

ABSTRACT
To gain insight into the mechanism of herpesvirus entry into cells, the four glycoproteins that are necessary for herpes simplex virus (HSV) fusion were cloned from the saimiriine herpesvirus 1 (SaHV-1) genome, a primate member of the alphaherpesvirus family. Cell-cell fusion assays indicate that SaHV-1 entry glycoproteins function with the previously identified alphaherpesvirus entry receptors nectin-1 and CD155 but not with herpesvirus entry mediator (HVEM) or paired immunoglobulin-like type 2 receptor alpha (PILRα). Replacement of HSV-1 gD with the SaHV-1 gD homolog resulted in a complete loss of fusion function when coexpressed with HSV-1 gB and gH/gL. HSV-1 gD was also unable to substitute for SaHV-1 gD when coexpressed with SaHV-1 gB and gH/gL. Similarly, the gH/gL heterodimers from HSV-1 and SaHV-1 were not interchangeable. In contrast, both the HSV-1 and SaHV-1 gB homologs retained function in a heterotypic context. These results suggest that an essential interaction between homotypic gD and gH/gL occurs during both HSV-1 and SaHV-1 entry. To map the site of this homotypic interaction, we created a series of gD chimeras, focusing on the “profusion domain” (PFD) that consists of HSV-1 gD residues 261 to 305 or SaHV-1 gD residues 264 to 307. We identified a seven-amino-acid stretch (264 RTLPPPK 270) at the N terminus of the SaHV-1 gD PFD that contributes to homotypic fusion. Finally, we found that the gD receptor-binding region and PFD cannot function independently but that both can inhibit the function of wild-type gD.
IMPORTANCE The herpesvirus entry machinery requires the concerted action of at least four glycoproteins; however, details of the interactions among these glycoproteins are not well understood. Like HSV-1, SaHV-1 belongs to the alphaherpesvirus subfamily. Using cell-cell fusion experiments, we found that SaHV-1 uses the entry receptors nectin-1 and CD155 but not HVEM or PILRα. By swapping the entry glycoproteins between HSV-1 and SaHV-1, we revealed a functional interaction between gD and gH/gL. To examine the homotypic interaction site on gD, we evaluated the function of a panel of HSV-1/SaHV-1 gD chimeras and identified a small region in the SaHV-1 gD profusion domain that is critical for SaHV-1 fusion. This study contributes to our understanding of the molecular mechanisms of herpesvirus entry and membrane fusion.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is an alphaherpesvirus that causes recurrent mucocutaneous lesions on the mouth, face, or genitalia and occasionally meningitis or encephalitis. Saimiriine herpesvirus 1 (designated SaHV-1 in this work) is a primate herpesvirus that belongs to the alphaherpesvirus subfamily and has high sequence similarity to human HSV-1 and HSV2. SaHV-1 was originally isolated from tamarins (Saguinus spp.) that succumbed to lethal generalized disease; however, squirrel monkeys (Saimiri spp.) are the natural host for SaHV-1 (1, 2). There have been no confirmed cases of human SaHV-1 infection (3). The recent completion of the SaHV-1 genome sequence (3) provided both an impetus to investigate the function of SaHV-1 entry glycoproteins and a tool to compare the entry of this alphaherpesvirus to HSV-1 at a molecular level.
Entry of HSV-1 into cells and virus-induced cell fusion require coordinated interactions among four viral glycoproteins (gD, gB, gH, and gL). The binding of gD to a cellular entry receptor triggers the entry process, and the final fusion of the viral envelope with the cellular membrane is executed by the viral fusion protein gB (4, 5). gH and gL exist as a heterodimer (gH/gL) and also are required for entry; however, their role in entry is less clear. gH/gL may act as a bridge between gD and gB and/or bind to cellular receptors (6).
Details of the interactions among the glycoproteins required for entry are not well understood. Coprecipitation experiments have suggested that gD can interact with either gH/gL or gB via independent binding sites (7). Bimolecular fluorescence complementation studies have demonstrated interactions between all of the entry glycoprotein combinations (gD with gH/gL, gD with gB, and gH/gL with gB) (8–10). Different studies disagree over whether the interaction between gH/gL and gB requires the presence of gD (7). Complicating the matter further is the fact that the detection of a physical complex does not necessitate that an interaction be a functionally relevant one that contributes to fusion promotion.
HSV-1 gD is a 369-amino-acid (aa) protein after signal peptidase cleavage. The structures of the HSV-1 gD ectodomain crystallized alone or in complex with receptor have been described (11–13). The core of HSV-1 gD (residues T56 to R184) adopts an Ig-like V-type fold that is flanked by an N-terminal region containing receptor contact sites and a large C-terminal extension. Structural and mutational studies support a model in which the C terminus of the gD ectodomain auto-inhibits the receptor-binding site on gD (12). Upon receptor binding, the gD C terminus moves to unveil the receptor-binding site. This conformational change may also serve as a signal to trigger fusion.
The C-terminal region of the gD ectodomain is critical for gD function. When this region of HSV-1 gD was replaced with a homologous region from another alphaherpesvirus (pseudorabies virus [PRV]), the chimeric gD retained receptor-binding activity but lost the ability to promote fusion (14). Similarly, the infectivity of a gD-null HSV-1 could be rescued by the addition of truncated soluble gD, but only if the soluble gD included a C-terminal gD region, dubbed the profusion domain (PFD) (residues P261 to P305) (15). The previously mentioned coprecipitation experiments propose that the sites of gB and gH/gL interaction partially map to the PFD (7). Most of the proline-rich PFD is a flexible structural element which is not resolved in the crystal structures (11–13).
Sequence alignments demonstrate that HSV-1 and SaHV-1 gD have 50% sequence identity. After the predicted signal peptidase cleavage, SaHV-1 gD is a 371-aa protein. The predicted Ig-like core of SaHV-1 gD lies between residues T54 and K187, and its predicted PFD lies within residues R264 and P307 (see Fig. 4B). Several entry receptors bind to HSV-1 gD, including nectin-1 (16–20), herpesvirus entry mediator (HVEM) (21), and modified heparan sulfate (22, 23). Nectin-1 is a cell adhesion molecule that belongs to the immunoglobulin superfamily and is widely expressed by a variety of cell types, including epithelial cells and neurons, critical cell types for HSV-1 pathogenesis (24, 25). A related member of the Ig superfamily, CD155 (also called poliovirus receptor), can function as a receptor for the animal alphaherpesviruses PRV and bovine herpesvirus (BHV-1) (17). HVEM is a member of the tumor necrosis factor receptor family (26). Heparan sulfate that is modified by 3-O-sulfotransferases can also serve as a gD-binding entry receptor (23). Receptor usage of SaHV-1 has not been determined.
FIG 4.

Mutant gD constructs examining the profusion domain (PFD). (A) Schematic representation of the HSV-1 and SaHV-1 gD constructs. Chimeras, point mutants, and deletion mutants were generated. Gray bars represent HSV-1 gD sequence, whereas black bars represent SaHV-1 sequence. The construct names begin with “QF.” For all constructs, the native signal sequence (dashed box) was replaced by an exogenous signal sequence and an N-terminal FLAG tag. Amino acids for each construct are numbered. Regular type indicates HSV-1 gD numbering, whereas italicized type with an asterisk indicates SaHV-1 gD numbering. The HSV-1 gD PFD residues are denoted P261-P305, and the SaHV-1 gD PFD residues are denoted R264-P307. The table on the right summarizes the ability of the constructs to mediate fusion with nectin-1-expressing cells when coexpressed with gB, gH, and gL from either HSV-1 or SaHV-1. A minus sign indicates that the construct failed to mediate fusion, and a plus sign indicates that it mediated fusion at near wild-type levels. Arrows indicate that the construct mediated fusion at levels higher (up arrow) or lower (down arrow) than that of wild-type gD. (B) Sequence alignment of the C termini of the HSV-1 and SaHV-1 gD ectodomains. The upper numbers refer to HSV-1 gD, and the lower numbers refer to SaHV-1 gD. The PFD is enclosed in a box. Conserved residues are highlighted in gray. The N-terminal seven residues of the PFD are in bold.
Receptors that bind to HSV-1 gB also have been reported to mediate HSV-1 fusion, including the paired immunoglobulin-like type 2 receptor alpha (PILRα) (27), myelin-associated glycoprotein (MAG) (28, 29), and nonmuscle myosin heavy chain IIA (NMHC-IIA) (28). PILRα is expressed on many cells of the immune system as well as neurons (27, 30–35). MAG is a cell surface molecule that is preferentially expressed in neural tissues, especially on the myelin sheath, and plays a role in the regulation of axonal growth (36–39). NMHC-IIA is expressed in a wide variety of cultured cell lines and various tissues and cell types in vivo (40, 41).
Previously, we used the entry glycoproteins of two gammaherpesviruses (Epstein-Barr virus [EBV] and rhesus lymphocryptovirus [LCV]) to demonstrate that LCV gL requires a homotypic interaction with LCV gB for function (42). Furthermore, we mapped a presumed gB functional interaction site to two residues in gL by creating EBV/LCV gL chimeras and site-specific mutants of gL. Similarly, in this study, we use HSV-1 and SaHV-1 entry glycoproteins to explore other homotypic requirements for fusion. The results indicate a functional interaction between gD and gH/gL. This evidence for a functional interaction between these proteins builds on previous studies that report a physical interaction (7, 8, 43). To examine the homotypic interaction site on gD, we evaluated the function of a panel of HSV-1/SaHV-1 gD chimeras. We also report the receptor usage of SaHV-1 using cell-cell fusion and explore the function of the profusion domain of gD.
MATERIALS AND METHODS
Cells.
Chinese hamster ovary (CHO-K1; ATCC) cells were grown in Ham's F12 medium supplemented with 10% fetal bovine serum (FBS).
Plasmids.
Sequence alignments of HSV-1 and SaHV-1 were performed using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Plasmids expressing HSV-1 strain KOS gB (pPEP98), gD (pPEP99), gH (pPEP100), and gL (pPEP101) were previously described (44), as were plasmids expressing human nectin-1 (pBG38) (17), human HVEM (pBEC10) (21), CD155 (kindly provided by Eckard Wimmer), and human PILRα (pQF003) (45). Table 1 and Fig. 4A depict the constructs generated for this study. In Table 1, pQF077-080 from open reading frames UL27 (gB), US6 (gD), UL22 (gH), and UL1 (gL) were cloned into pCAGGS using SaHV-1 viral DNA kindly provided by Alberto Severini. FLAG-tagged gB, gD, gH, and gL for HSV-1 (pQF112-115) and SaHV-1 (pQF081-084) were generated by subcloning the HSV-1 glycoproteins or cloning SaHV-1 viral DNA into a pFLAG-myc-CMV-21 expression vector (E5776; Sigma) downstream of the FLAG epitope. The native signal sequences were removed, including HSV-1 gB aa 1 to 30, SaHV-1 gB aa 1 to 39, HSV-1 gD aa 1 to 25, SaHV-1 gD aa 1 to 36, HSV-1 gH aa 1 to 18, SaHV-1 gH aa 1 to 29, HSV-1 gL aa 1 to 19, and SaHV-1 gL aa 1 to 23. The signal peptides from the SaHV-1 glycoproteins were predicted by using SignalP 4.0 Server (http://www.cbs.dtu.dk/services/SignalP/). FLAG-tagged HSV-1 gB, gD, gH, and gL were generated by PCR amplification from pPEP98, pPEP99, pPEP100, and pPEP101, respectively.
TABLE 1.
Plasmids generated for this studya
| Construct | Protein |
|---|---|
| pQF077* | SaHV-1 gB |
| pQF078* | SaHV-1 gD |
| pQF079* | SaHV-1 gH |
| pQF080* | SaHV-1 gL |
| pQF081 | SaHV-1 FLAG-tagged gB |
| pQF082 | SaHV-1 FLAG-tagged gD |
| pQF083 | SaHV-1 FLAG-tagged gH |
| pQF084 | SaHV-1 FLAG-tagged gL |
| pQF112 | HSV-1 FLAG-tagged gB |
| pQF113 | HSV-1 FLAG-tagged gD |
| pQF114 | HSV-1 FLAG-tagged gH |
| pQF115 | HSV-1 FLAG-tagged gL |
*, cloned into pCAGGS. Other constructs were cloned into pFLAG-myc-CMV-21 (E5776; Sigma).
Figure 4A illustrates the FLAG-tagged gD chimeras (pQF122-125, pQF137-139, pQF146, pQF147, pQF152) and deletion mutants (pQF151, pQF153-161) generated by PCR or QuikChange site-directed mutagenesis. pQF144 and pQF148-150 are HSV-1 gD PFD point mutants generated by QuikChange site-directed mutagenesis (Agilent Technologies). The amino acids in HSV-1 gD and SaHV-1 gD were numbered after putative signal peptide cleavage sites. Plasmids made for this study were sequenced by the Northwestern Genomic core facility.
Western blots.
Western blots of whole lysates were performed to check overall expression of HSV-1 and SaHV-1 glycoproteins. CHO-K1 cells seeded in 6-well plates were transfected with 1.5 μg of empty vector or plasmids expressing FLAG-tagged gB, gD, gH, and gL from HSV-1 and SaHV-1 using 5 μl of Lipofectamine 2000. After 24 h of incubation, the cells were detached using EDTA (0.2 g EDTA/liter in phosphate-buffered saline [PBS]), washed with PBS, and lysed with 200 μl of lysis buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1 mM Na3VO3, 1% Nonidet P-40) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN). Proteins were separated by SDS-PAGE on 4 to 20% gels after being boiled for 5 min under reducing conditions. Western blot analyses were performed using rabbit anti-FLAG antibodies (F7425; Sigma) at a 1:1,000 dilution for 1 h at room temperature. Anti-rabbit secondary antibody coupled to horseradish peroxidase (HRP) and enhanced chemiluminescence Western blotting detection reagents (GE Healthcare) were used.
CELISA.
A cell-based enzyme-linked immunosorbent assay (CELISA) was used to evaluate the cell surface expression of the FLAG-tagged HSV-1 and SaHV-1 glycoproteins. CHO-K1 cells seeded in 96-well plates were transfected with 60 ng of empty vector or plasmids expressing FLAG-tagged gB, gD, or gH/gL (30 ng DNA for gH and 30 ng DNA for gL) from HSV-1 and SaHV-1 using 0.15 μl of Lipofectamine 2000 (Invitrogen) diluted in Opti-MEM (Invitrogen). The cells were washed once with PBS 24 h after transfection, and CELISA was performed as described previously (46). For other gD chimeras and mutants, CHO cells were transfected with all of the plasmids used to prepare effector cells for the cell fusion assays as described below, so that levels of gD expression could be assessed in replicates of the cell populations used in the fusion assays. Six h after transfection, the cells were detached with EDTA and suspended in 1.5 ml of F12 medium supplemented with 10% FBS. Fifty μl of cell suspension was replated in triplicate in 96-well plates and incubated for 16 h. CELISA was performed using an anti-FLAG monoclonal antibody (Sigma F1084). The cells were washed, fixed, and incubated with biotinylated goat anti-mouse IgG (Sigma), followed by streptavidin-HRP (GE Healthcare) and HRP substrate (BioFX).
Cell fusion assay.
In parallel with CELISA, cell fusion activity was measured using a quantitative luciferase-based cell fusion assay as previously described (44). CHO-K1 cells were seeded in 6-well plates 1 day before transfection for both effector and target cells. One set of CHO-K1 cells (effector cells) was transfected with 400 ng (each) of plasmids encoding T7 RNA polymerase, gB, gD, gH, and gL using 5 μl of Lipofectamine 2000. In experiments testing heterotypic glycoprotein function, combinations of glycoproteins from HSV-1 or SaHV-1 were transfected; however, gH and gL were always derived from the same virus. In experiments testing gD function, wild-type (WT) gD was replaced with a gD chimera or mutant. A separate set of CHO-K1 cells (target cells) was transfected with 400 ng of a plasmid encoding the firefly luciferase gene under the control of the T7 promoter plus 1.5 μg of empty vector (pcDNA3) or plasmid carrying receptor (PILRα, nectin-1, HVEM, or CD155) using 5 μl of Lipofectamine 2000. Six h after transfection, the cells were detached with EDTA and suspended in 1.5 ml of F12 medium supplemented with 10% FBS. Effector and target cells were mixed in a 1:1 ratio and replated in 96-well plates for 18 h. Luciferase activity was quantitated by a luciferase reporter assay system (Promega) using a Wallac-Victor luminometer (PerkinElmer).
RESULTS
Expression of HSV-1 and SaHV-1 entry glycoproteins.
The entry glycoproteins from HSV-1 and SaHV-1 were cloned into an expression vector replacing the glycoprotein signal sequence with a heterologous signal sequence and with the addition of an N-terminal FLAG epitope. Cell surface expression of the glycoproteins was evaluated by transfecting CHO-K1 cells with the construct and using CELISA to detect the FLAG epitope (46, 47). Since gH requires gL as a chaperone, the two proteins were cotransfected such that only one of the proteins was FLAG tagged in each transfection. For gD and gB, the FLAG-tagged constructs were transfected alone. High levels of expression of FLAG-tagged gB, gD, gH/gL from HSV-1 and SaHV-1 were readily detected on the cell surface (Fig. 1A). A Western blot of total cell lysate also confirmed expression of the glycoproteins (Fig. 1B). FLAG-tagged gH and FLAG-tagged gL were transfected together, and FLAG-tagged gD and gB were transfected separately. Each glycoprotein tested migrated to the expected size. Western blots of all gD chimera, point, and deletion mutants shown in Table 1 and Fig. 4 were performed, and the proteins migrated at the expected sizes (data not shown).
FIG 1.

Expression of entry glycoproteins from HSV-1 and SaHV-1. (A) Cell surface expression measured by CELISA. CHO cells in a 96-well plate were transfected overnight with plasmids encoding FLAG-tagged gB, FLAG-tagged gD, gL plus FLAG-tagged gH, gH plus FLAG-tagged gL, or empty vector. The “F-” indicates the constructs that were FLAG tagged. The cells were washed and incubated with an anti-FLAG M2 antibody. After extensive washing, cells were fixed and incubated with an anti-mouse secondary antibody for detection. Each bar shows the mean and standard deviation of three independent determinations. Background signals detected after transfection with the vector alone were subtracted from the values. Data for each set of glycoproteins were normalized to the expression level of HSV-1 F-gD or SaHV-1 F-gD. (B) Total protein expression measured by Western blot of cell lysates. CHO cells expressing the constructs above were lysed, and proteins were resolved by SDS-PAGE. FLAG-tagged gH was coexpressed with FLAG-tagged gL. Proteins were transferred to nitrocellulose and probed with rabbit anti-FLAG antibody followed by goat anti-rabbit IgG. gB, gD, gH, and gL migrated to their expected molecular weights (shown [in thousands] at left). V, empty vector.
SaHV-1 can use CD155 and nectin-1, but not HVEM and PILRα, in fusion.
A quantitative cell-cell fusion assay was used to examine the receptor usage of the SaHV-1 entry glycoproteins (Fig. 2). Target CHO-K1 cells were transfected with a plasmid encoding luciferase under a T7 promoter and either PILRα, nectin-1, HVEM, CD155, or empty vector. Effector CHO-K1 cells were transfected with a plasmid encoding T7 RNA polymerase and gB, gD, gH, and gL from either HSV-1 or SaHV-1. Effector and target cells were mixed, and luciferase activity was recorded as a measure of cell-cell fusion. Wild-type and flag-tagged constructs exhibited similar levels of fusion (data not shown) and thus the flag-tagged versions were used for subsequent experiments.
FIG 2.
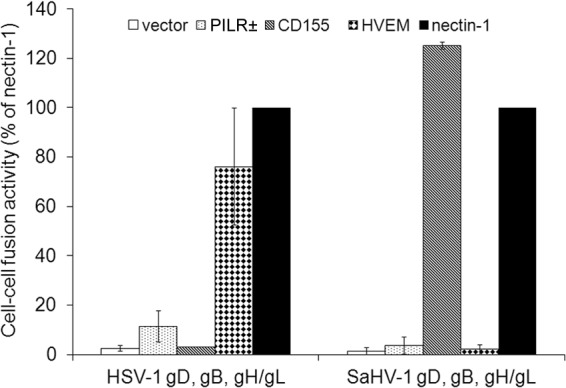
Receptor usage for HSV-1 and SaHV-1 entry glycoproteins. Target CHO cells were transfected with a reporter plasmid encoding luciferase under the control of the T7 promoter along with plasmids encoding PILRα, CD155, HVEM, nectin-1, or empty vector. The transfected cells were replated with effector CHO cells that had been transfected with plasmids encoding T7 polymerase and the complete set of either WT HSV-1 or FLAG-tagged SaHV-1 entry glycoproteins (gD, gB, gH, gL). After coincubation overnight, luciferase activity was measured as an indication of cell-cell fusion. For each set of viral glycoproteins, data were normalized to the fusion activity mediated by nectin-1, which was set at 100%. The relative light units after fusion with nectin-1 expressing cells averaged 110,000 for SaHV-1 and 187,000 for HSV-1. Each bar shows the mean and standard deviation of three independent determinations.
As expected, HSV-1 glycoproteins mediated fusion with cells expressing nectin-1, HVEM, and PILRα, but not CD155 (17, 47) (Fig. 2). As previously reported, fusion with PILRα-expressing cells was markedly lower than fusion with HVEM- or nectin-1-expressing cells (45). Cells expressing the SaHV-1 glycoproteins fused with nectin-1-expressing cells nearly as well as did those expressing the HSV-1 glycoproteins. SaHV-1 glycoproteins mediated fusion with cells expressing either nectin-1 or the related poliovirus receptor CD155, but not HVEM. SaHV-1 glycoprotein-mediated fusion with PILRα-expressing cells was greatly reduced compared to that for nectin-1 or CD155 fusion. Interestingly, CD155 mediated fusion of SaHV-1 glycoprotein-expressing cells at greater levels than did nectin-1. These fusion data indicate that nectin-1 and CD155 can function as receptors for SaHV-1 but HVEM and PILRα cannot.
HSV-1 and SaHV-1 entry glycoproteins exhibit homotypic requirements for function.
The entry glycoproteins from HSV-1 and SaHV-1 have high sequence similarity, suggesting that the homologous glycoproteins may be able to functionally substitute for one another. To identify any homotypic glycoprotein requirements for HSV-1 and SaHV-1 fusion, we performed the fusion assay while individually replacing glycoproteins with their heterotypic counterparts. Since gL is required for proper gH folding, gH/gL was treated as a unit and always derived from the same virus. Nectin-1 target cells were used because HSV-1 and SaHV-1 use this receptor to similar extents (Fig. 2).
Substitution of HSV-1 gD with SaHV-1 gD (or vice versa) resulted in a loss of fusion function (Fig. 3). Similarly, substitution of HSV-1 gH/gL with SaHV-1 gH/gL (or vice versa) also resulted in a loss of fusion function (Fig. 3). In contrast, HSV-1 gB and SaHV-1 gB retained some level of fusion function in a heterotypic context. When HSV-1 gB was coexpressed with SaHV-1 gD and gH/gL, fusion was observed at 66% of the level seen with the complete set of wild-type SaHV-1 glycoproteins. Similarly, when SaHV-1 gB was coexpressed with HSV-1 gD and gH/gL, fusion was observed at 15% of wild-type HSV-1 levels (Fig. 3).
FIG 3.
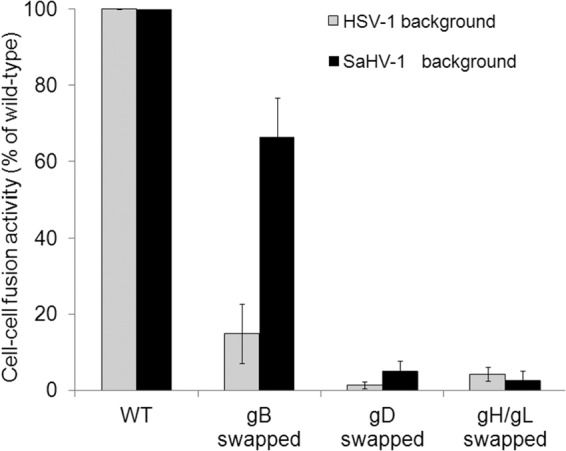
Heterotypic fusion activity of the HSV-1 and SaHV-1 entry glycoproteins. Target CHO cells were transfected with a reporter plasmid encoding luciferase under the control of the T7 promoter along with plasmids encoding nectin-1 or empty vector. Effector CHO cells were transfected with a plasmid encoding T7 polymerase along with a combination of plasmids encoding HSV-1 or SaHV-1 entry glycoproteins. Glycoproteins were swapped for heterotypic counterparts, as indicated. Target and effector cells were coincubated overnight, and luciferase activity was measured as an indication of cell-cell fusion. Data were normalized to the fusion activity measured when a homotypic set of glycoproteins was coexpressed (either all HSV-1 or all SaHV-1 glycoproteins). Each bar shows the mean and standard deviation of three independent determinations.
The ability of gB to function in a heterotypic context may be explained in part by high gB sequence conservation between the viruses (65% amino acid identity). The observation that HSV-1 gD does not mediate fusion in an SaHV-1 background (and vice versa) suggests that the heterotypic gD fails to functionally interact with one or more of the entry glycoproteins. The same can be said for heterotypic gH/gL. Since the two gB proteins are interchangeable to some extent, these results suggests that a homotypic gH/gL-gD functional interaction is required for fusion (Fig. 3).
The gD homotypic requirement for function does not map exclusively to the PFD.
To investigate why HSV-1 gD failed to mediate fusion when coexpressed with SaHV-1 gH/gL (and vice versa), we created chimeras of HSV-1 gD and SaHV-1 gD to map potential sites of interaction with gH/gL (Fig. 4A). We designed the chimeras to examine the PFD, because it has been implicated in interactions with other entry glycoproteins (12, 14, 15). The HSV-1 gD C terminus (residues 261 to 369) was replaced with the SaHV-1 gD C terminus (residues 264 to 371) including the SaHV-1 profusion domain to generate the construct pQF122. Conversely, the SaHV-1 gD C terminus was replaced with the HSV-1 gD C terminus to generate the construct pQF123. Chimeras that swap just the profusion domain were also generated. Construct pQF124 replaces the SaHV-1 gD PFD (residues 264 to 307) with the HSV-1 gD PFD (residues 261 to 305), and construct pQF125 replaces the HSV-1 gD PFD with the SaHV-1 gD PFD.
Surface expression of the chimeras was examined by CELISA and Western blotting (data not shown), confirming that all of the constructs were expressed at levels similar to that of wild-type HSV-1 gD (Fig. 5). The ability of the chimeras to mediate fusion with nectin-1-expressing cells was then determined, with coexpression of either HSV-1 gH/gL and gB or SaHV-1 gH/gL and gB (Fig. 5).
FIG 5.
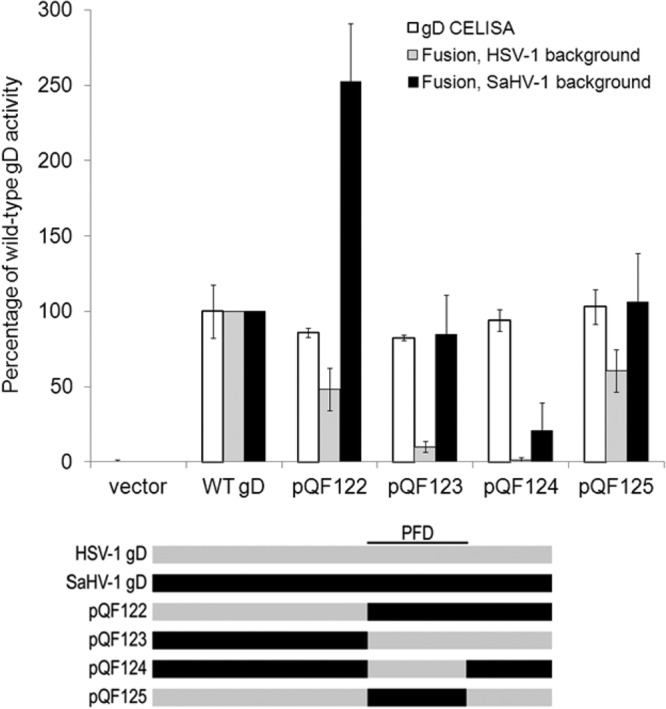
Cell surface expression and fusion activities of the HSV-1 and SaHV-1 gD chimeras. Target cells were transfected with a plasmid carrying the luciferase gene under the control of the T7 promoter and either nectin-1 or empty vector. Effector CHO cells were transfected with plasmids encoding T7 polymerase, gB, gH, and gL from either SaHV-1 or HSV-1, and either wild-type gD or a chimeric gD. “HSV-1 background” refers to HSV-1 gB, gH, and gL coexpression, whereas “SaHV-1 background” refers to SaHV-1 gB, gH, and gL coexpression. One set of effector cells was used for CELISA, and the rest were coincubated with target cells for the cell-cell fusion assay. CELISA data are presented as a percentage of wild-type HSV-1 gD expression. The fusion results are expressed as a percentage of wild-type HSV-1 gD or wild-type SaHV-1 gD activity after subtraction of background values (luciferase activity after coincubation with target cells transfected with vector). Means and standard deviations of results of three independent experiments are shown. For clarity, a schematic representation of the constructs is included, with HSV-1 sequence in gray and SaHV-1 sequence in black. Refer to Fig. 4 for a more detailed representation.
When the chimeras were coexpressed with HSV-1 gH/gL and gB, fusion was reduced for all of the chimeras. Unexpectedly, the two chimeras that contain the SaHV-1 PFD (pQF122 and pQF125) retained function, whereas the two chimeras with the HSV-1 PFD (pQF123 and pQF124) lost function. These results indicate that the HSV-1 PFD and C terminus are insufficient to support the homotypic HSV-1 requirement. Elements in HSV-1 gD N-terminal to the PFD must contribute to a functional HSV-1 homotypic interaction.
When the chimeras were coexpressed with SaHV-1 gH/gL and gB, fusion was retained for all of the constructs except pQF124, the chimera that replaces the SaHV-1 PFD with the HSV-1 PFD. A comparison of the results for chimeras pQF124 and pQF123 suggests that the SaHV-1 gD region C-terminal to the PFD (present in pQF123 but not pQF124) contributes to the homotypic interaction required for SaHV-1 fusion. Alternatively, the pQF124 chimera may fail to function due to improper folding. The near wild-type levels of cell surface expression for pQF124 suggest that the protein was not globally malfolded; however, more modest changes to its folding might impact its function. pQF124 was the only chimera that failed to mediate fusion when coexpressed with either HSV-1 or SaHV-1 glycoproteins, indicating that the majority of the constructs were properly processed.
A seven-amino-acid stretch in the SaHV-1 gD PFD (RTLPPPK) contributes to SaHV-1 fusion.
Interestingly, both of the chimeras containing the SaHV-1 PFD (pQF122 and pQF125) mediated fusion when coexpressed with either HSV-1 or SaHV-1 glycoproteins. Based on our intial observations, we expected that the chimeric proteins would function exclusively with HSV-1 or SaHV-1 glycoproteins.
To investigate this further, we created additional gD chimeras based on pQF125. The N terminus of the SaHV-1 PFD present in pQF125 was progressively replaced by the corresponding HSV-1 sequence in constructs pQF137, pQF138, and pQF139 (Fig. 4A). The chimeras showed near wild-type surface expression by Western blotting (data not shown) and CELISA (Fig. 6A).
FIG 6.
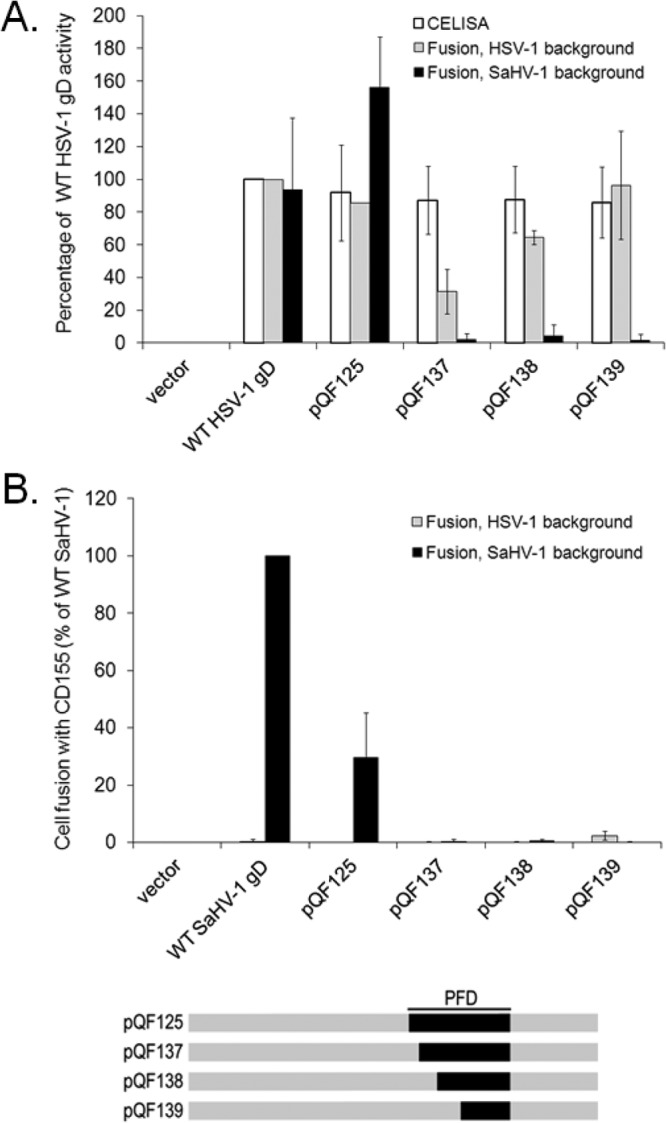
Defining a region in the SaHV-1 gD PFD that is important for fusion. (A) Cell surface expression and nectin-1 usage by pQF125 and its derivatives. Target cells were transfected with a plasmid carrying luciferase under the control of the T7 promoter along with nectin-1 or empty vector. Effector CHO cells were transfected with plasmids encoding T7 polymerase, gB, gH, and gL from either SaHV-1 or HSV-1, and either wild-type gD or a chimeric gD. One set of effector cells was used for CELISA, and the rest were coincubated with target cells for the cell-cell fusion assay. The fusion results are expressed as a percentage of wild-type HSV-1 gD or wild-type SaHV-1 gD activity after subtraction of background values (luciferase activity after coincubation with target cells transfected with vector). Means and standard deviations of results of three independent experiments are shown. (B) CD155 usage by pQF125 and its derivatives. The procedures from Fig. 6A were repeated, except that the target cells were transfected with CD155 instead of nectin-1. For clarity, a schematic representation of the constructs is included, with HSV-1 sequence in gray and SaHV-1 sequence in black. Refer to Fig. 4 for a more detailed representation.
When coexpressed with SaHV-1 gH/gL and gB, all three of these chimeras failed to function in fusion (Fig. 6A). All three chimeras mediated fusion when coexpressed with HSV-1 gH/gL and gB, indicating that they were properly folded. This suggests that the residues at the N terminus of the PFD are critical for the SaHV-1 homotypic interaction. A comparison of the results for chimeras pQF125 and pQF137 indicates that the seven SaHV-1 residues present in pQF125 but absent from pQF137 (SaHV-1 gD residues 264 to 270) are required for homotypic function in SaHV-1 fusion.
CD155 mediated fusion with cells expressing glycoproteins from SaHV-1 but not from HSV-1 (Fig. 2). Surprisingly, when the HSV-1 PFD was replaced with the SaHV-1 PFD (pQF125), fusion activity with CD155-expressing cells increased to 30% of the fusion activity of wild-type SaHV-1 gD (Fig. 6B). The homology between CD155 and nectin-1 implies that their binding sites on gD likely overlap. The nectin-1-binding site maps outside the PFD, and the PFD was not resolved in the crystal structure of gD bound to nectin-1 (11). These results suggest that substitution of the PFD has an indirect effect on the conformation of the CD155/nectin-1-binding site on gD. This effect maps to the N-terminal seven amino acids of the SaHV-1 PFD, since pQF125 mediates fusion with CD155-expressing cells but pQF137 does not.
Additional mutants confirm the importance of the SaHV-1 gD RTLPPPK sequence.
The loss of function we observed with pQF137 led us to construct the inverse chimera, pQF146. In pQF146, the SaHV-1 PFD is replaced with HSV-1 PFD except for the first seven amino acids, which remain SaHV-1 (Fig. 4A). In addition, we generated chimeras in which only the first seven amino acids of the PFD were swapped, pQF147 and pQF152 (Fig. 4A). Single or double mutations in the first seven HSV-1 PFD amino acids were also constructed, focusing on the prolines in this stretch. Residues were mutated either to alanine or the corresponding SaHV-1 residue (Fig. 4A).
Cell surface expression of the chimeras and point mutations was similar to that of WT HSV-1 gD (Fig. 7A). pQF146 mediated fusion when coexpressed with glycoproteins from SaHV-1 but not HSV-1 (Fig. 7B). This result supports the notion that the first seven amino acids of the SaHV-1 PFD are critical for function. As determined earlier, pQF124 fails to functionally interact with SaHV-1 glycoproteins. When the first seven amino acids of the HSV PFD were added back to pQF124 to create the pQF146 chimera, function with SaHV-1 glycoproteins was restored (Fig. 7B).
FIG 7.
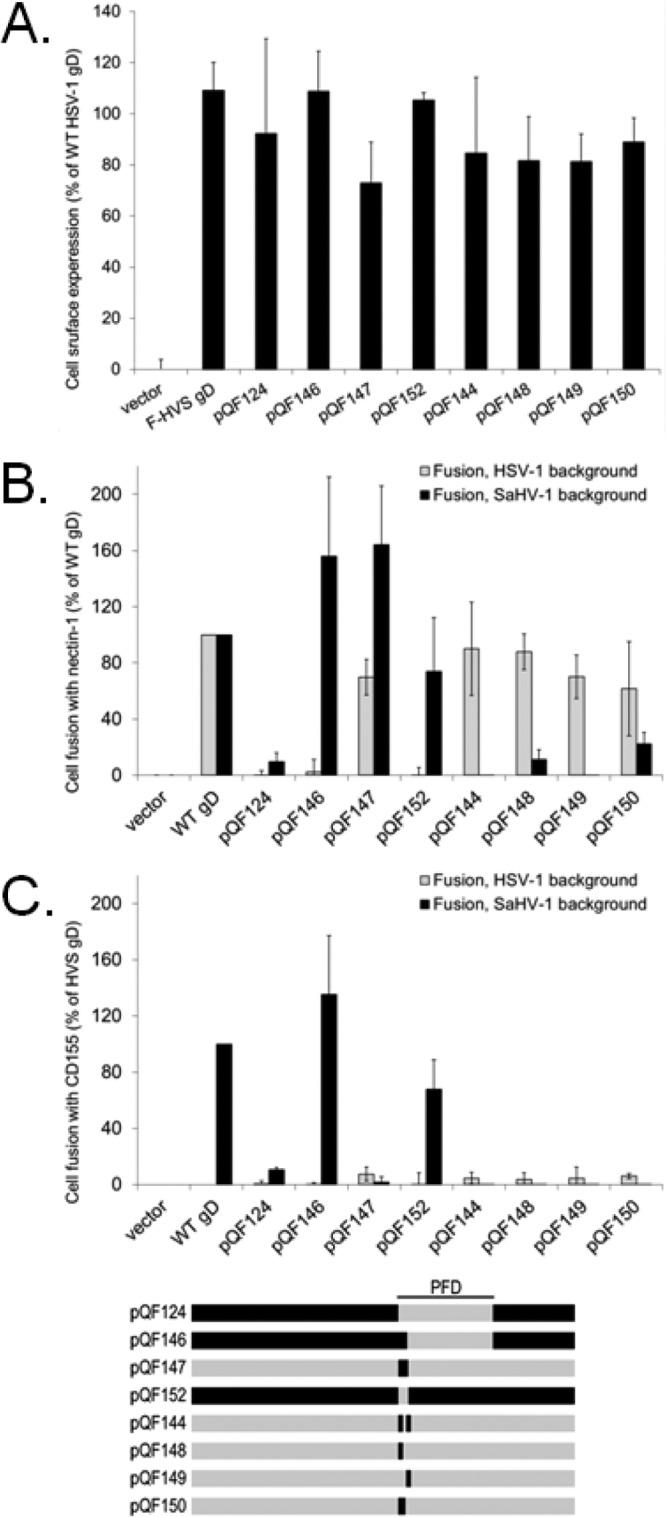
The seven N-terminal amino acids of the SaHV-1 gD PFD are critical for SaHV-1 fusion. (A) Cell surface expression of gD chimeras and HSV-1 gD point mutants. The level of surface expression of gD mutants was determined by CELISA with effector cells used in the fusion assays below. (B and C) Cell fusion activity of gD mutants. Target cells were transfected with a plasmid carrying luciferase under the control of the T7 promoter along with nectin-1 (B), CD155 (C), or empty vector. Effector CHO cells were transfected with plasmids encoding T7 polymerase, gB, gH, and gL from either SaHV-1 or HSV-1, and either wild-type gD or a chimeric gD. Effector and target cells were coincubated, and luciferase activity was determined as an indication of fusion activity. The results are expressed as a percentage of wild-type HSV-1 gD or wild-type SaHV-1 gD activity, after subtraction of background (vector). Means and standard deviations of results of three independent experiments are shown. For clarity, a schematic representation of the constructs is included, with HSV-1 sequence in gray and SaHV-1 sequence in black. Refer to Fig. 4 for a more detailed representation.
Substitution of the first seven amino acids of HSV-1 PFD alone with the residues from the SaHV-1 PFD (pQF147) provided the strongest support for the importance of these residues for SaHV-1 homotypic fusion. pQF147 mediated fusion when coexpressed with SaHV-1 gH/gL and gB (Fig. 7B) despite the fact that only seven of the residues in pQF147 originate from the SaHV-1 sequence. The requirements for HSV-1 fusion maps differently. The chimera pQF152, consisting of SaHV-1 gD with the first seven residues of the PFD from HSV-1, failed to mediate fusion when coexpressed with HSV-1 gH/gL and gB. Curiously, pQF152 retained function with SaHV-1 glycoproteins, indicating that although the first seven residues of the SaHV-1 PFD are sufficient to drive interaction with SaHV-1 glycoproteins, they are not essential.
All of the point mutants (pQF144, pQF148, pQF149, pQF150) retained fusion activity when coexpressed with HSV-1, but none of the mutants gained the ability to functionally interact with SaHV-1 glycoproteins. Thus, the ability of pQF147 to interact with SaHV-1 glycoproteins maps to more than just these residues.
As expected, chimeras pQF146 and pQF152 can mediate fusion with CD155-expressing cells, since both chimeras carry SaHV-1 sequence at the gD-receptor-binding site (Fig. 7C). Since neither chimera functionally interacted with HSV-1 gH/gL and gB, the failure of these chimeras to mediate fusion with CD155-expressing cells when coexpressed with HSV-1 glycoproteins is not surprising.
Deletion of the profusion domain abrogates fusion of both HSV-1 and SaHV-1.
To further explore the importance of the PFD and regions around the PFD, we made a number of deletion mutants (Fig. 4A). The first seven residues of the PFD were deleted from HSV-1 gD (pQF151) and SaHV-1 gD (pQF156). The entire PFD also was deleted from HSV-1 and SaHV-1 gD (pQF153 and pQF157, respectively, herein termed PFD-deleted mutants). To examine the importance of the short stretch of residues that link the PFD to the transmembrane region (Fig. 4B), these membrane-proximal residues were deleted from HSV-1 and SaHV-1 gD alone (pQF154 and pQF159) or in combination with the PFD deletion (pQF155 and pQF158).
The deletion mutants were all tested for cell surface expression by CELISA. Most of the deletion mutants were expressed at wild-type HSV-1 gD levels, with the exceptions of pQF153, pQF155 and pQF158 (Fig. 8A). As expected for misfolded proteins, pQF153, pQF155, and pQF158 did not mediate fusion (Fig. 8B and C).
FIG 8.
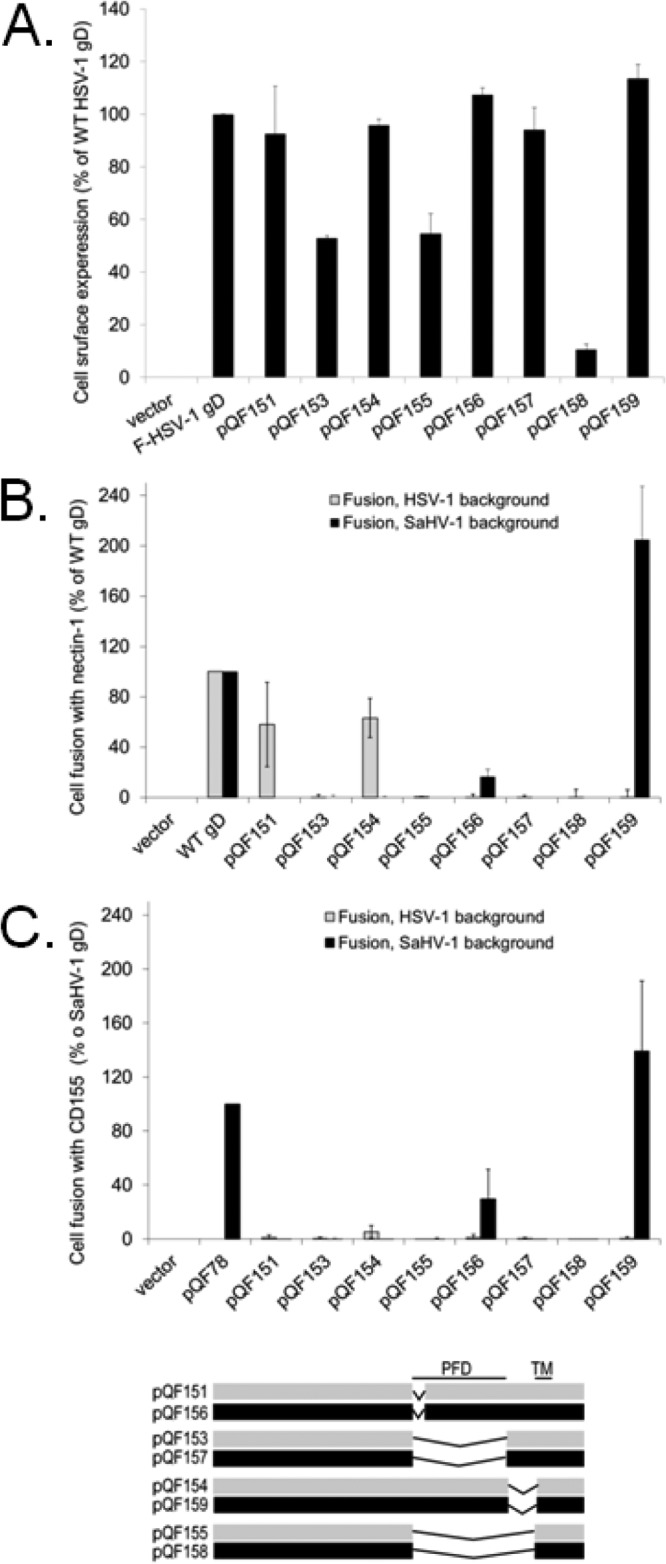
Deletion of the PFD abrogates fusion. (A) Cell surface expression of gD deletion mutants. The level of surface expression of gD mutants was determined by CELISA with effector cells used in the fusion assays below. (B and C) Cell fusion activity of gD deletion mutants. Target cells were transfected with a plasmid carrying luciferase under the control of the T7 promoter along with nectin-1 (B), CD155 (C), or empty vector. Effector CHO cells were transfected with plasmids encoding T7 polymerase, gB, gH, and gL from either SaHV-1 or HSV-1, and either wild-type gD or a chimeric gD. Effector and target cells were coincubated, and luciferase activity was determined as an indication of fusion activity. The results are expressed as a percentage of wild-type HSV-1 gD or wild-type SaHV-1 gD activity, after subtraction of background (vector). Means and standard deviations of results of three independent experiments are shown. For clarity, a schematic representation of the constructs is included, with HSV-1 sequence in gray and SaHV-1 sequence in black. Refer to Fig. 4 for a more detailed representation. TM, transmembrane domain.
Deletion of the SaHV-1 gD PFD (pQF157) did not alter cell surface expression (Fig. 8A), but it completely abrogated cell fusion (Fig. 8B and C), providing further support for the critical role of this region. Deletion of the first seven PFD residues decreased fusion activity for both HSV-1 (pQF151) and SaHV-1 gD (pQF156), suggesting that these residues contribute to function. Surprisingly, deletion of the membrane-proximal residues decreased HSV-1 gD function (pQF154) but enhanced SaHV-1 gD fusion function (pQF159). Deletion of these sequences may impact the conformation of the PFD. None of the deletion mutants gained the ability to function with heterotypic glycoproteins.
The gD receptor-binding domain and PFD cannot function independently, but both can inhibit the function of wild-type gD.
Previous work has demonstrated that soluble forms of gD can trigger fusion, indicating that the transmembrane domain and cytoplasmic tail of gD are dispensable for function (15). A recent study demonstrated that the receptor-binding domain of the paramyxovirus HN protein is dispensable for activation of the paramyxovirus fusion protein (48). In that study, an HN mutant with the receptor-binding domain deleted retained function. To determine if gD might trigger fusion similarly in the absence of a receptor-binding domain, we generated additional HSV-1 and SaHV-1 gD mutants encoding the PFD in the absence of a receptor-binding region (pQF160 and pQF161, respectively) (Fig. 4A).
These PFD-only mutants included the wild-type transmembrane and cytoplasmic domains and were expressed on the cell surface at levels similar to that for wild-type HSV-1 gD (Fig. 9A); however, neither mutant mediated fusion when coexpressed with homotypic entry glycoproteins (Fig. 9B and C). Thus, unlike the case for paramyxovirus HN, gD lacking a receptor-binding domain was insufficient to trigger fusion.
FIG 9.
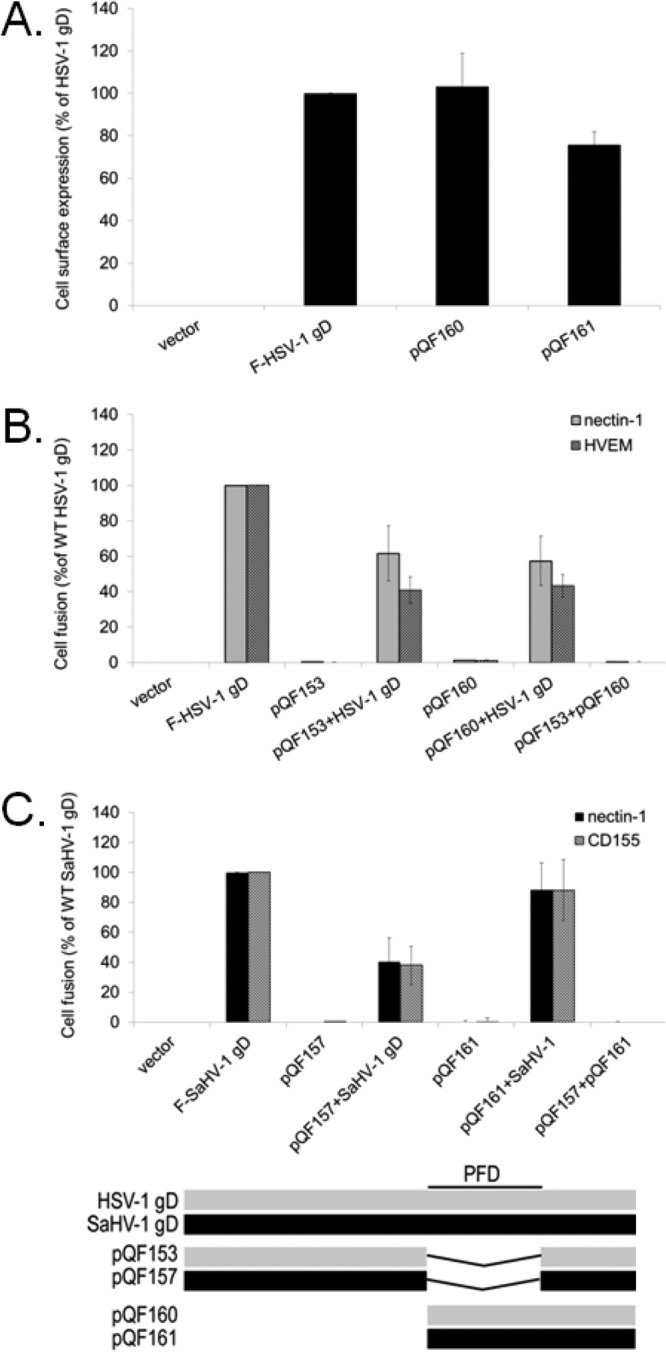
The gD PFD inhibits fusion activity of wild-type gD. (A) Cell surface expression of the PFD-only constructs. The level of surface expression of the HSV-1 and SaHV-1 gD PFD-only mutants was determined by CELISA with effector cells used in the fusion assays below. (B) Target cells were transfected with a plasmid carrying luciferase under the control of the T7 promoter along with nectin-1, HVEM, or empty vector. Effector cells were transfected with plasmids encoding luciferase, gB, gH, and gL from HSV-1, and combinations of wild-type HSV-1 gD, HSV-1 gD with a deletion of the PFD (pQF153), and HSV-1 gD lacking a receptor-binding domain (pQF160). After coincubation of target and effector cells, luciferase activity was measured as an indication of cell-cell fusion activity. (C) Target cells were transfected with a plasmid carrying luciferase under the control of the T7 promoter along with nectin-1, CD155, or empty vector. Effector cells were transfected with plasmids encoding luciferase, gB, gH, and gL from SaHV-1, and combinations of wild-type SaHV-1 gD, SaHV-1 gD with a deletion of the PFD (pQF157), or SaHV-1 gD lacking a receptor-binding domain (pQF161). After coincubation of target and effector cells, luciferase activity was measured as an indication of cell-cell fusion activity. Means and standard deviations of results of three independent experiments are shown for all panels. For clarity, a schematic representation of the constructs is included, with HSV-1 sequence in gray and SaHV-1 sequence in black. Refer to Fig. 4 for a more detailed representation.
To investigate whether the receptor-binding region and PFD of gD can function independently or whether they must be carried on the same protein, we tested whether these PFD-only mutants (pQF160 and pQF161) could rescue the fusion activity of the previously examined gD mutants with a deleted PFD (pQF153 and pQF157). Coexpression of the PFD-only and PFD-deleted gD mutants did not rescue fusion for either HSV-1 or SaHV-1 proteins (Fig. 9B and C), indicating that these regions of gD cannot function independently when carried on separate proteins.
To demonstrate functional activity of these PFD-only and PFD-deleted gD mutants, we tested whether they inhibited fusion mediated by wild-type gD. The mutants were coexpressed with wild-type gD during the fusion assay. For HSV-1, both the PFD-only and PFD-deleted gD mutants inhibited fusion (Fig. 9B). Although PFD-deleted HSV-1 gD (pQF153) was poorly expressed at the cell surface (Fig. 8A), expression was sufficient to inhibit fusion. For SaHV-1, the PFD-deleted mutant (pQF157) inhibited fusion more efficiently than the PFD-only mutant (pQF161). Inhibition of fusion by the PFD-deleted mutants (pQF153 and pQF157) may be due to competition for receptors. Inhibition of fusion by the PFD-only mutants (pQF160 and pQF161) is more interesting and may reflect competition for interaction sites with the other entry glycoproteins, such as gH/gL.
DISCUSSION
The recently published genome sequence of SaHV-1 (3) allowed us to clone gD, gH, gL, and gB from SaHV-1 viral DNA and examine the expression and function of these proteins (Fig. 1). Cell-cell fusion experiments demonstrated that SaHV-1 uses the entry receptors nectin-1 and CD155 but not PILRα or HVEM (Fig. 2).
Nectin-1 serves as a receptor for many alphaherpesviruses, including HSV-1, HSV-2, PRV, BHV-1, B virus, cercopithecine herpesvirus 2 (CeHV-2), and now SaHV-1 (16, 17, 47, 49). Nectin-1 homologs are highly conserved (50), suggesting that squirrel monkey nectin-1 is a natural receptor for SaHV-1. Although residues at the gD–nectin-1 interface (gD β2 and β3) are only 40% conserved between SaHV-1 and HSV-1 gD, these changes did not prevent nectin-1 usage. Amino acid substitutions downstream of the Ig fold in HSV-1 gD (D215, R222, F223) have been shown to abrogate interactions with nectin-1 and nectin-2 (51). Substitutions at position Y38 also reduce interactions with nectin-1, but not with HVEM (52). Compared to HSV-1 gD, SaHV-1 gD has conservative changes in two of these residues (Y38F and F223Y).
Interestingly, SaHV-1 gD gains the ability to use CD155 as a receptor (Fig. 2). CD155 is an entry receptor for PRV and BHV-1 but not HSV-1 or HSV-2 (17). The structural similarity of CD155 and nectin-1 suggests that they would bind to the same site in gD. The divergent residues in SaHV-1 gD may account for its ability to bind to CD155. In unpublished cell-cell fusion experiments, we have shown that B virus and CeHV-1 gD, which are more similar in sequence to HSV-1 gD than SaHV-1 gD, also cannot use CD155 as a receptor.
The evolutionary relationships of seven alphaherpesviruses are depicted in a phylogenic tree of the gD sequences (Fig. 10A). HSV-1 and HSV-2 are very closely related, and entry glycoproteins of these two viruses are interchangeable. CeHV-1 (cercopithecine herpesvirus type 1 or B virus) and CeHV-2 gD are more closely related to HSV-1 gD than is SaHV-1 gD, but SaHV-1 gD is more closely related to HSV-1 gD than either PRV and BHV-1 gD. An amino acid sequence alignment of these seven gD proteins further demonstrates that PRV and BHV-1 are more distantly related than the other five gD homologs (Fig. 10B).
FIG 10.

(A) Phylogenetic tree of gD from seven alphaherpesviruses, generated using Phylogeny.fr (http://www.phylogeny.fr). (B) Amino acid alignment of gD from seven alphaherpesviruses, generated using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Proteins are ordered by relatedness to HSV-1 gD. Minor variations in the HSV-1 and SaHV-1 alignments compared to Fig. 4 are a consequence of performing a multiple alignment versus a pairwise alignment. The HSV-1 gD V-like Ig fold (K1 to R184), profusion domain (P261 to P305), and transmembrane domain (L317 to M339) are in bold italics, as are the corresponding homologous regions in SaHV-1 gD. The N-terminal loop of HSV-1 gD that binds to HVEM is underlined, and the PFDs are boxed based on to HSV-1 gD sequence. HSV-1 numbering begins after the signal sequence. Cysteines involved in disulfide bonds are highlighted in gray.
Our results demonstrate that SaHV-1 cannot use HVEM as a receptor (Fig. 2). Sequence alignment indicates that the lack of HVEM usage is likely due to differences between HSV-1 gD and SaHV-1 gD within the receptor-binding region. HVEM binds to gD within an N-terminal hairpin loop on gD (gD residues 7 to 15 and residues 24 to 32) (13). Previous work has demonstrated that an HSV-1 gD Q27P mutation eliminates HVEM receptor usage and that an HSV-1 gD L25P mutation reduces HVEM usage (13, 21, 53, 54). Relative to HSV-1 gD, SaHV-1 gD carries multiple changes in residues shown to contribute to HVEM binding, including M11P, N15A, L25T, and Q27P. The SaHV-1 gD Q27P residue alone would account for the lack of HVEM usage. The pathogenic consequence of HVEM not serving as a receptor remains to be determined.
The cell-cell fusion assays demonstrate that SaHV-1 cannot use human PILRα as a receptor (Fig. 2). Rather than binding to gD like the receptors discussed above, PILRα binds to gB. The O-glycosylation sites T53 and T480 on HSV-1 gB are important for association with PILRα (55). Concurrent mutation of both residues abrogates the association of HSV-1 gB with PILRα. Sequence alignment of HSV-1 and SaHV-1 gB indicate that T53 and T480 are conserved in SaHV-1 gB. The lack of SaHV-1 gB functional interaction with PILRα may map to other structural differences in SaHV-1 gB. Alternatively, PILRα-mediated SaHV-1 fusion may be too weak to be detected by the cell-cell fusion assay. For HSV-1 fusion, PILRα is used significantly less efficiently than is nectin-1 or HVEM. In addition, the possibility that a Saimiri PILRα homolog would function in SaHV-1 entry remains.
Similarities between the SaHV-1 and HSV-1 sequences led us to examine whether these alphaherpesvirus entry glycoproteins would be able to interact with one another functionally. Fusion assays that individually swapped entry glycoproteins between HSV-1 and SaHV-1 demonstrated that both gD and gH/gL have homotypic requirements for fusion, i.e., they cannot function with glycoproteins from the other virus (Fig. 3). In contrast, gB from either virus was able to mediate fusion when coexpressed with either HSV-1 or SaHV-1 entry glycoproteins. These results implicate a functional homotypic interaction between gD and gH/gL, in agreement with current models of HSV entry (6). This finding provides a framework to examine the sites of the homotypic interaction.
To map the sites on gD responsible for its homotypic requirement, a panel of HSV-1 and SaHV-1 gD mutants was created (Fig. 4). The mutations focused on the gD PFD, a region previously suggested to be the site of gH/gL interaction (15). Of the 45 residues in the HSV-1 gD PFD, only 11 residues are identical between HSV-1 and SaHV-1 gD, and 24 residues have nonconservative changes (Fig. 4B). The results demonstrate that the PFD sequence influences the homotypic interaction (Fig. 5 and 6). For SaHV-1 fusion, seven amino acids at the N terminus of the PFD were critical for homotypic fusion (Fig. 6 and 7). Whereas wild-type HSV-1 gD could not mediate fusion when coexpressed with SaHV-1 gH/gL and gB (Fig. 3), substitution of just the first seven amino acids of the PFD with the corresponding SaHV-1 gD sequence (pQF147) completely rescued its ability to mediate fusion with SaHV-1 gH/gL and gB coexpression (Fig. 7). Point mutations in this region did not refine the site further (Fig. 7). Deletion of the HSV-1 or SaHV-1 PFD or deletion of just the seven N-terminal residues of the SaHV-1 gD PFD (pQF156) inhibited fusion, supporting the critical nature of this region for gD function (Fig. 8). The PFD may be the site of physical interaction with gH/gL, or the PFD may support the structure of a gH/gL interaction site on gD.
Although these residues at the N terminus of the PFD play an important role in the homotypic requirements of gD, the homotypic specificity did not map exclusively to this site. The context of the gD molecule influenced the outcome. For example, when the seven N-terminal residues of the PFD of SaHV-1 gD were replaced by the corresponding HSV-1 gD residues (pQF152), the gD chimera did not gain the ability to functionally interact with HSV-1 entry glycoproteins and, in fact, retained the ability to functionally interact with SaHV-1 entry glycoproteins (Fig. 7). In our unpublished experiments, peptides derived from the HSV-1 and SaHV-1 PFD failed to inhibit HSV-1 and SaHV-1 fusion. The local environment of the PFD likely influences its structure and function.
The receptor-binding region of the parainfluenza virus 5 HN protein is dispensable for F protein activation (48). In contrast, we found that deletion of the receptor-binding region of gD abrogates fusion (Fig. 9). The gD PFD alone was insufficient to promote fusion. Coexpression of PFD-deleted gD constructs did not rescue the function of the gD molecules lacking a receptor-binding region. In these experiments, the gD receptor-binding function could not be carried on a protein separate from the PFD, possibly because of the conformational changes that must occur after receptor binding. Interestingly, coexpression of the PFD-only constructs with wild-type gD inhibited fusion, suggesting that the PFD may compete with the wild type for a critical binding site on one of the entry glycoproteins.
This study provides evidence for a functional interaction between gD and gH/gL, involving residues within the PFD. Future experiments will attempt to demonstrate a physical interaction between gD and gH/gL that maps to the site defined in this work.
ACKNOWLEDGMENTS
We thank Eckard Wimmer for providing the CD155 expression plasmid and Alberto Severini for providing SaHV-1 viral DNA. We thank N. Susmarski for timely and excellent technical assistance and members of the Longnecker laboratory for their help in these studies. Sequencing services were performed at the Northwestern University Genomics Core Facility. R.L. is the Dan and Bertha Spear Research Professor in Microbiology-Immunology.
This work was supported by NIH grants CA021776 and AI067048 to R.L.
Footnotes
Published ahead of print 26 March 2014
REFERENCES
- 1.Daniel MD, Karpas A, Melendez LV, King NW, Hunt RD. 1967. Isolation of herpes-T virus from a spontaneous disease in squirrel monkeys (Saimiri sciureus). Arch. Gesamte Virusforsch. 22:324–331. 10.1007/BF01242953 [DOI] [PubMed] [Google Scholar]
- 2.King NW, Hunt RD, Daniel MD, Melendez LV. 1967. Overt herpes-T infection in squirrel monkeys (Saimiri sciureus). Lab. Anim. Care 17:413–423 [PubMed] [Google Scholar]
- 3.Tyler S, Severini A, Black D, Walker M, Eberle R. 2011. Structure and sequence of the saimiriine herpesvirus 1 genome. Virology 410:181–191. 10.1016/j.virol.2010.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nature Rev. Microbiol. 9:369–381. 10.1038/nrmicro2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. 10.3390/v4050800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299. 10.1128/JVI.01700-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 284:17370–17382. 10.1074/jbc.M109.005728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723. 10.1073/pnas.0707452104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825–3834. 10.1128/JVI.02687-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532–11537. 10.1128/JVI.01343-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein d bound to the human receptor nectin-1. PLoS Pathog. 7:e1002277. 10.1371/journal.ppat.1002277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 24:4144–4153. 10.1038/sj.emboj.7600875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169–179. 10.1016/S1097-2765(01)00298-2 [DOI] [PubMed] [Google Scholar]
- 14.Zago A, Jogger CR, Spear PG. 2004. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 101:17498–17503. 10.1073/pnas.0408186101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G. 2004. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. U. S. A. 101:7445–7450. 10.1073/pnas.0401883101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cocchi F, Menotti L, Mirandola P, Lopez M, Campadelli-Fiume G. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 72:9992–10002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. 10.1126/science.280.5369.1618 [DOI] [PubMed] [Google Scholar]
- 18.Shukla D, Dal Canto M, Rowe CL, Spear PG. 2000. Striking similarity of murine nectin-1a to human nectin-1a (HveC) in sequence and activity as a gD receptor for alphaherpesvirus entry. J. Virol. 74:11773–11781. 10.1128/JVI.74.24.11773-11781.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menotti L, Avitabile E, Dubreuil P, Lopez M, Campadelli-Fiume G. 2001. Comparison of murine and human nectin-1 binding to herpes simplex virus glycprotein D (gD) reveals a weak interaction of murine nectin-1 to gD and a gD-dependent pathway of entry. Virology 282:256–266. 10.1006/viro.2001.0850 [DOI] [PubMed] [Google Scholar]
- 20.Menotti L, Lopez M, Avitabile E, Stefan A, Cocchi F, Adelaide J, Lecocq E, Dubreuil P, Campadelli-Fiume G. 2000. The murine homolog of human Nectin1delta serves as a species nonspecific mediator for entry of human and animal alpha herpesviruses in a pathway independent of a detectable binding to gD. Proc. Nat. Acad. Sci. U. S. A. 97:4867–4872. 10.1073/pnas.97.9.4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. 10.1016/S0092-8674(00)81363-X [DOI] [PubMed] [Google Scholar]
- 22.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. 10.1016/S0092-8674(00)80058-6 [DOI] [PubMed] [Google Scholar]
- 23.Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510. 10.1172/JCI13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takai Y, Irie K, Shimizu K, Sakisaka T, Ikeda W. 2003. Nectins and nectin-like molecules: roles in cell adhesion, migration, and polarization. Cancer Sci. 94:655–667. 10.1111/j.1349-7006.2003.tb01499.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takai Y, Miyoshi J, Ikeda W, Ogita H. 2008. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 9:603–615. 10.1038/nrm2457 [DOI] [PubMed] [Google Scholar]
- 26.Ware CF. 2008. Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol. Rev. 223:186–201. 10.1111/j.1600-065X.2008.00629.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. 10.1016/j.cell.2008.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. 10.1038/nature09420 [DOI] [PubMed] [Google Scholar]
- 29.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 107:866–871. 10.1073/pnas.0913351107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiratori I, Ogasawara K, Saito T, Lanier LL, Arase H. 2004. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J. Exp. Med. 199:525–533. 10.1084/jem.20031885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mousseau DD, Banville D, L'Abbe D, Bouchard P, Shen S-H. 2000. PILRa, a novel immunoreceptor tyrosine-based inhibitory motif-bearing protein, recruits SHP-1 upon tyrosine phosphorylation and is paired with the truncated counterpart PILRb. J. Biol. Chem. 275:4467–4474. 10.1074/jbc.275.6.4467 [DOI] [PubMed] [Google Scholar]
- 32.Fournier N, Chalus L, Durand I, Garcia E, Pin JJ, Churakova T, Patel S, Zlot C, Gorman D, Zurawski S, Abrams J, Bates EE, Garrone P. 2000. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J. Immunol. 165:1197–1209 [DOI] [PubMed] [Google Scholar]
- 33.Kubagawa H, Chen CC, Ho LH, Shimada TS, Gartland L, Mashburn C, Uehara T, Ravetch JV, Cooper MD. 1999. Biochemical nature and cellular distribution of the paired immunoglobulin-like receptors, PIR-A and PIR-B. J. Exp. Med. 189:309–318. 10.1084/jem.189.2.309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uehara T, Blery M, Kang DW, Chen CC, Ho LH, Gartland GL, Liu FT, Vivier E, Cooper MD, Kubagawa H. 2001. Inhibition of IgE-mediated mast cell activation by the paired Ig-like receptor PIR-B. J. Clin. Invest. 108:1041–1050. 10.1172/JCI12195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munitz A, McBride ML, Bernstein JS, Rothenberg ME. 2008. A dual activation and inhibition role for the paired immunoglobulin-like receptor B in eosinophils. Blood 111:5694–5703. 10.1182/blood-2007-12-126748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. 2002. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417:941–944. 10.1038/nature00867 [DOI] [PubMed] [Google Scholar]
- 37.McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. 2005. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 309:2222–2226. 10.1126/science.1114362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu BP, Fournier A, GrandPre T, Strittmatter SM. 2002. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science 297:1190–1193. 10.1126/science.1073031 [DOI] [PubMed] [Google Scholar]
- 39.Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M. 2008. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322:967–970. 10.1126/science.1161151 [DOI] [PubMed] [Google Scholar]
- 40.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. 2009. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 10:778–790. 10.1038/nrm2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. 2004. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 279:2800–2808. 10.1074/jbc.M309981200 [DOI] [PubMed] [Google Scholar]
- 42.Plate AE, Smajlović J, Jardetzky TS, Longnecker R. 2009. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J. Virol. 83:768–7689. 10.1128/JVI.00457-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescence protein. J. Virol. 81:11532–11537. 10.1128/JVI.01343-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pertel P, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. 10.1006/viro.2000.0713 [DOI] [PubMed] [Google Scholar]
- 45.Fan Q, Lin E, Satoh T, Arase H, Spear PG. 2009. Differential effects on cell fusion activity of mutations in herpes simplex virus 1 glycoprotein B (gB) dependent on whether a gD receptor or a gB receptor is overexpressed. J. Virol. 83:7384–7390. 10.1128/JVI.00087-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin E, Spear PG. 2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 104:13140–13145. 10.1073/pnas.0705926104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan Q, Amen M, Harden M, Severini A, Griffiths A, Longnecker R. 2012. Herpes B virus utilizes human nectin-1 but not HVEM or PILRalpha for cell-cell fusion and virus entry. J. Virol. 86:4468–4476. 10.1128/JVI.00041-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. U. S. A. 109:E2625–2634. 10.1073/pnas.1213813109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan Q, Longnecker R. 2012. Is nectin-1 the “master” receptor for deadly herpes B virus infection? Virulence 3:405. 10.4161/viru.20587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milne RSB, Connolly SA, Krummenacher C, Eisenberg RJ, Cohen GH. 2001. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 281:315–328. 10.1006/viro.2000.0798 [DOI] [PubMed] [Google Scholar]
- 51.Manoj S, Jogger CR, Myscofski D, Yoon M, Spear PG. 2004. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl. Acad. Sci. U. S. A. 101:12414–12421. 10.1073/pnas.0404211101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC, Cohen GH, Eisenberg RJ. 2005. Potential nectin-1 binding site on herpes simplex virus glycoprotein D. J. Virol. 79:1282–1295. 10.1128/JVI.79.2.1282-1295.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2 and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 77:9221–9231. 10.1128/JVI.77.17.9221-9231.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, Peng T, Nicola AV, Montgomery RI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ. 1997. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J. Virol. 71:6083–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, Fan Q, Satoh T, Arii J, Lanier LL, Spear PG, Kawaguchi Y, Arase H. 2009. Binding of herpes simplex virus glycoprotein B (gB) to paired immunoglobulin-like type 2 receptor alpha depends on specific sialylated O-linked glycans on gB. J. Virol. 83:13042–13045. 10.1128/JVI.00792-09 [DOI] [PMC free article] [PubMed] [Google Scholar]