpnI selection, as described previously (55). These deletions were confirmed by DNA sequencing and, in each case, a small fragment containing the deletion was introduced back into parental pNoV1(0,0) with SapI and Mlul restriction enzymes, resulting in plasmids pT7-N1Δ12-34, pT7-N1Δ42-64, and pT7-N1Δ12-64. To facilitate immunodetection of wild-type (WT) and mutant forms of the NoV RdRp, we inserted an in-frame C-terminal influenza virus hemagglutinin (HA) epitope tag between the last amino acid of the RdRp and its stop codon via circular PCR-based mutagenesis with overlapping oligonucleotide primers (Table 1) followed by DpnI selection, as described previously (55). The insertion was confirmed by DNA sequencing, and a small DNA fragment containing the insertion was cloned back into the WT and mutant versions of pNoV1(0,0), resulting in plasmids pT7-N1-HA, pT7-N1Δ12-34-HA, pT7-N1Δ42-64-HA, and pT7-N1Δ12-64-HA.
TABLE 1.
Oligonucleotide primers used for circular PCR-based mutagenesis
| Primer | Sequence (5′–3′) |
|---|---|
| ΔMAR1plus | CGAGACAATCATCAACGGCGCAGTCGCGGGGTCCTGCGTGGTG |
| ΔMAR1minus | CACCACGCAGGACCCCGCGACTGCGCCGTTGATGATTGTCTCG |
| ΔMAR2plus | GTCGCGGGGTCCTGCGTGGTGCAGCACCGTCTGCCGATGGCC |
| ΔMAR2minus | GGCCATCGGCAGACGGTGCTGCACCACGCAGGACCCCGCGAC |
| ΔMAR1 + 2plus | CGAGACAATCATCAACGGCGCACAGCACCGTCTGCCGATGGCC |
| ΔMAR1 + 2minus | GGCCATCGGCAGACGGTGCTGTGCGCCGTTGATGATTGTCTCG |
| NA-HA-QCplus | CCCAGCTGGGTCGCGTGGGCGTGGTAAAGGCTACCCATACGACGTGCCAGACTACGCCTGAGTGATTCATCGTCCCATCTGACGAAACCC |
| NA-HA-QCminus | GGGTTTCGTCAGATGGGACGATGAATCACTCAGGCGTAGTCTGGCACGTCGTATGGGTAGCCTTTACCACGCCCACGCGACCCAGCTGGG |
Immunofluorescence confocal microscopy.
BSR-T7/5 cells were mock transfected or transfected with plasmids as described above. In some experiments (where indicated), MitoTracker Red CM-H2XRos (MTR; Life Technologies) was used as a vital dye to stain the mitochondria of living cells prior to immunofluorescence (500 nM MTR in serum-free Opti-MEM for 1 h at 37°C), as described previously (32). After MTR treatment, cells were fixed, permeabilized in 100% methanol, and blocked at room temperature in phosphate-buffered saline (PBS) supplemented with 1% bovine serum albumin, 1% nonfat milk, 0.1% sodium azide, and 0.1% Tween 20. For the selective membrane permeabilization assays, cells were fixed with 4% paraformaldehyde in PBS at 4°C overnight and permeabilized at room temperature for 10 min with either 0.002% (wt/vol) saponin (Sigma-Aldrich) or 0.002% saponin and 0.2% Triton X-100 (TX100; Fisher Scientific) as previously described (32). After permeabilization, cells were blocked and washed with PBS lacking Tween 20.
Immunofluorescence was performed as described previously (32) with the following modifications. Antigens were detected within blocked BSR-T7/5 cells with primary antibodies specific for the HA epitope tag (mouse monoclonal IgG3; Santa Cruz Biotechnology), newly synthesized viral RNA (bromodeoxyuridine [BrU]; mouse monoclonal IgG1; Sigma-Aldrich), the outer mitochondrial membrane marker monoamine oxidase (MAO; rabbit polyclonal IgG; Santa Cruz Biotechnology), or the inner mitochondrial membrane marker cytochrome c oxidase subunit III (COX3; goat polyclonal IgG; Santa Cruz Biotechnology). Binding of primary antibodies to their respective antigens was detected with the following fluorescently labeled secondary antibodies (Santa Cruz Biotechnology): fluorescein isothiocyanate (FITC)-labeled goat anti-mouse to detect either HA or BrU, Texas Red (TR)-labeled goat anti-rabbit to detect MAO, or TR-labeled rabbit anti-goat to detect COX3.
To minimize photobleaching effects, all staining steps were performed in the dark. Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies). Coverslips were mounted with fluorescence mounting medium (Dako) and sealed to prevent drying and fluorescence fading. High-resolution digital fluorescent images were captured using a Zeiss LSM 700 confocal microscope equipped with a 63× immersion oil objective and ZEN 2009 software (Zeiss, New York, NY) for acquisition and processing of confocal images. Single-plane images were sequentially scanned using a 1-Airy unit pinhole setting for each channel and acquired at a 1,024- by 1,024-pixel resolution. The same software was used to visualize colocalization of the red and green signals and to export the images as TIF files. Adobe Photoshop software was used to crop, rotate, and resize panels where needed to ensure the images were all the same size and scale, using the scale bars embedded in each image as a guide.
ZEN 2009 software was also used to evaluate the grade of colocalization between two fluorescently labeled intracellular targets. Regions of interest (ROIs) were demarcated manually within captured images to reduce background contribution. The degree of colocalization of two selected signals was determined as described previously (56), using a squared Manders' overlap coefficient (OC) of dual-color images (specifically, the green and red emission fluorescence signals), executed on a pixel-by-pixel basis. OC values are expressed as the percentage of overlap between red pixels (MTR or TR) and green pixels (FITC) and are shown in Table 2. An OC of 0 indicates a lack of overlap between pixels of two defined signals within the selected ROI, whereas a value of 1 represents perfectly colocalized signals of all pixels in a selected ROI (56).
TABLE 2.
| Confocal micrograph by expt | Overlapa |
|---|---|
| Time course (Fig. 3) | |
| 4 hpt | 0 |
| 8 hpt | 88 ± 2.6 |
| 12 hpt | 86 ± 3.3 |
| 16 hpt | 89 ± 2.9 |
| 20 hpt | 82 ± 6.1 |
| 24 hpt | 83 ± 3.1 |
| Selective permeabilization (Fig. 4) | |
| Saponin alone, MAO, merge | 82 ± 2.1 |
| Saponin Alone, COX3, merge | 0 |
| Saponin + Triton X-100, COX3, merge | 88 ± 2.9 |
| MAR deletion mutants (Fig. 6) | |
| WT, merge | 86 ± 2.3 |
| Δ12-34, merge | 88 ± 12.4 |
| Δ42-64, merge | 85 ± 5.7 |
| Δ12-64, merge | 47 ± 4.2 |
| BrU labeling (Fig. 9) | |
| Mock, merge | 0 |
| WT, merge | 94 ± 1.5 |
| Δ12-64, merge | 0 |
Overlap was defined as the percent overlap between red (MTR or TR) and green (FITC) pixels (means ± standard deviations), as described in Materials and Methods and in reference 56.
Cell fractionation and differential centrifugation.
Cells were harvested by scraping, and crude subcellular fractions were prepared as previously described (57–60), with the following minor modifications. Transfected BSR-T7/5 cells were washed twice with ice-cold PBS, harvested by scraping into PBS, and collected by centrifugation. Cell pellets were gently resuspended in hypotonic lysis buffer (1 mM Tris-HCl [pH 7.4], 0.1 mM EDTA, 15 mM NaCl) (35) supplemented with 1 mM each benzamidine (Sigma-Aldrich) and phenylmethylsulfonyl fluoride (PMSF; Sigma-Adrich). Cells were homogenized with a glass Dounce instrument and unbroken cells, large cell debris, and nuclei were removed by centrifugation. The resulting postnuclear lysate (PNL) was centrifuged at 20,000 × g for 30 min to pellet intracellular membranes. The supernatant was carefully removed and centrifuged at 100,000 × g for 1 h to pellet microsomal membranes, and the supernatant was carefully collected as the cytosolic fraction. All centrifugation steps were performed at 4°C. Membrane pellets were resuspended in 2× Laemmli sample buffer, resolved by SDS-polyacrylamide gel electrophoresis (PAGE), and analyzed by immunoblotting as described below.
Membrane flotation and membrane disassociation assays.
The 20,000 × g pellet from transfected BSR-T7/5 cells was extracted as described above and resuspended in TED buffer (50 mM Tris-HCl [pH 8.0], 10 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], and 5% glycerol) (60, 61), supplemented with 1 mM each benzamidine and PMSF. Nycodenz was added to membrane suspensions to a final concentration of 37.5% (wt/vol). Membrane suspensions were loaded under 5 to 35% (wt/vol) discontinuous Nycodenz gradients as previously described (36) and centrifuged to equilibrium in a swinging-bucket rotor at 100,000 × g for 20 h. After centrifugation, 10 500-μl fractions were collected; in this case, the five fractions from the top of each gradient were designated the low-density (LD) fractions, while the bottom five fractions were designated the high-density (HD) fractions. Samples were mixed with 2× Laemmli sample buffer, and proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
Membrane disassociation assays were performed as previously described (36, 62, 63), with the following minor modifications. The 20,000 × g membrane pellets from transfected BSR-T7/5 cells were collected as described above and resuspended in either 1 M NaCl, 100 mM sodium carbonate (Na2CO3; pH 11.0), or 1 M NaCl with 1.5% Triton X-100 and incubated on ice for 30 min. Nycodenz was added to the treated membrane suspensions to 37.5%, and the samples were centrifuged on discontinuous Nycodenz gradients as described above. After centrifugation, we collected 10 500-μl fractions as described above. In this case, the five LD fractions were pooled, as were the five HD fractions. Equal volume samples from each pooled fraction were mixed with 2× Laemmli sample buffer, resolved by SDS-PAGE, and analyzed by immunoblotting.
SDS-PAGE and immunoblot analysis.
All samples were prepared and resolved by SDS-PAGE as previously described (64). Transferred proteins were subjected to immunodetection with mouse anti-HA monoclonal or rabbit anti-MAO A/B polyclonal antibodies followed by incubation with goat anti-mouse or goat anti-rabbit polyclonal antibodies (Santa Cruz Biotechnology) conjugated to either horseradish peroxidase (HRP) or alkaline phosphatase (AP). Blots were developed using either the Amersham enhanced chemiluminescence (ECL) Prime Western blotting detection reagent (GE Healthcare Life Sciences) for HRP-conjugated secondary antibodies or Immun-Star AP substrate (Bio-Rad) for AP-conjugated secondary antibodies; blots were then exposed to X-ray film. Developed films were photographed with a Gel Doc XR Molecular Imager running Quantity One 1-D analysis software (Bio-Rad) to generate the digital images shown.
Visualization of RNA replication complexes.
Newly synthesized NoV RNA was labeled in transfected BSR-T7/5 cells by liposome-mediated introduction of 5-bromouridine 5′-triphosphate (BrUTP; Sigma-Aldrich), as previously described (65–67) but with the following modifications. At 19.5 hpt, cellular transcription was inhibited by a 30-min pretreatment with 20 μg/ml actinomycin D-mannitol (Act D; Sigma-Aldrich). Lipofectamine 2000 was mixed with serum-free Opti-MEM containing 10 mM BrUTP, and the mixture was incubated at room temperature to allow BrUTP-containing liposomes to form. The BrUTP-liposome complexes were diluted in Opti-MEM with 1% fetal bovine serum and 20 μg/ml Act D, applied to the cells, and incubated for 4 h (20 to 24 hpt). At 24 hpt, cellular mitochondria were stained with MTR for 1 h, fixed in 100% methanol, and processed for immunostaining and confocal microscopy as described above.
RNA isolation and Northern blot hybridization analysis.
Total cellular RNA was isolated from transfected BSR-T7/5 cells by using an RNeasy minikit (Qiagen) as described previously (68). RNA samples (0.5 μg for detection of positive strands or 2 μg for detection of negative strands) were separated on denaturing formaldehyde-agarose gels, stained with ethidium bromide, and transferred to charged nylon membranes as previously described (69). Northern blot hybridization was performed as described previously (70, 71), using 32P-labeled riboprobes specific for the positive or negative strand of NoV RNA3 that also detect RNA1 (47, 69, 72). The blots were visualized with a Personal Molecular Imager (Bio-Rad) and quantitated using Quantity One 1-D analysis software (Bio-Rad). Levels of RNA1 and RNA3 replication products were normalized to those of cellular 28S rRNA (visualized by ethidium bromide staining of the gel before transfer) and are presented as the percentages of the WT values. The relative RNA values from three independent experiments are presented as mean values ± standard deviations.
RESULTS
Expression and localization kinetics of the NoV RdRp in transfected mammalian cells.
Previous studies of NoV-infected tissues showed close association of NoV particles with ultrastructurally malformed mitochondria, implicating this organelle in the NoV infectious cycle (1, 43, 44). Additionally, the FHV RdRp associates with mitochondrial membranes in infected Drosophila and yeast cells (32, 36). To determine whether the NoV RdRp localizes to mitochondria in cells, we used a well-defined reverse genetics system in which NoV RNA replication can be initiated in mammalian cells from cloned cDNA copies of the NoV genomic RNAs (14, 47, 69, 72). For example, the entire replicative cycle can be initiated on expression of the NoV RNA1 and RNA2 cDNAs from T7 promoters in plasmid-transfected baby hamster kidney BSR-T7/5 cells (46) that constitutively express cytoplasmic T7 RNA polymerase (14, 47). The replicative cycle launched from this system mirrors the kinetics seen during NoV infection (47).
To facilitate detection of the NoV RdRp, we constructed a version of the full-length RNA1 cDNA in which the RdRp was engineered to contain a 10-amino-acid C-terminal influenza virus HA epitope tag. To ensure that the HA tag did not hinder the polymerase activity of the RdRp, we transfected BSR-T7/5 cells with plasmids pT7-N1 and pT7-N1-HA, which express WT or HA-tagged versions of the RdRp, respectively. At 24 hpt, we isolated total cellular RNA and analyzed accumulation of viral RNA replication products by Northern blotting hybridization using probes specific for the positive or negative strands of RNA1 that also could detect subgenomic RNA3. The HA-tagged version of the RdRp synthesized negative-strand RNA replication intermediates (Fig. 1B, lanes 5 and 6) and positive-strand RNA replication products (Fig. 1A, lanes 2 and 3) to levels that matched or exceeded that of the WT (Fig. 1E compared with H). Therefore, we concluded that the tagged RdRp was as functional as the WT in catalyzing viral RNA replication when expressed in transfected BSR-T7/5 cells and that the 30-nucleotide insertion encoding the tag did not affect the ability of RNA1 to serve as a template for RNA replication (Fig. 1).
FIG 1.
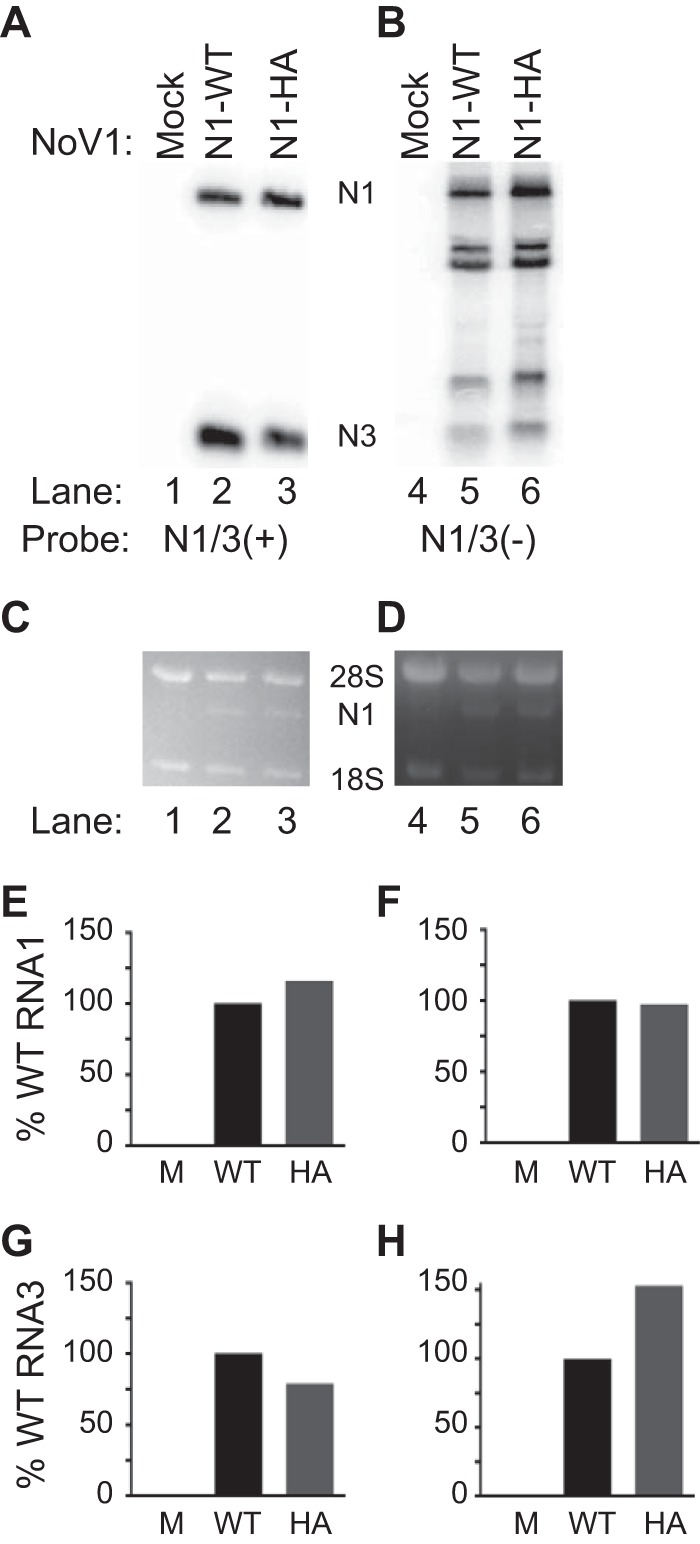
NoV RNA replication is unaffected by the presence of a C-terminal HA epitope tag. (A and B) Total cellular RNAs were isolated from BSR-T7/5 cells transfected with either pT7-N1 (WT) or tagged pT7-N1-HA, separated on denaturing gels, and subjected to Northern blot hybridization analysis using probes specific for the positive (A) or negative (B) strands of RNA1, which also detect subgenomic RNA3. (C and D) Prior to transfer, gels were stained with ethidium bromide to allow visualization of 18S and 28S rRNAs for use as loading controls. (E to H) Quantitation of positive (+) and negative (-) strands of NoV RNA1 (N1) and NoV RNA3 (N3) relative to WT levels are shown: N1(+) (E); N1(-) (F); N3(+) (G); N3 (-) (H). The relative RNA values for a single representative experiment are shown.
We explored the kinetics of RdRp expression in transfected mammalian cells by immunoblotting (Fig. 2) and by indirect immunofluorescence and confocal microscopy (Fig. 3). Duplicate sets of BSR-T7/5 cells were mock transfected or transfected with pT7-N1-HA and analyzed at 4-h intervals between 4 and 24 hpt. Cell lysates were prepared at each time point as described in Materials and Methods, and HA-tagged RdRp was analyzed by immunoblotting with a monoclonal antibody directed against the HA epitope tag (Fig. 2). As shown in Fig. 2, the RdRp was already detectable at 4 hpt, and its levels increased up to 20 hpt, with maximal expression maintained through at least 24 hpt.
FIG 2.
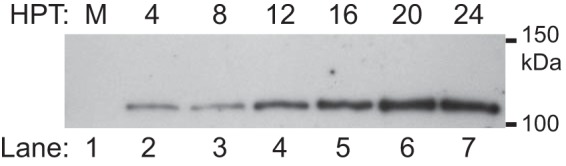
NoV RdRp expression was detected at early times posttransfection and reached maximal levels by 20 hpt. BSR-T7/5 cells were transfected with pT7-N1-HA, and PNLs were prepared at 4-h intervals between 4 and 24 hpt. Proteins were separated on an SDS-PAGE gel, and the HA-tagged RdRp was detected by immunoblotting with anti-HA antibodies.
FIG 3.

NoV RdRp localizes to mitochondria, and expression of the RdRp induces mitochondrial clustering in transfected BSR-T7/5 cells. BSR-T7/5 cells were transfected with pT7-N1-HA and incubated as described in the legend to Fig. 1. At each time point, the RdRp was detected by immunostaining with anti-HA primary and FITC-labeled secondary antibodies (green). Nuclei were visualized by staining with DAPI (blue), and the morphology of mitochondrial networks was visualized by staining with MitoTracker Red CM-H2XRos (red). Each panel represents a different time point: A, 4 hpt; B, 8 hpt; C, 12 hpt; D, 16 hpt; E, 20 hpt; F, 24 hpt. Immunofluorescence confocal microscopy images with merged signals are shown; the yellow signal in the merge images indicates colocalization. Closed arrowheads indicate RdRp expression in transfected cells; open arrowheads indicate untransfected cells. Bar, 10 μm.
The duplicate set of transfected cells was analyzed by confocal microscopy over the same time period. At each time point, mitochondria were stained with MTR, and the cells were fixed and permeabilized. The viral RdRp was visualized by immunostaining with anti-HA primary and FITC-labeled secondary antibodies followed by confocal microscopy, as described in Materials and Methods. For each panel, colocalization of the red and green signals was determined by using a squared Manders' OC of dual-color images, expressed as the percentage of overlap between the red (MTR) and green (FITC) pixels, as described previously (56). The OC values calculated for the images shown in Fig. 3 are summarized in Table 2.
In mock-transfected cells (data not shown) and in untransfected cells (Fig. 3B, cell in upper right corner designated by an open arrowhead), MTR treatment consistently resulted in a diffuse cytoplasmic staining pattern characteristic of mitochondria (red signal). A similar pattern was observed at 4 hpt in cells transfected with pT7-N1-HA (Fig. 3A). In both cases, MTR and FITC were not observed to colocalize, exhibiting an OC value of 0% (Table 2).
However, in contrast to the untransfected cells, at 8 hpt some of the mitochondria had begun to clump or cluster together (Fig. 3B, cell in lower left designated by a closed arrowhead). This clustering progressed over time thereafter, such that by 24 hpt all of the visible mitochondria appeared in clustered networks (Fig. 3B to F compared with A). In these same cells, immunofluorescence staining of the NoV RdRp showed the appearance of clustered structures in the cytoplasm that were excluded from the nucleus (Fig. 3, green signal designated by the closed arrowhead) and localized with MTR (Fig. 3F, yellow signal designated by the closed arrowhead), with high levels of red (MTR) and green (FITC) pixel overlap detected between 8 and 24 hpt, reaching a maximal OC of nearly 90% at 16 hpt (Table 2). Interestingly, the colocalization of the viral RdRp with MTR depended on the presence of actively replicating RNA1, since localization was diminished when the RdRp was expressed from a nonreplicating template (data not shown). These results suggest that NoV RdRp localized to mitochondria in transfected mammalian cells, and this interaction induced clustering of mitochondria into networks. Since maximal RdRp expression and mitochondrial clustering were observed at 24 hpt (Fig. 2 and 3, respectively), we selected this time point for all of the subsequent analyses presented here.
NoV RdRp localizes to the mammalian outer mitochondrial membrane.
The localization of NoV RdRp to mitochondria (Fig. 3) prompted us to determine the specific mitochondrial compartment (outer or inner membrane or matrix) that was involved, using a selective membrane permeabilization procedure. These experiments relied on the affinity of the detergent saponin for cholesterol in membranes. The plasma membrane contains a higher concentration of cholesterol than do mitochondrial membranes (53, 73). Therefore, at the low concentration used in this study, saponin permeabilizes the plasma membrane without compromising the integrity of the mitochondrial membranes. However, when cells are treated with the same concentration of saponin in the presence of Triton X-100, their mitochondrial membranes become permeabilized as well.
Therefore, BSR-T7/5 cells were transfected with pT7-N1-HA as before and we performed selective membrane permeabilization prior to analysis by immunofluorescence confocal microscopy (Fig. 4). Following appropriate incubation, the transfected cells were fixed with paraformaldehyde and permeabilized with saponin alone (Fig. 4A to F) or saponin and Triton X-100 (TX100) together (Fig. 4G to I), as described in Materials and Methods. They were then subjected to immunofluorescence confocal microscopy as before. The positions of the blue DAPI-stained nuclei are shown in the merged images (Fig. 4C, F, and I).
FIG 4.

NoV RdRp localizes to the surface of mitochondria in transfected BSR-T7/5 cells where its C terminus is exposed to the cytoplasm. BSR-T7/5 cells were transfected with pT7-N1-HA as before and incubated for 24 h. Cells were fixed with paraformaldehyde and permeabilized with saponin, either alone (A to F) or together with Triton X-100 (G to I). HA-tagged RdRp was immunostained with mouse anti-HA monoclonal primary and FITC-labeled goat anti-mouse secondary antibodies (green) (B, E, and H), as described in the legend to Fig. 2. MAO was immunostained with rabbit anti-MAO polyclonal primary and Texas Red (TR)-labeled goat anti-rabbit secondary antibodies (red) (A). COX3 was immunostained with goat anti-COX3 polyclonal primary and TR-labeled rabbit anti-goat secondary antibodies (red) (D and G). Merged signals, including DAPI staining of the nuclei, are shown in panels C, F, and I; the yellow signal in the merge image indicates colocalization. Closed arrowheads indicate RdRp expression in transfected cells; open arrowheads indicate untransfected cells. Bar, 10 μm.
When the cells were permeabilized with saponin alone, RdRp immunofluorescence appeared in clustered structures (Fig. 4B and C, closed arrowheads) that localized to the outer mitochondrial membrane marker MAO, as evidenced by an OC value of 82% (Table 2). Under these permeabilization conditions, the inner mitochondrial membrane marker COX3 was not detected (Fig. 4D to F), and the corresponding OC value was 0 (Table 2). When cells were treated with both saponin and TX100 to permeabilize the mitochondrial membranes, COX3 immunofluorescence became visible (Fig. 4G) and localized with RdRp immunofluorescence (Fig. 4H and I, closed arrowheads), with an OC value of 88% (Table 2). As before, the mitochondria in the cells expressing the RdRp exhibited clustering (Fig. 4, closed arrowheads), in marked contrast to the diffuse cytoplasmic staining pattern seen in the surrounding untransfected cells (Fig. 4, open arrowheads). These results suggest that the NoV RdRp interacted with the outer surface of mitochondria and was oriented such that its C terminus, which contains the HA tag, was exposed to the cytoplasm in transfected mammalian cells.
Predicted membrane-associated regions of the NoV RdRp.
As a first step in characterizing the interaction of the NoV RdRp with mitochondria, we used proteomic in silico prediction software to examine the likelihood of the RdRp interacting with mitochondrial or other cellular membranes. The N-terminal 123 amino acids of NoV protein A (39, 47) are shown schematically in Fig. 5A. Several of these programs (Psipred, TMpred, and SOSUI) predict the presence of two potential MARs at the RdRp N terminus, although the exact boundaries of these regions varied slightly among the programs used to predict them (50–52, 74). For example, Psipred's MEMSAT-SVM topology analysis predicted two MARs in the NoV RdRp, at aa 28 to 46 and aa 42 to 57; TMpred predicted potential transmembrane helices at aa 26 to 46 and aa 44 to 65; the SOSUI prediction tool predicted two MARs at aa 12 to 34 and aa 42 to 64. We show the SOSUI prediction in Fig. 5B, since it encompasses the other two predictions. These regions were designated MAR1 (aa 12 to 34), which contains the sequence SSALNIVSRALGYRVPLAKSLAL, and MAR2 (aa 42 to 64), which contains the sequence YKIIVHRRTLVAFLVIGPYATVV. Furthermore, TopPred II topology prediction software (49) predicted the presence of two hydrophobic regions near the N terminus of the RdRp (Fig. 5C) that overlapped the predicted MARs and are likely to interact with membranes.
FIG 5.
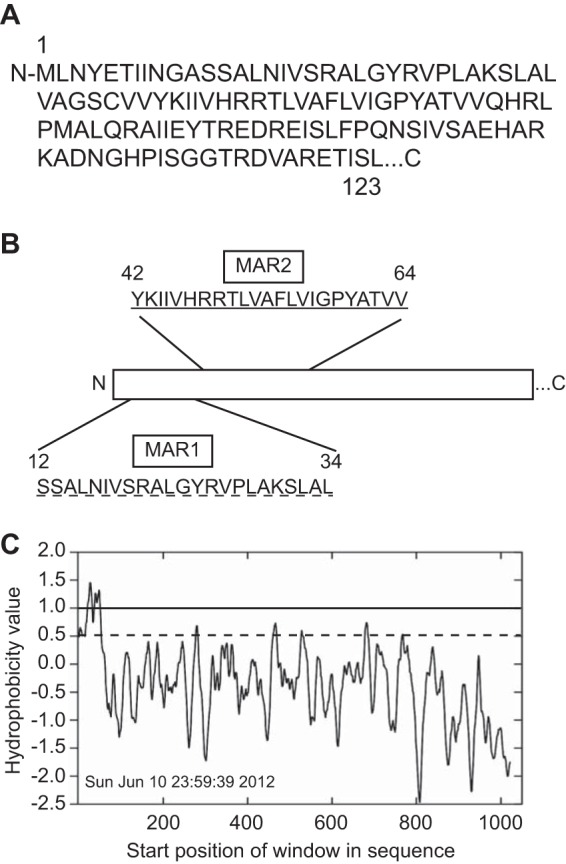
NoV RdRp is predicted to contain two membrane-associated regions. (A) The primary sequence of the N-terminal 123 amino acids of NoV protein A (accession number NP_077730.1) (39, 47). (B) SOSUI software (52) predicted two potential 23-aa transmembrane domains (underlined), located near the N terminus of the NoV RdRp, corresponding to aa 12 to 34 (MAR1) and 42 to 64 (MAR2), respectively. (C) The TopPred II topology prediction program identified a large hydrophobic region at the N terminus of the NoV RdRp that overlaps the predicted MARs. Hydrophobicity was calculated using the method of Kyte and Doolittle, with a core window size of 11 and a full window of 21 aa, and plotted as a function of amino acid position using GraphPad Prism software. Solid line, upper cutoff; dashed line, lower cutoff.
The MARs predicted for NoV do not share homology with those found within the FHV, GGNNV, or AHNV RdRp sequences (35, 36, 38), which also fail to overlap one another. This is not surprising, since the RdRps of alpha- and betanodaviruses share little (less than 30%) sequence identity at the amino acid level (39). While the RdRps of FHV and GGNNV are both predicted to contain helices that span the membrane (35, 36), the MARs in the NoV RdRp are unlikely to be membrane-spanning α-helices, due to the presence of charged residues within the predicted helical regions (Fig. 5A) not found for FHV and GGNNV. Nevertheless, the observation that the N-terminal region of the NoV RdRp encompasses two hydrophobic regions and contains multiple predicted MARs warranted further investigation into the RdRp's potential for association with membranes.
Localization of NoV RdRp MAR deletion mutants in transfected mammalian cells.
To determine whether the predicted MARs (Fig. 5) play a direct role in localizing the RdRp to mitochondria, we deleted the MAR1 and MAR2 regions from our HA-tagged RdRp expression constructs, either singly (N1Δ12-34-HA or N1Δ42-64-HA, respectively) or both of them together (N1Δ12-64-HA) and tested the effects of these deletions on RdRp localization in mammalian cells. BSR-T7/5 cells were transfected with plasmids expressing either WT or the MAR deletions, and at 24 hpt the cells were stained with MTR, fixed, and processed for immunofluorescence confocal microscopy as described in Materials and Methods.
For both single MAR deletion mutants (Δ12-34 and Δ42-64), the majority of the RdRp staining pattern (green signal designated by closed arrowheads) still localized to mitochondria, with OC values of 88% and 85%, respectively (Table 2), but many mitochondria now no longer appeared to be associated with the RdRp, judging by the reduction in yellow signal (closed arrowheads) in the merged images (Fig. 6D to I compared with A to C). In contrast, deletion of both MARs resulted in reduction of mitochondrial localization, with the level of overlap between MTR and FITC reduced to 47% (Table 2) and a loss of mitochondrial clustering (Fig. 6). Instead, the double deletion mutant exhibited altered localization, such that the RdRp was now detected in discrete sites within the cell (Fig. 6J to L compared with A to C). These results suggested that complete localization of the NoV RdRp to mitochondria requires both predicted MARs, raising the possibility that the RdRp may interact directly with mitochondrial membranes.
FIG 6.

Subcellular localization of the NoV RdRp is dependent on both MARs. BSR-T7/5 cells were transfected with pT7-N1-HA (B), pT7-N1Δ12–34-HA (E), pT7-N1Δ42–64-HA (H), or pT7-N1Δ12–64-HA (K). At 24 hpt, mitochondria were labeled with MTR (red), and cells were fixed and permeabilized. HA-tagged RdRp was detected by immunostaining (green), as described in the legend to Fig. 2. Merged signals, including DAPI staining of the nuclei (blue), are shown; the yellow signal of the merge image indicates colocalization. Closed arrowheads indicate RdRp expression in transfected cells; open arrowheads indicate untransfected cells. Bar, 10 μm.
Membrane association of the NoV RdRp in transfected mammalian cells.
To determine whether the NoV RdRp interacts with membranes in a MAR-dependent manner, we transfected BSR-T7/5 cells with either pT7-N1-HA or pT7-N1Δ12-64-HA. After incubation, we lysed the cells and performed differential centrifugation to collect intracellular membranes as described in Materials and Methods. PNLs were subjected to centrifugation at 20,000 × g to pellet the majority of intracellular membranes, including the mitochondrial membranes (the P20 fraction). The resulting supernatant (the S20 fraction) was subjected to further centrifugation at 100,000 × g to pellet microsomal membranes (the P100 fraction); cytosolic proteins will remain in the supernatant (the S100 fraction). The PNLs and the P20, P100, and S100 fractions were analyzed by immunoblotting for the presence of the RdRp by using the anti-HA antibodies (Fig. 7A).
FIG 7.
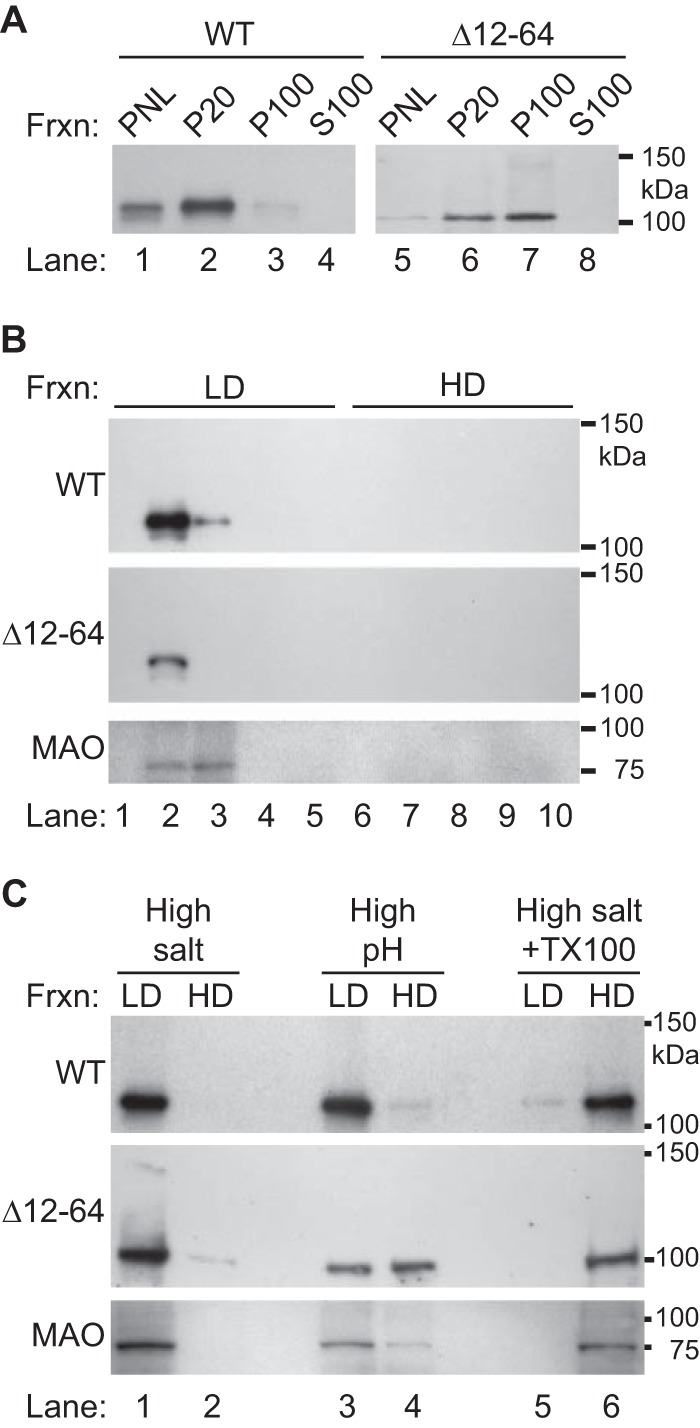
NoV RdRp cosediments with intracellular membranes, associates with the intracellular membrane fraction, and behaves as an integral membrane protein. Intracellular membranes were harvested from BSR-T7/5 cells transfected with either pT7-N1-HA or pT7-N1Δ12–64-HA by differential centrifugation. Proteins were separated on SDS-PAGE gels and detected by immunoblotting with antibodies specific for the HA tag or for the mitochondrial outer membrane protein MAO, as indicated in the figure. The relative positions of the protein standards are indicated at the right. (A) Proteins were examined in PNLs, membrane fractions P20 and P100 (pellets from the 20,000 and 100,000 × g spins, respectively), and the cytosolic fraction S100 (supernatant from 100,000 × g spins). The blot for pT7-N1Δ12–64-HA (right) was an overexposure necessitated by the production of lower levels of the mutant protein than the WT. (B) Intracellular membranes were fractionated on an equilibrium density gradient, as described in Materials and Methods, and proteins present in the LD and HD fractions were examined. (C) Membrane fractions were treated with high salt, high pH, or high salt with TX-100 prior to fractionation by equilibrium density gradient.
Under these conditions, the WT NoV RdRp was found to sediment mostly in the P20 fraction, although some was detected in the P100 fraction as well (Fig. 7A, lanes 2 and 3). It was not detected in the cytosolic fraction (Fig. 7A, lane 4). These results suggest that the RdRp may be a membrane-associated protein that sediments with the cellular fraction containing the majority of mitochondrial membranes (57–59).
Since the results of our confocal microscopy experiments suggested that both MARs were required for mitochondrial localization (Fig. 6), we hypothesized that deletion of the MARs from the RdRp would prevent its association with membranes. However, the Δ12-64 RdRp mutant was detected in both the P20 and P100 membrane pellets rather than in the cytosolic S100 fraction, as we had predicted (Fig. 7A, lanes 5 to 8). In contrast to the WT RdRp, however, the mutant was more evenly distributed between the low- and high-speed pellets. These results suggested that Δ12-64 RdRp mutant remained associated with membrane-containing fractions even in the absence of the MARs, although it no longer associated with mitochondria (Fig. 6).
To further explore whether the NoV RdRp specifically interacts with intracellular membranes, we used equilibrium density gradients to analyze the flotation profiles of the WT and mutant RdRps isolated from transfected BSR-T7/5 cells (Fig. 7B), as described in Materials and Methods. We collected fractions of equal volumes beginning at the top of the gradient and analyzed the distribution of the NoV RdRp and the outer mitochondrial membrane protein MAO by immunoblotting. Under these conditions, intracellular membranes float to the LD fractions closer to the top of the gradient, where they form a membrane “wafer” that also contains membrane-associated proteins (32, 36, 75). In contrast, if a protein is not specifically associated with a membrane, it will be present in the HD fractions in the bottom portion of the gradient.
The WT NoV RdRp floated to fractions 2 and 3 (Fig. 7B, top panel), which corresponded to the fractions harboring the membrane wafer. MAO, which is an integral membrane protein, was also found in the same two fractions (Fig. 7B, bottom panel). These results support our previous finding that the NoV RdRp specifically interacts with intracellular membranes in transfected BSR-T7/5 cells. In contrast, the Δ12-64 mutant protein also specifically floated with intracellular membranes and was detected in fraction 2 of the gradient (Fig. 7B, middle panel). Together, the data presented in Fig. 6 and 7A and B suggest that, while the WT RdRp associated with mitochondrial membranes, the Δ12-64 version of the protein associated with another cosedimenting membrane present in the P20 fraction but was no longer associated with mitochondria. The implications of the mutant's altered localization will be explored further in the Discussion. Nevertheless, we further characterized the nature of the NoV RdRp's interaction with mitochondrial membranes by examining the conditions required to disrupt the interaction and remove it from the membranes.
Membrane dissociation suggests the NoV RdRp is an integral membrane protein.
The nature of the NoV RdRp membrane association was examined by using extraction reagents known to separate integral from peripheral membrane proteins. Under high salt or alkaline pH conditions, peripheral membrane proteins are released from the membrane, while integral membrane proteins remain bound to intact membrane sheets; removal of integral membrane proteins is facilitated by solubilization with detergents (62, 63). Therefore, the P20 membrane fractions from cells transfected with WT or mutant versions of the RdRp were extracted, treated with high salt, high pH, or high salt with TX-100, and fractionated on an equilibrium density gradient as described in Materials and Methods. The gradients were divided into equal volume fractions, and MAO and RdRp proteins were detected by immunoblotting. As before, fractions from the upper portion of the gradient containing the membrane wafer were designated the LD fractions, and those from the bottom portion containing the soluble proteins were designated the HD fractions.
Membrane association of the NoV RdRp was not disrupted by the presence of high salt or high pH (Fig. 7C, top panel, lanes 1 to 4), but the protein was removed from the LD membrane fraction by treatment with high salt and detergent together (Fig. 7C, top panel, lanes 5 and 6). Similar results were obtained for the outer mitochondrial integral membrane protein MAO under the same conditions (Fig. 7C, bottom panel, lanes 1 to 6). These results suggest the NoV RdRp is associated with intracellular membranes from transfected mammalian cells, with its behavior most resembling that of an integral membrane protein.
In contrast, some of the Δ12-64 mutant RdRp was removed from membranes by high salt (Fig. 7C, middle panel, lanes 1 and 2), and just over half of it was dissociated by treatment with alkaline pH (Fig. 7C, middle panel, lanes 3 and 4). All of the detectable mutant RdRp was solubilized by treatment with high salt and TX-100 (Fig. 7C, middle panel, lanes 5 and 6). The partial release of the mutant RdRp by high salt and high pH suggests that deletion of aa 12 to 64 alters the RdRp mechanism of membrane association. Clearly, sequences within the 52-aa MAR play a major role in anchoring the RdRp to intracellular membranes, although the exact nature of that interaction remains to be determined.
The levels of the mutant RdRp proteins detected by confocal microscopy (Fig. 6) or by immunoblotting (Fig. 7) were decreased relative to the WT. However, it was unclear whether this was due to its being synthesized at lower levels or whether it was being degraded at an increased rate after synthesis. To test whether the mutant RdRp was being degraded via a proteasome-mediated pathway, we examined the effect of the proteasome inhibitor MG132 (48) on WT and mutant RdRp accumulation. Cells were transfected with the WT or Δ12-64 RdRp expression plasmids as before and left untreated or treated with MG132 for the 8 h prior to harvest (starting at 16 hpt), and proteins were subjected to immunoblot analysis with the anti-HA antibodies as before (Fig. 8). In the presence of MG132, the levels of RdRp increased for both WT and the Δ12-64 mutant (Fig. 8, lanes 4 and 5, respectively). These data suggest that, while there is turnover of the RdRp protein during the NoV replicative cycle, the Δ12-64 deletion mutant does not appear to exhibit increased proteasomal degradation compared to the WT RdRp (Fig. 8). Since there was no apparent increase in degradation of the mutant RdRp, we concluded that it is being synthesized at lower levels. Since RNA1 serves as both the mRNA for RdRp synthesis and RNA replication template, lower levels of RdRp synthesis could result from an inability of the mutant to establish RNA replication complexes or to replicate RNA1. We examine each possibility in the following sections.
FIG 8.
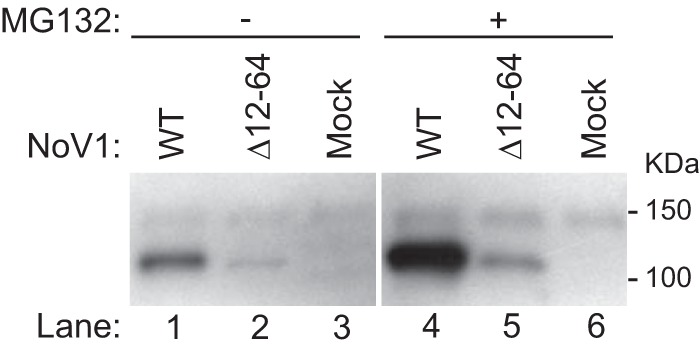
Deletion of the MARs reduced RdRp expression levels but did not appear to increase proteasome-mediated degradation. BSR-T7/5 cells were transfected with pT7-N1-HA or pT7-N1Δ12–64-HA as described for Fig. 1. Cells in duplicate wells were left untreated (lanes 1 to 3) or treated with the proteasome inhibitor MG132 for the 8 h prior to harvest (lanes 4 to 6). Proteins were detected by immunoblotting with antibodies specific for the HA tag as described in the legend to Fig. 2. The relative positions of the Precision Plus (Bio-Rad) protein standards are indicated on the right.
NoV RdRp-catalyzed RNA synthesis localizes to mitochondria in transfected mammalian cells.
In light of the localization of the RdRp to mitochondrial membranes, we wondered whether this also represented the site of RC formation. We therefore transfected BSR-T7/5 cells with pT7-N1-HA and labeled the RdRp-catalyzed viral RNA synthesis products with BrUTP as described in Materials and Methods. Labeling was performed in the presence of Act D to inhibit transcription by cellular RNA polymerase II; under these conditions, only the products of the viral RdRp were labeled. Mitochondria were also labeled with MTR, and cells were fixed and permeabilized. BrUTP-labeled viral RNA was immunostained with monoclonal antibromodeoxyuridine primary and FITC-labeled secondary antibodies, as described in Materials and Methods.
Mock-transfected cells (Fig. 9A) and untransfected cells (Fig. 9D; the cell on the right, indicated by an open arrowhead) showed no BrUTP labeling of newly synthesized RNA (green signal) in the presence of Act D and resulted in OC values of 0 (Table 2); these cells served as internal negative controls. In cells transfected with pT7-N1-HA, the MTR staining pattern (red signal) displayed the distinct clustered networks we observed previously (Fig. 9D and F; the cell on the left, indicated by the closed arrowheads). In these same cells, we detected newly synthesized BrUTP-labeled RNA (Fig. 9E, green signal indicated by closed arrowheads), which appeared in clustered cytoplasmic structures that localized with MTR in the merged image (Fig. 9F, yellow signal indicated by closed arrowheads), with an OC value of 94% for MTR and FITC (Table 2). These results suggested that NoV RNA synthesis occurs in close proximity to the observed clustered mitochondrial networks, which also harbor the RdRp, and that these clusters represent membrane-associated RCs.
FIG 9.

NoV RdRp-catalyzed RNA synthesis localizes to mitochondria in transfected BSR-T7/5 cells. BSR-T7/5 cells were transfected with pT7-N1-HA and incubated for 19.5 h. Cells were pretreated with Act D for 30 min, then newly synthesized viral RNA was labeled with BrUTP for 4 h in the presence of Act D. At 24 hpt, mitochondria were stained with MTR (red), and cells were fixed and permeabilized. BrU-labeled viral RNA was detected by immunostaining with monoclonal anti-BrdU primary and FITC-labeled secondary antibodies (green; BrU). Merged signals, including DAPI staining of the nuclei (blue), are shown; the yellow signal in the merge image indicates colocalization. Closed arrowheads indicate BrU-labeled viral RNA in transfected cells; open arrowheads indicate untransfected cells. Bar, 10 μm.
Deletion of one or both MARs results in severe defects in RNA replication.
Since deletion of the MARs disrupted mitochondrial localization of the RdRp and the RCs, we tested the effects of these mutations on viral RNA replication. BSR-T7/5 cells were transfected with plasmids expressing the WT RdRp, the single MAR mutations N1Δ12-34 and N1Δ42-64, or the double mutation with both MARs deleted (N1Δ12-64). After incubation, total RNA was isolated and separated on denaturing formaldehyde-agarose gels, and viral RNA replication products were detected by Northern blotting hybridization by using probes specific for the positive (Fig. 10A) or negative (Fig. 10B) strands of RNA1 and RNA3 as described for Fig. 1 and in references 47, 69, and 72. The positive and negative strands of RNA1 and RNA3 were quantitated by densitometry, as shown in Fig. 10E to H.
FIG 10.
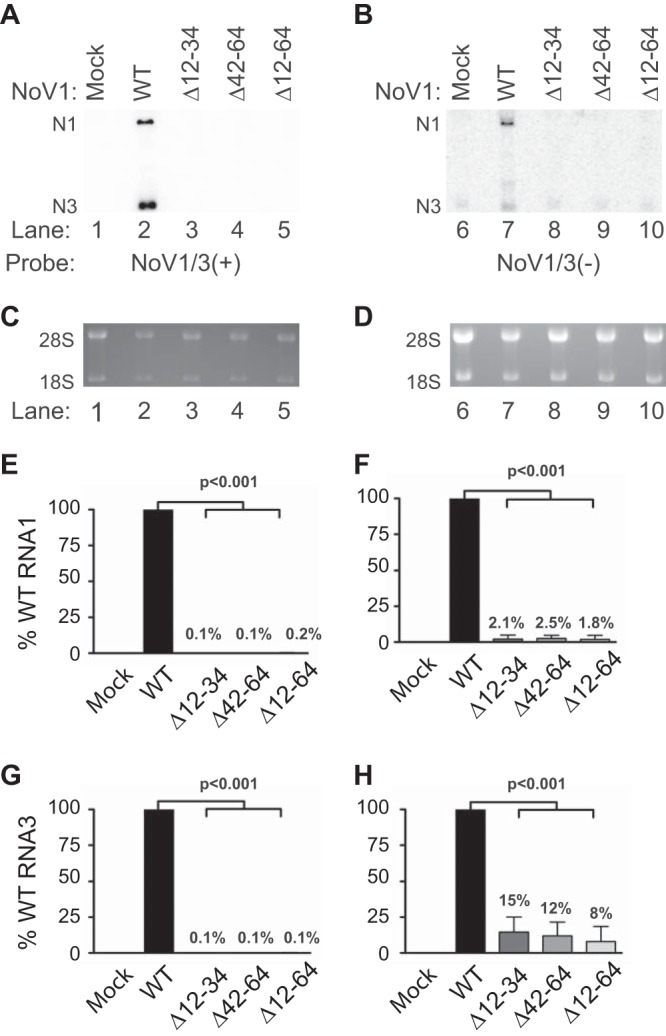
Deletion of the MARs of NoV RdRp results in a severe defect in RNA replication. Total cellular RNAs were isolated from BSR-T7/5 cells transfected with either pT7-N1 (WT), pT7-N1Δ12–34 (ΔMAR1), pT7-N1Δ42–64 (ΔMAR2), or pT7-N1Δ12–64 (ΔMAR1 + 2), separated on denaturing gels, and subjected to Northern blot hybridization analysis as described in the legend to Fig. 1. The positive (A) or negative (B) strands of RNA1 and RNA3 were detected as described; as before, rRNAs were stained with ethidium bromide for use as loading controls (C and D, respectively). (E to H) Quantitation of (+) and (-) strands of N1 and N3 relative to WT is shown for N1 (+) (E), N1 (-) (F), N3 (+) (G), and N3 (-) (H). The relative RNA values from three independent experiments are presented as mean values ± standard deviations.
All three mutations resulted in severe defects in RNA1 replication and RNA3 synthesis (Fig. 10). In some experiments, we detected low levels of the negative strand of RNA3 for each mutant (Fig. 10H). However, the presence of a cross-reactive RNA species in the mock-transfected sample (Fig. 10B, lane 6) complicated interpretation of this result, so it is possible that this apparent synthesis of an RNA replication intermediate represents a technical artifact. The density from the mock-transfected lane was therefore subtracted from the other lanes to reflect this background species (Fig. 10H). The inability of the double deletion to replicate was not unexpected in light of our observation that this mutant fails to form RCs (Fig. 9). However, even the single deletions, which appeared to retain some ability to interact with mitochondria (Fig. 6), failed to synthesize detectable RNA replication products.
DISCUSSION
Localization of RCs to intracellular membranes is a hallmark of positive-strand RNA viruses. In this study, we showed that the NoV RdRp localized to the outer mitochondrial membrane in mammalian cells at 37°C, consistent with the presence of two computer-predicted MARs. Expression of the RdRp in cells induced mitochondria to cluster into distinct cytoplasmic networks between 4 and 8 hpt, but only when the RdRp was expressed from a replicable template (data not shown). While not yet visualized at the ultrastructural level, we hypothesize that the clustered phenotype seen in cultured cells represents the same altered mitochondrial morphology and mitochondrial aggregation previously described for NoV-infected muscle tissue (1, 43, 44). We further showed that NoV RNA synthesis also localizes to these RdRp-induced mitochondrial networks, further establishing mitochondria as the site of viral RC formation.
Our data suggest that the RdRp interacts with the mitochondrial outer membrane, although it remains possible that the RdRp localizes to mitochondria-associated endomembranes instead. The results of biochemical assays favor our original interpretation, although additional experiments may be required to resolve the issue. Nevertheless, the NoV RdRp cosedimented with the intracellular membrane fraction (P20), which that contains the mitochondria, rather than in the P100 fraction, where endosomal proteins would be found. The RdRp's specific interaction with membranes in this fraction was confirmed by a flotation assay and mimicked that of a mitochondrial integral membrane protein, MAO.
While the topological data presented here suggest that the NoV RdRp is an outer mitochondrial membrane protein, we pondered the nature of its interaction with these membranes. We show in Fig. 5 that the NoV RdRp contains two extremely hydrophobic regions at its N terminus. Several secondary structure prediction programs (Psipred, TMpred, and SOSUI) predicted the presence of two MARs within the bounds of aa 12 to 65 of the NoV RdRp N terminus. We decided to pursue the SOSUI predictions, since they overlapped the predictions of the other programs and because the SOSUI-predicted MARs are separate stretches of amino acids.
Proper localization of viral nonstructural proteins (including RdRps) to membranes and membrane association are dependent on the integrity of the primary and secondary structures of the protein. For the NoV RdRp, these properties are mediated by the MARs, since their deletion from the RdRp results in a multitude of deleterious phenotypes. Specifically, their deletion resulted in defects in localization of the RdRp to mitochondria and RdRp-induced clustering of the mitochondria (Fig. 6), in RdRp protein levels (Fig. 7 and 8), in localization of RCs (Fig. 9), and in RNA replication (Fig. 10). Interestingly, the double deletion mutant retained its ability to associate with intracellular membranes (Fig. 7). However, localization of the mutant RdRp changed from mitochondrial to unidentified but discrete sites in the cytoplasm (Fig. 6). The presence of the mutant RdRp in both the P20 and P100 fractions in the differential centrifugation experiments and its retention in the LD portion of the density gradients (Fig. 7) lend credence to the idea that these discrete sites are associated with cellular membranes.
Our observation that the mutant RdRp exhibits characteristics of a peripheral membrane protein (Fig. 7) suggest that the MARs play a role in the interaction of the NoV RdRp with mitochondrial membranes as an integral membrane protein. Yet, the question remains as to why an RdRp that lacks the MARs remains associated with membranes rather than becoming soluble, as we had expected. In light of the observation that many viral proteins contain amphipathic α-helices important for association with membranes and for RNA replication (19), similar structural elements may play a role in the interaction between the NoV RdRp and the outer mitochondrial membrane. We hypothesize that these interactions remain intact in the MAR deletion mutants. It is also possible that the mutant (and probably the WT) RdRps interact with one or more membrane proteins that help to direct them to membranes.
As noted above, deletion of the MARs also appeared to result in a decrease in RdRp protein expression levels (Fig. 7 and 8). However, it remained unclear whether less of the mutant RdRp was detected in these experiments because it was being degraded after synthesis or because it was being synthesized at lower levels. To test whether the mutant RdRp was being degraded via a proteasome-mediated pathway, we examined the effect of the proteasome inhibitor MG132 (48) on accumulation of the mutant protein. In the presence of MG132, the levels of RdRp accumulation increased for both the WT and the Δ12-64 mutant (Fig. 8). These data suggest that, while there is turnover of the RdRp protein during the NoV replicative cycle, the Δ12-64 deletion mutant does not appear to exhibit increased proteasomal degradation compared to the WT RdRp (Fig. 8). Since there was no apparent increase in degradation of the mutant RdRp, we conclude that it is being synthesized at lower levels. This interpretation is supported by the failure of the mutant RNA1 to replicate (Fig. 10), resulting in greatly reduced levels of the mRNA from which the mutant RdRp is translated.
Although the NoV RdRp was shown here to be an integral membrane protein with MAR structures that play critical roles in RC formation, it is not yet clear how this interaction is mediated. The RdRps of the related nodaviruses FHV and GGNNV are integral membrane proteins that interact with mitochondrial membranes via transmembrane α-helices (35, 36). However, these RdRps are markedly different from that of NoV, with the NoV and FHV RdRps sharing 44% amino acid sequence identity (39) and those of NoV and GGNNV sharing only 26% amino acid sequence identity, as we calculated using ClustalW2 software (data not shown). We therefore hypothesized that the mechanism of interaction between the NoV RdRp and mitochondrial membranes differs from those shared by the FHV and GGNNV RdRps. The presence of charged residues and prolines within both NoV MARs makes it unlikely that the NoV RdRp actually spans the outer mitochondrial membrane via transmembrane α-helices within this region. Instead, it may remain on the outer membrane as a monotopic integral membrane protein, nesting in the outer leaflet with parallel hydrophobic helices, as has been described for other membrane-associated proteins (76, 77), including the endoplasmic reticulum-localized ATPase torsin A (78).
Likely as a direct consequence of disrupting mitochondrial integrity (79), FHV and GGNNV have been previously shown to induce apoptosis in infected tissues (80–83). FHV infection induces apoptosis in cultured Drosophila DL-1 cells by inhibiting cellular translation, thereby resulting in depletion of Drosophila inhibitor-of-apoptosis protein (DIAP1) (83), while GGNNV-infected sea bass and COS-7 cells undergo apoptosis through activation of a caspase-dependent pathway (80–82). In contrast, NoV infection in cultured mammalian cells does not appear to induce apoptosis or, in fact, any apparent cytopathic effect (CPE). For example, no morphological changes were observed in BHK21 cells transfected with purified NoV genomic RNA1 and RNA2 even at 24 hpt, when abundant RNA replication and progeny virus particles were detected (11). Furthermore, during the time course of this and our previous studies (14, 47), cells expressing the NoV RdRp did not exhibit any of the morphological hallmarks of apoptosis, including cytoplasmic vacuolization, membrane blebbing, rounding, nuclear fragmentation, or detachment from the growth surface. Instead, in this study the cells appeared healthy for up to 24 hpt, and in a previous study they remained healthy for 12 days postinfection (47). Furthermore, since previous studies showed that caspase-3 can cleave the HA tag from expressed proteins (84), our ability to detect the HA-tagged NoV RdRp in transfected mammalian cells suggests that activation of an intrinsic, caspase-mediated apoptotic pathway (79) in these cells is unlikely. Therefore, while it is clear that NoV establishes RCs in conjunction with mitochondrial membranes via a direct interaction with its RdRp, the mitochondria appear to retain their integrity. This is unique among the nodaviruses studied to date and may represent a unique adaptation in a virus whose natural host range has expanded to include mammals. Clearly, further study of the role of mitochondria in the NoV life cycle is warranted.
These results may also shed new light on the mechanism of NoV pathogenesis in infected mammals and insects. NoV replicates in the musculature of the lower back and hind limbs of suckling mice and suckling hamsters, leading to flaccid paralysis of the hind limbs and death (44, 85, 86). Similarly, viral replication in the muscle tissues of greater wax moth (Galleria mellonella) larvae results in fatal paralysis of the hind segments (1, 43, 44). In infected muscle cells from mice and G. mellonella larvae, the mitochondria clustered around the nucleus and, late in infection, exhibited altered architecture. Strikingly, the tissues exhibited progressive disorganization of the muscle fibers and fibrils, as the mitochondria became altered and the cells filled with paracrystalline arrays of progeny viral particles (44). It has been hypothesized that the paralysis seen in these animals is a result of this disruption of the muscle fibrils by altered mitochondria (1, 43, 44).
Our results support the hypothesis that the tissue damage seen in NoV-infected animals is the direct result of interaction between the RdRp and the outer mitochondrial membrane, leading to clustering of mitochondria into large networks, thereby reorganizing a vital organelle responsible for energizing the cell—all secondary effects of NoV replication complex formation. The work described here establishes a basis for further characterization of the interaction between the NoV RdRp and membranes in cultured mammalian cells, leading to formation of viral RCs in the outer mitochondrial membrane. Furthermore, this report contributes to elucidation of the strategic genome replication mechanisms used by other positive-strand RNA viruses for localization of RdRps and other viral nonstructural proteins to membranes and establishment of RCs. We are in the process of further characterizing the mechanisms by which NoV RCs are formed.
ACKNOWLEDGMENTS
This work was supported by NSF grant DMS0800272 to Ming-Ying Leung (UTEP), Michela Taufer (University of Delaware), and K.L.J. and by a UTEP University Research Institute grant to K.L.J. V.U.G. and S.M. were supported by NIH-funded Research Scholars Program grant 2R25GM069621-10 to Renato J. Aguilera (UTEP). This project was also supported by grant 8G12MD007592 from the National Institute on Minority Health and Health Disparities (NIMHD), a component of the National Institutes of Health (NIH), which also supported the salary of A.V.-R.
We thank the UTEP Border Biomedical Research Center (BBRC) Genomic Analysis, Biomolecule Analysis, and Cytometry, Screening, and Imaging Core facilities. We also thank Ana Betancourt for DNA sequencing. We thank Alexandra Navarro and Debarko Banerji for assisting with critical experiments. We also thank other members of the Johnson laboratory for many helpful discussions and Ricardo Bernal, Siddhartha Das, and Arshad Khan (all from UTEP) for critical reviews of the manuscript.
The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official views of NIMHD or NIH.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.Garzon S, Charpentier G. 1991. Nodaviridae, p 351–370 In Adams JR, Bonami JR. (ed), Atlas of invertebrate viruses. CRC Press, Boca Raton, FL [Google Scholar]
- 2.Scherer WF, Buescher EL, Flemings MB, Noguchi A, Scanlon J. 1959. Ecologic studies of Japanese encephalitis virus in Japan. III. Mosquito factors, zootropism and vertical flight of Culex tritaeniorhynchus with observations on variations in collections from animal-baited traps in different habitats. Am. J. Trop. Med. Hyg. 8:665–677 [PubMed] [Google Scholar]
- 3.Scherer WF, Buescher EL, McClure HE. 1959. Ecologic studies of Japanese encephalitis virus in Japan. V. Avian factors. Am. J. Trop. Med. Hyg. 8:689–697 [DOI] [PubMed] [Google Scholar]
- 4.Scherer WF, Moyer JT, Izumi T, Gresser I, McCown J. 1959. Ecologic studies of Japanese encephalitis virus in Japan. VI. Swine infection. Am. J. Trop. Med. Hyg. 8:698–706 [DOI] [PubMed] [Google Scholar]
- 5.Scherer WF, Hurlbut HS. 1967. Nodamura virus from Japan: a new and unusual arbovirus resistant to diethyl ether and chloroform. Am. J. Epidemiol. 86:271–285 [DOI] [PubMed] [Google Scholar]
- 6.Bailey L, Scott HA. 1973. The pathogenicity of Nodamura virus for insects. Nature 241:545. [DOI] [PubMed] [Google Scholar]
- 7.Tesh RB. 1980. Infectivity and pathogenicity of Nodamura virus for mosquitoes. J. Gen. Virol. 48:177–182 [Google Scholar]
- 8.Ball LA, Johnson KL. 1998. Nodaviruses of insects, p 225–267 In Miller LK, Ball LA. (ed), The insect viruses. Plenum Publishing Corporation, New York, NY [Google Scholar]
- 9.Ball LA, Johnson KL. 1999. Reverse genetics of nodaviruses. Adv. Virus Res. 53:229–244 [DOI] [PubMed] [Google Scholar]
- 10.Ball LA. 1992. Cellular expression of a functional nodavirus RNA replicon from vaccinia virus vectors. J. Virol. 66:2335–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ball LA, Amann JM, Garrett BK. 1992. Replication of Nodamura virus after transfection of viral RNA into mammalian cells in culture. J. Virol. 66:2326–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallagher TM, Friesen PD, Rueckert RR. 1983. Autonomous replication and expression of RNA1 from Black beetle virus. J. Virol. 46:481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Li WX, Ding SW. 2002. Induction and suppression of RNA silencing by an animal virus. Science 296:1319–1321. 10.1126/science.1070948 [DOI] [PubMed] [Google Scholar]
- 14.Johnson KL, Price BD, Eckerle LD, Ball LA. 2004. Nodamura virus nonstructural protein B2 can enhance viral RNA accumulation in both mammalian and insect cells. J. Virol. 78:6698–6704. 10.1128/JVI.78.12.6698-6704.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W-X, Li H, Lu R, Li F, Dus M, Atkinson P, Brydon EWA, Johnson KL, Garcia-Sastre A, Ball LA, Palese P, Ding S-W. 2004. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. U. S. A. 101:1350–1355. 10.1073/pnas.0308308100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan CS, Ganem D. 2005. A virus-encoded inhibitor that blocks RNA interference in mammalian cells. J. Virol. 79:7371–7379. 10.1128/JVI.19.72.7371-7379.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahlquist P, Noueiry AO, Lee W-M, Kushner DB, Dye BT. 2003. Host factors in positive-strand RNA virus genome replication. J. Virol. 77:8181–8186. 10.1128/JVI.77.15.8181-8186.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6:363–374. 10.1038/nrmicro1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139–173. 10.1007/3-540-26764-6_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bienz K, Bienz-Isler G, Egger D, Weiss M, Loeffler H. 1970. Coxsackie virus infection in skeletal muscles of mice. An electron microscopic study. II. Appearance and fate of virus progeny. Arch. Gesamte Virusforsch. 31:257–265 [DOI] [PubMed] [Google Scholar]
- 21.Bienz K, Egger D, Pfister T. 1994. Characteristics of the poliovirus replication complex. Arch. Virol. Suppl. 9:147–157 [DOI] [PubMed] [Google Scholar]
- 22.Bienz K, Egger D, Pfister T, Troxler M. 1992. Structural and functional characterization of the poliovirus replication complex. J. Virol. 66:2740–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bienz K, Egger D, Troxler M, Pasamontes L. 1990. Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J. Virol. 64:1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bienz-Isler G, Bienz K, Weiss M, Loeffler H. 1970. Coxsackie virus infection in skeletal muscles of mice. An electron microscopic study. I. Cell and nucleus alterations. Arch. Gesamte Virusforsch. 31:247–256 [DOI] [PubMed] [Google Scholar]
- 25.Schlegel A, Giddings TH, Jr, Ladinsky MS, Kirkegaard K. 1996. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70:6576–6588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlegel A, Kirkegaard K. 1995. Cell biology of enterovirus infection, p 135–154 In Rotbart HA. (ed), Human enterovirus infections. ASM Press, Washington, DC [Google Scholar]
- 27.Schwartz M, Chen J, Janda M, Sullivan M, den Boon J, Ahlquist P. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505–514. 10.1016/S1097-2765(02)00474-4 [DOI] [PubMed] [Google Scholar]
- 28.Song BH, Yun SI, Choi YJ, Kim JM, Lee CH, Lee YM. 2008. A complex RNA motif defined by three discontinuous 5-nucleotide-long strands is essential for flavivirus RNA replication. RNA 14:1791–1813. 10.1261/rna.993608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJM, Locker JK. 1999. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 73:7641–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Franco A, Russo M, Martelli GP. 1984. Ultrastructure and origin of cytoplasmic multivesicular bodies induced by Carnation Italian ringspot virus. J. Gen. Virol. 65:1233–1237 [Google Scholar]
- 31.Magliano D, Marshall JA, Bowden DS, Vardaxis N, Meanger J, Lee J-Y. 1998. Rubella virus replication complexes are virus-modified lysosomes. Virology 240:57–63 [DOI] [PubMed] [Google Scholar]
- 32.Miller DJ, Schwartz MD, Ahlquist P. 2001. Flock House virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol. 75:11664–11676. 10.1128/JVI.75.23.11664-11676.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubino L, Russo M. 1998. Membrane targeting sequences in tombusvirus infections. Virology 252:431–437 [DOI] [PubMed] [Google Scholar]
- 34.Russo M, Di Franco A, Martelli GP. 1983. The fine structure of Cymbidium ringspot virus infections in host tissues. III. Role of peroxisomes in the genesis of multivesicular bodies. J. Ultrastruct. Res. 82:52–63 [DOI] [PubMed] [Google Scholar]
- 35.Guo YX, Chan S-W, Kwang J. 2004. Membrane association of greasy grouper nervous necrosis virus protein A and characterization of its mitochondrial localization targeting signal. J. Virol. 78:6498–6508. 10.1128/JVI.78.12.6498-6508.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller DJ, Ahlquist P. 2002. Flock House virus RNA polymerase is a transmembrane protein with amino-terminal sequences sufficient for mitochondrial localization and membrane insertion. J. Virol. 76:9856–9867. 10.1128/JVI.76.19.9856-9867.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller DJ, Schwartz MD, Dye BT, Ahlquist P. 2003. Engineered retargeting of viral RNA replication complexes to an alternative intracellular membrane. J. Virol. 77:12193–12202. 10.1128/JVI.77.22.12193-12202.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mezeth KB, Nylund S, Henriksen H, Patel S, Nerland AH, Szilvay AM. 2007. RNA-dependent RNA polymerase from Atlantic halibut nodavirus contains two signals for localization to the mitochondria. Virus Res. 130:43–52. 10.1016/j.virusres.2007.05.014 [DOI] [PubMed] [Google Scholar]
- 39.Johnson KN, Johnson KL, Dasgupta R, Gratsch T, Ball LA. 2001. Comparisons among the larger genome segments of six nodaviruses and their encoded RNA replicases. J. Gen. Virol. 82:1855–1866 http://vir.sgmjournals.org/content/82/8/1855.long. [DOI] [PubMed] [Google Scholar]
- 40.Johnson KN, Johnson KL, Dasgupta R, Gratsch T, Ball LA. 2001. Corrigendum. Comparisons among the larger genome segments of six nodaviruses and their encoded RNA replicases. J. Gen. Virol. 82:3119. [DOI] [PubMed] [Google Scholar]
- 41.Gallagher TM. 1987. Synthesis and assembly of nodaviruses. Ph.D. thesisUniversity of Wisconsin—Madison, Madison, WI [Google Scholar]
- 42.Price BD, Ahlquist P, Ball LA. 2002. DNA-directed expression of an animal virus RNA for replication-dependent colony formation in Saccharomyces cerevisiae. J. Virol. 76:1610–1616. 10.1128/JVI.76.4.1610-1616.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garzon S, Charpentier G, Kurstak E. 1978. Morphogenesis of the Nodamura virus in the larvae of the lepidopteran Galleria mellonella (L.). Arch. Virol. 56:61–76 [DOI] [PubMed] [Google Scholar]
- 44.Garzon S, Strykowski H, Charpentier G. 1990. Implication of mitochondria in the replication of Nodamura virus in larvae of the lepidoptera, Galleria mellonella (L.) and in suckling mice. Arch. Virol. 113:165–176 [DOI] [PubMed] [Google Scholar]
- 45.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1994. Current protocols in molecular biology. John Wiley & Sons, Inc., Somerset, NJ [Google Scholar]
- 46.Buchholz UJ, Finke S, Conzelmann K-K. 1999. Generation of Bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson KL, Price BD, Ball LA. 2003. Recovery of infectivity from cDNA clones of Nodamura virus and identification of small nonstructural proteins. Virology 305:436–451. 10.1006/viro.2002.1769 [DOI] [PubMed] [Google Scholar]
- 48.Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. 1996. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 119:572–576 [DOI] [PubMed] [Google Scholar]
- 49.Claros MG, von Heijne G. 1994. TopPred II: an improved software for membrane protein structure predictions. Comput. Appl. Biosci. 10:685–686 [DOI] [PubMed] [Google Scholar]
- 50.McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server. Bioinformatics (Oxford, England) 16:404–405. 10.1093/bioinformatics/16.4.404 [DOI] [PubMed] [Google Scholar]
- 51.Buchan DW, Ward SM, Lobley AE, Nugent TC, Bryson K, Jones DT. 2010. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 38:W563–W568. 10.1093/nar/gkq427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirokawa T, Boon-Chieng S, Mitaku S. 1998. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics (Oxford, England) 14:378–379 [DOI] [PubMed] [Google Scholar]
- 53.Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132 [DOI] [PubMed] [Google Scholar]
- 54.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 55.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 56.Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of object in dual-colour confocal images. J. Microsc. 169:375–382 [DOI] [PubMed] [Google Scholar]
- 57.Graham JM. 2002. Homogenization of mammalian cultured cells. ScientificWorldJournal 2:1630–1633. 10.1100/tsw.2002.848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Graham JM. 2002. Preparation of crude subcellular fractions by differential centrifugation. ScientificWorldJournal 2:1638–1642. 10.1100/tsw.2002.851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Graham JM. 2001. Isolation of mitochondria from tissues and cells by differential centrifugation. Curr. Protoc. Cell Biol. Chapter 3:Unit 3.3. 10.1002/0471143030.cb0303s04 [DOI] [PubMed] [Google Scholar]
- 60.Wu S-X, Kaesberg P. 1991. Synthesis of template-sense, single-stranded Flock House virus RNA in a cell-free replication system. Virology 183:392–396 [DOI] [PubMed] [Google Scholar]
- 61.Saunders K, Kaesberg P. 1985. Template-directed RNA polymerase from Black beetle virus-infected Drosophila melanogaster cells. Virology 147:373–381 [DOI] [PubMed] [Google Scholar]
- 62.Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93:97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okamoto T, Schwab RB, Scherer PE, Lisanti MP. 2001. Analysis of the association of proteins with membranes. Curr. Protoc. Cell Biol. Chapter 5:Unit 5.4. 10.1002/0471143030.cb0101s05 [DOI] [PubMed] [Google Scholar]
- 64.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 65.Haukenes G, Szilvay A-M, Brokstad KA, Kanestrom A, Kalland K-H. 1997. Labeling of RNA transcripts of eukaryotic cells in culture with BrUTP using a liposome transfection reagent (DOTAP). Biotechniques 22:308–312 [DOI] [PubMed] [Google Scholar]
- 66.van der Meer Y, van Tol H, Locker JK, Snijder EJ. 1998. ORF1a-encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J. Virol. 72:6689–6698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vanderlaan M, Thomas CB. 1985. Characterization of monoclonal antibodies to bromodeoxyuridine. Cytometry 6:501–505 [DOI] [PubMed] [Google Scholar]
- 68.Albarino CG, Price BD, Eckerle LD, Ball LA. 2001. Characterization and template properties of RNA dimers generated during Flock House virus RNA replication. Virology 289:269–282. 10.1006/viro.2001.1125 [DOI] [PubMed] [Google Scholar]
- 69.Rosskopf JJ, Upton JH, III, Rodarte L, Romero TA, Leung M-Y, Taufer M, Johnson KL. 2010. A 3′ terminal stem-loop structure in Nodamura virus RNA2 forms an essential cis-acting signal for RNA replication. Virus Res. 150:12–21. 10.1016/j.virusres.2010.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leeds P, Peltz SW, Jacobson A, Culbertson MR. 1991. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 5:2303–2314 [DOI] [PubMed] [Google Scholar]
- 71.Newman TC, Ohme-Takagi M, Taylor CB, Green PJ. 1993. DST sequences, highly conserved among plant SAUR genes, target reporter transcripts for rapid decay in tobacco. Plant Cell 5:701–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Price BD, Eckerle LD, Ball LA, Johnson KL. 2005. Nodamura virus RNA replication in Saccharomyces cerevisiae: heterologous gene expression allows replication-dependent colony formation. J. Virol. 79:495–502. 10.1128/JVI.79.1.495-502.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saks VA, Veksler VI, Kuznetsov AV, Kay L, Sikk P, Tiivel T, Tranqui L, Olivares J, Winkler K, Wiedemann F, Kunz WS. 1998. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol. Cell. Biochem. 184:81–100 [PubMed] [Google Scholar]
- 74.Hofmann K, Stoffel W. 1993. TMBASE: a database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 374:166 [Google Scholar]
- 75.Stapleford KA, Rapaport D, Miller DJ. 2009. Mitochondrion-enriched anionic phospholipids facilitate Flock house virus RNA polymerase membrane association. J. Virol. 83:4498–4507. 10.1128/JVI.00040-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.von Heijne G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225:487–494 [DOI] [PubMed] [Google Scholar]
- 77.Petty HR. 2001. Overview of the physical state of proteins within cells. Curr. Protoc. Cell Biol. Chapter 5:Unit 5.1. 10.1002/0471143030.cb0501s00 [DOI] [PubMed] [Google Scholar]
- 78.Vander Heyden AB, Naismith TV, Snapp EL, Hanson PI. 2011. Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum. EMBO J. 30:3217–3231. 10.1038/emboj.2011.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang C, Youle RJ. 2009. The role of mitochondria in apoptosis. Annu. Rev. Genet. 43:95–118. 10.1146/annurev-genet-102108-134850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen S-P, Wu J-L, Su Y-C, Hong J-R. 2007. Anti-Bcl-2 family members, zfBcl-x(L) and zfMcl-1a, prevent cytochrome c release from cells undergoing betanodavirus-induced secondary necrotic cell death. Apoptosis 12:1043–1060. 10.1007/s10495-006-0032-x [DOI] [PubMed] [Google Scholar]
- 81.Chen S-P, Yang H-L, Her GM, Lin H-Y, Jeng M-F, Wu J-L, Hong J-R. 2006. Betanodavirus induces phosphatidylserine exposure and loss of mitochondrial membrane potential in secondary necrotic cells, both of which are blocked by bongkrekic acid. Virology 347:379–391. 10.1016/j.virol.2005.11.052 [DOI] [PubMed] [Google Scholar]
- 82.Chen SP, Yang HL, Lin HY, Chen MC, Wu JL, Hong JR. 2006. Enhanced viability of a nervous necrosis virus-infected stable cell line over-expressing a fusion product of the zfBcl-xL and green fluorescent protein genes. J. Fish Dis. 29:347–354. 10.1111/j.1365-2761.2006.00725.x [DOI] [PubMed] [Google Scholar]
- 83.Settles EW, Friesen PD. 2008. Flock house virus induces apoptosis by depletion of Drosophila inhibitor-of-apoptosis protein DIAP1. J. Virol. 82:1378–1388. 10.1128/JVI.01941-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brown-Bryan TA, Leoh LS, Ganapathy V, Pacheco FJ, Mediavilla-Varela M, Filippova M, Linkhart TA, Gijsbers R, Debyser Z, Casiano CA. 2008. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/p75. Mol. Cancer Res. 6:1293–1307. 10.1158/1541-7786.MCR-08-0125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murphy FA, Scherer WF, Harrison AK, Dunne HW, Gary GW., Jr 1970. Characterization of Nodamura virus, an arthropod transmissible picornavirus. Virology 40:1008–1021 [DOI] [PubMed] [Google Scholar]
- 86.Scherer WF, Verna JE, Richter GW. 1968. Nodamura virus, an ether- and chloroform-resistant arbovirus from Japan. Physical and biological properties, with ecologic observations. Am. J. Trop. Med. Hyg. 17:120–128 [DOI] [PubMed] [Google Scholar]