

ABSTRACT
Prime-boost immunization regimens have proven efficacious at generating robust immune responses. However, whether the level of replication of the boosting antigen impacts the magnitude and protective efficacy of vaccine-elicited immune responses remains unclear. To evaluate this, we primed mice with replication-defective adenovirus vectors expressing the lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP), followed by boosting with either LCMV Armstrong, which is rapidly controlled, or LCMV CL-13, which leads to a more prolonged exposure to the boosting antigen. Although priming of naive mice with LCMV CL-13 normally results in T cell exhaustion and establishment of chronic infection, boosting with CL-13 resulted in potent recall CD8 T cell responses that were greater than those following boosting with LCMV Armstrong. Furthermore, following the CL-13 boost, a greater number of anamnestic CD8 T cells localized to the lymph nodes, exhibited granzyme B expression, and conferred improved protection against Listeria and vaccinia virus challenges compared with the Armstrong boost. Overall, our findings suggest that the replicative capacity of the boosting antigen influences the protective efficacy afforded by prime-boost vaccine regimens. These findings are relevant for optimizing vaccine candidates and suggest a benefit of robustly replicating vaccine vectors.
IMPORTANCE The development of optimal prime-boost vaccine regimens is a high priority for the vaccine development field. In this study, we compared two boosting antigens with different replicative capacities. Boosting with a more highly replicative vector resulted in augmented immune responses and improved protective efficacy.
INTRODUCTION
A major challenge in the development of T cell-based vaccines is the generation of CD8 T cell responses of sufficient quantity and quality. Following immunization, cytotoxic CD8 T cells undergo extensive division and differentiation into long-lived memory cells. The number and function of antigen-specific T cells are influenced by several factors, including the route of antigen delivery (1), distinct triggering of innate responses (2–4), the degree of costimulation (5–10), the prime-boost time interval (11, 12), the level of preexisting immunity to the vaccine vector (13–15), and the recall history of responding T cells (16–18). Although the magnitude and duration of the priming stimulus have been shown to impact the function, numbers, and differentiation of CD8 T cells (1, 19–27), it is currently not known how the nature of the boosting stimulus influences anamnestic responses and immune protection by memory CD8 T cells.
Alternative serotype adenovirus (Ad) vectors, such as adenovirus type 26 (Ad26) and Ad35, have been shown to induce functionally improved memory T cell responses than that induced by Ad5 (11, 28). We have also demonstrated the protective efficacy of vaccine regimens based on alternative serotype Ad vectors against high-dose intravenous (i.v.) and repetitive low-dose intrarectal simian immunodeficiency virus (SIV) challenges in rhesus monkeys (29, 30).
In this study, we investigated whether the replicative capacity of the boosting antigen influences the protective efficacy of vaccine-elicited CD8 T cell responses. Mice were first primed with an alternative serotype Ad vector (Ad26) expressing the lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP) (Ad26-GP) and were then boosted with either LCMV Armstrong or LCMV CL-13. These two strains differ by only two amino acids that render CL-13 able to replicate faster, but all T cell epitopes between Armstrong and CL-13 remain identical (31, 32). Although CL-13 induces a chronic infection in naive mice, prior immunization with Ad vectors expressing the LCMV GP results in immune-mediated control following systemic CL-13 challenge (28). We boosted Ad26-GP-primed mice with LCMV Armstrong or LCMV CL-13 and challenged them with high doses of Listeria monocytogenes or vaccinia virus expressing the LCMV GP33-41 (glycoprotein 33, residues 33 to 41) CD8 T cell epitope. Compared to boosting with LCMV Armstrong, boosting with LCMV CL-13 resulted in enhanced adaptive immune responses and improved immune protection. These results suggest a benefit for boosting vectors with enhanced replicative capacity, thus providing a rationale for the development of replicating vaccine vectors.
MATERIALS AND METHODS
Mice and infections.
Six- to 8-week-old female C57BL/6 mice (Jackson Laboratory) were used. Mice were immunized intramuscularly with 1010 viral particles (VP) of replication-incompetent E1/E3 deleted adenovirus serotype 26 (33) expressing LCMV GP as described previously (28). LCMV Armstrong and LCMV CL-13 were administered by i.v. injection (2 × 106 PFU) via the lateral tail vein. To assess for reduced induction of primary (nucleoprotein [NP]-specific) T cell responses in Ad26-GP-primed mice, Ad26-GP-primed mice were sacrificed on day 5 following an intraperitoneal (i.p.) LCMV Armstrong boost, and the spleens were harvested and stimulated with NP or GP peptides for 5 h (a similar pattern was observed if the spleens were harvested at day 7). Detailed Listeria and confirmatory vaccinia virus challenges were performed as described previously (12). To assess immune protection, we first titrated the minimum dose of Listeria monocytogenes gp33 (LMgp33) that resulted in discernible survival outcomes in mice that were primed with Ad26-GP and boosted with LCMV. Intravenous challenge with LMgp33 at a dose of ≥108 CFU resulted in 100% mortality, whereas challenge with a ≤106 CFU dose resulted in 100% survival in all primed-boosted mice (data not shown). We thus used an intermediate challenge dose (107 CFU), and bacterial titers were determined at day 2 postchallenge as previously described (28). For viral challenges, vaccinia virus expressing the LCMV GP33-41 epitope (VVgp33) was injected i.p. (108 PFU), and viral titers were determined at day 5 postchallenge in the ovaries. All experiments were performed with approval of the Institutional Animal Care and Use Committee (IACUC), Beth Israel Deaconess Medical Center (BIDMC).
Viral titration.
Titration of LCMV was performed on Vero cell monolayers by standard plaque assays as previously described (28). In brief, 10-fold dilutions from serum samples were added on top of Vero E6 monolayers. The plates were incubated for 60 min and rocked every 15 min. A 1:1 solution of 1% agarose in 2X199 medium (Invitrogen catalog no. 31100-35) was aliquoted on top, and 4 days later, a 1:1 solution of 1% agarose in 2X199 medium with 1:50 neutral red was added on top. The number of plaques was counted on day 5. Listeria and vaccinia virus plaque assays were performed as previously described (34, 35).
Histology.
Tissues were fixed for 1 day in Bouin's fixative (PolySciences, Inc.) and delivered to the Harvard Mouse Histopathology Core for sectioning and hematoxylin and eosin (H&E) staining.
Reagents and flow cytometry.
Single-cell suspensions were prepared from blood and tissue samples. During staining, dead cells were excluded by gating out cells positive for Live/Dead fixable dead cell stain (Invitrogen). Major histocompatibility complex (MHC) class I monomers were obtained from the NIH Tetramer Core Facility (Emory University). LCMV peptides were purchased from Anaspec (Fremont, CA). Samples were acquired with a Becton, Dickinson LSRII and analyzed using FlowJo (Treestar).
Statistical analysis.
Statistical analyses were performed using Prism version 6 (Graphpad). Statistical significance for survival Kaplan-Meier plots was determined by Mantel-Cox tests. All other data were analyzed using Mann-Whitney tests. The absolute numbers of antigen-specific T cells were calculated by multiplying the total number of lymphocytes isolated in each tissue by the fraction of live CD8+ lymphocytes that were either tetramer positive or gamma interferon (IFN-γ) positive (IFN-γ+) as shown previously (36).
RESULTS
LCMV CL-13 boosting results in increased magnitude of anamnestic CD8 T cell responses compared with LCMV Armstrong boosting.
We initiated studies by priming C57BL/6 mice with Ad26 vectors expressing lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP) as reported previously (28). After 60 days, we boosted mice with 2 × 106 PFU of LCMV Armstrong (which is rapidly controlled) or LCMV CL-13 (which is more slowly controlled) (Fig. 1A). In naive mice, LCMV Armstrong infection results in a transient acute infection that is typically cleared within 8 to 10 days, whereas LCMV CL-13 establishes a systemic infection that lasts 60 to 90 days (20). Thus, we hypothesized that boosting Ad26-GP-primed mice with LCMV Armstrong would provide only a brief antigenic stimulus, whereas boosting with LCMV CL-13 would result in a more durable antigenic stimulus.
FIG 1.

Boosting with LCMV CL-13 results in transiently increased viral loads and transiently augmented interferon responses compared to boosting with LCMV Armstrong. (A) Experimental design. Mice were primed with Ad26-GP, and after 60 days, they received an LCMV Armstrong (Arm) or LCMV CL-13 boost. (B) Clearance kinetics of LCMV Armstrong or LCMV CL-13 boosts. (C) Weight loss following LCMV Armstrong or LCMV CL-13 boosting. (D) IFN-α levels in serum on day 1 postboost. (E) IFN-γ levels in serum on day 1 postboost. Data are from Ad26-GP-primed mice that were boosted with LCMV Armstrong or CL-13. Day 10 was selected as the last time point in panel B because it corresponded to the day of complete viral control in all mice. Error bars represent the standard errors of the means (SEM). Data are from three experiments (four mice per group per experiment). Values that were statistically significant are indicated by asterisks as follows: *, P = 0.0009; **, P = 0.0002; ***, P < 0.0001.
As expected, the LCMV Armstrong boost was cleared significantly faster than the LCMV CL-13 boost in Ad26-GP-primed mice. On day 3 following the boost, there were 79-fold-higher LCMV titers in the mice that received the LCMV CL-13 boost than in the mice that received the LCMV Armstrong boost (P < 0.0001) (Fig. 1B). Moreover, the LCMV Armstrong boost was cleared within 5 days, whereas the LCMV CL-13 boost was cleared within 10 days (Fig. 1B). Thus, the use of LCMV Armstrong versus LCMV CL-13 allowed a comparison of the impact of boost duration on immunogenicity and protective efficacy.
By day 7 after the boost, the mice that received the LCMV Armstrong boost lost only an average of 4% of their body weight, whereas those that received the LCMV CL-13 boost exhibited a trend toward increased weight loss (11%). All mice recovered by day 14 (Fig. 1C). Boosting with LCMV CL-13 also resulted in significantly greater IFN-α levels in sera compared to boosting with LCMV Armstrong on day 1 postboost (Fig. 1D), likely reflective of augmented innate stimulation as a result of increased LCMV replication. IFN-α levels declined to baseline levels in both groups after day 3 postboost (data not shown). IFN-γ levels were also higher after the CL-13 boost than after the Armstrong boost (Fig. 1E).
We next evaluated GP-specific CD8 T cell responses in these mice. Mice that received the LCMV CL-13 boost showed increased frequencies (Fig. 2A) and numbers (Fig. 2B) of GP-specific memory CD8 T cells in blood compared to mice that received the LCMV Armstrong boost (P = 0.05) on day 60 postboost. This enhancement in the frequencies and numbers of memory CD8 T cells following the CL-13 boost was also evident in other tissues, such as liver, lung, and kidney (P = 0.05) (Fig. 2C and D).
FIG 2.

Boosting with LCMV CL-13 results in increased magnitude of anamnestic CD8 T cell responses compared to boosting with LCMV Armstrong. (A) Representative fluorescence-activated cell sorting (FACS) plots showing the percentage of CD8 T cells in blood that were DbGP33+. PBMCs, peripheral blood mononuclear cells. (B) Summary of DbGP33 CD8 T cell responses in blood. (C) Representative FACS plots showing DbGP33+ responses in several tissues. (D) Numbers of DbGP33+ CD8 T cell responses in several tissues. Data are from day 60 LCMV boost. Error bars represent SEM. Data are from three experiments (four mice per group per experiment). Values that were statistically significant (P = 0.05) are indicated by an asterisk.
There were also higher frequencies of polyfunctional (IFN-γ+ TNF-α+ [tumor necrosis factor alpha positive]) secondary CD8 T cells (Fig. 3A) and CD4 T cells (Fig. 3B) in mice that received the LCMV CL-13 boost than in the mice that received the LCMV Armstrong boost (day 60 postboost). The total numbers of GP-specific CD8 were also greater in mice that received the CL-13 boost than in the mice that received the Armstrong boost (Fig. 3C). Additionally, memory CD8 T cells from mice that received the CL-13 boost showed enhanced cytokine coexpression compared to the Armstrong boost (P = 0.05) (Fig. 3D). Memory CD4 T cell responses were also greater in mice that received the CL-13 boost compared with the Armstrong boost (Fig. 3E and F). We also analyzed humoral responses before and after boosting. Following priming with Ad26-GP, there were no detectable levels of LCMV-specific IgG (data not shown). However, following LCMV boosting, LCMV-specific antibodies were detected and mice that received the CL-13 boost had slightly increased titers of LCMV-specific IgG in sera relative to those that received the Armstrong boost (P = 0.05) (Fig. 3G).
FIG 3.

Boosting with LCMV CL-13 results in increased numbers of functional T cell responses and enhanced humoral responses compared to boosting with LCMV Armstrong. (A) Representative FACS plots showing the percentages of functional CD8 T cells in spleen that were specific for several GP-derived epitopes. (B) Representative FACS plots showing the percentages of functional CD4 T cells in spleen that were specific for GP61. (C) Numbers of functional GP33-specific CD8 T cells in spleen. (D) Numbers of several GP-specific CD8 T cell responses that coexpressed IFN-γ and TNF-α in spleen. (E) Numbers of functional GP61-specific CD4 T cells in spleen. (F) Numbers of functional GP61-specific CD4 T cell responses that coexpressed IFN-γ and TNF-α in spleen. (G) LCMV-specific antibody levels in sera at day 15 postboost (enzyme-linked immunosorbent assay [ELISA]). The dotted line represents the limit of detection (LOD). Prime immunizations were performed with Ad26-GP, and at day 60, mice were boosted with either LCMV Armstrong or LCMV CL-13. Data from panels A to F are from day 60 postboost following 5-h peptide stimulation. Error bars represent SEM. Data are from three experiments (four mice per group per experiment). Values that were statistically significant are indicated by asterisks as follows: *, P = 0.05; **, P = 0.02.
We then examined whether Ad26-GP priming, which generated only glycoprotein-specific T cell responses, would also result in enhanced elicitation of nucleoprotein (NP)-specific T cell responses following an LCMV boost. To test this, we primed mice with Ad26-GP, and after 60 days, we boosted mice with LCMV Armstrong and assessed GP versus NP T cell responses on day 5 postboost (Fig. 4A). Of note, NP antigens were not included in the Ad26-GP prime, and thus, all NP-specific T cells constituted de novo-generated (primary) T cell responses following the LCMV boost. Interestingly, the anamnestic GP-specific T cell responses in Ad26-GP-primed mice that received an LCMV Armstrong boost were associated with a decreased induction of NP-specific primary T cell responses (Fig. 4). The same pattern was observed after LCMV CL-13 boosting (data not shown). Ad26-GP-primed mice exhibited reduced nucleoprotein 396 (NP396)-specific CD8 (Fig. 4B to D) and NP309-specific CD4 (Fig. 4E to G) T cell responses compared to unimmunized mice after the LCMV boost. After boosting, a reduced induction of other NP-specific CD8 T cell responses (NP205) was also observed in Ad26-GP-primed mice relative to unprimed mice (data not shown). This could potentially be explained by a fitness advantage of secondary effectors over primary effectors, perhaps in their ability to compete for cytokines or access to antigen-presenting cells.
FIG 4.

Recall of memory GP-specific T cell responses is associated with reduced priming of NP-specific CD8 T cell responses following LCMV boost. (A) Experimental design. Mice were primed with Ad26-GP or not primed (unvaccinated [Unvax]), and after 60 days, they received an LCMV Armstrong boost. (B) Representative FACS plots showing the percentages of CD8 T cells that were GP or NP specific. (C) Numbers of GP-specific CD8 T cell responses. (D) Numbers of NP396-specific CD8 T cells. (E) Representative FACS plots showing the percentages of CD4 T cells that were GP or NP specific. (F) Numbers of GP61-specific CD4 T cell responses. (G) Numbers of NP309-specific CD4 T cell responses. Data are from spleens on day 5 after LCMV Armstrong boost, following 5-h peptide stimulation. Error bars represent SEM. Data are from two experiments (four mice per group per experiment). Values that were statistically significant (P = 0.05) are indicated by an asterisk.
LCMV CL-13 boosting results in greater granzyme B expression by memory CD8 T cells than LCMV Armstrong boosting.
We next compared granzyme B expression in memory CD8 T cells following Armstrong versus CL-13 boosting. CL-13 boosting resulted in increased percentages (Fig. 5A) and increased expression per cell (Fig. 5B) of granzyme B-expressing CD8 T cells relative to Armstrong boosting. The percentage of splenic memory CD8 T cells that expressed granzyme B was also significantly greater following CL-13 boost than Armstrong boost (P = 0.02) (Fig. 5C). Granzyme B expression was particularly higher in memory CD8 T cells from lymph nodes of mice boosted with CL-13 (47%) compared with Armstrong (18%) (P = 0.02) (Fig. 5C). The expression of granzyme B per cell was also higher in memory CD8 T cells of mice that received the CL-13 boost, especially in lymph nodes (Fig. 5D). In addition, we also noticed enhanced granzyme B expression by total splenic CD44+ CD4+ T cells following CL-13 boosting than following Armstrong boosting (data not shown). Taken together, these results suggested that the CL-13 boost improved both quantitative and qualitative features of the memory T cell response compared with the Armstrong boost. Although we observed differences in granzyme B expression following Armstrong or CL-13 boosting, we did not notice differences in the expression of other markers involved in T cell differentiation such as CD62L or CD44 (data not shown).
FIG 5.

Boosting with LCMV CL-13 enhances granzyme B expression on memory CD8 T cells relative to boosting with LCMV Armstrong. (A) Representative FACS plots showing the percentages of CD8 T cells that expressed granzyme B. LN, lymph nodes; GzB, granzyme B. (B) Representative histograms showing the mean fluorescence intensity (MFI) of granzyme B. (C) Summary of the percentages of granzyme B-expressing cells. (D) Summary of the MFI of granzyme B expression. Prime immunizations were performed with Ad26-GP followed by LCMV Armstrong or LCMV CL-13 boost. Panels B, C, and D show data from DbGP33-specific CD8 T cells from the indicated tissues on day 60 after LCMV boost. Error bars represent SEM. Data are from three experiments (four mice per group per experiment). Values that were statistically significant are indicated by asterisks as follows: *, P = 0.02; **, P = 0.01.
Boosting with LCMV CL-13 results in superior protective efficacy against Listeria and vaccinia virus challenges compared to boosting with LCMV Armstrong.
To assess protective efficacy of these LCMV boosting regimens, we challenged Ad26-GP-primed, LCMV-boosted mice with a high dose of Listeria (107 CFU of LMgp33) or vaccinia virus (108 PFU of VVgp33), which expressed the GP33-41 CD8 T cell epitope (Fig. 6A).
FIG 6.

Improvement in immune protection following boosting with LCMV CL-13 relative to boosting with LCMV Armstrong. (A) Experimental design. Mice were primed with Ad26-GP, and after 60 days, they received an LCMV Armstrong or LCMV CL-13 boost. After >30 days, mice were challenged with LMgp33 or VVgp33. (B) Survival after lethal LMgp33 challenge. (C) Bacterial loads in the spleen after lethal LMgp33 challenge (day 2). (D) Bacterial loads in liver after lethal LMgp33 challenge (day 2). (E) Hematoxylin and eosin stain of tissue sections on day 2 after LMgp33 challenge. The black arrow indicates areas of microabscesses. (F) Viral loads after vaccinia virus (VVgp33) challenge (day 5). Error bars represent SEM. Data are from two experiments (eight mice per group per experiment).
Ad26-GP-primed mice that received the LCMV CL-13 boost were fully protected against this lethal LMgp33 challenge (107 CFU), whereas Ad26-GP-primed mice that received the LCMV Armstrong boost were only partially protected (P = 0.03) (Fig. 6B). Unvaccinated (naive) mice died within 12 h following this LMgp33 challenge dose (Fig. 6B). Moreover, plaque assays from spleen (Fig. 6C) and liver (Fig. 6D) on day 2 following Listeria challenge revealed striking differences in the bacterial loads. Interestingly, Ad26-GP-primed mice that had received the CL-13 boost prior to Listeria challenge exhibited 91-fold-lower bacterial titers in spleen than those that received the Armstrong boost (P = 0.02) (Fig. 6C). Histopathological examination by H&E staining also revealed striking differences between groups. Mice that were primed with Ad26-GP and boosted with LCMV Armstrong showed areas of splenic apoptosis and hepatic microabscesses on day 2 following Listeria challenge, whereas mice that were boosted with LCMV CL-13 showed normal spleen and liver histology (Fig. 6E).
To confirm the superior protective efficacy conferred by the LCMV CL-13 boost in a different challenge system, we also analyzed the protective efficacy of analogous vaccine regimens against recombinant vaccinia virus challenge. Mice primed with Ad26-GP that received the LCMV Armstrong or LCMV CL-13 boost were challenged with 108 PFU of vaccinia virus expressing gp33 (VVgp33), and viral titers were measured on day 5 in ovaries (Fig. 6A). Mice that received the CL-13 boost showed enhanced control of vaccinia virus compared to mice that received the Armstrong boost (P = 0.01) (Fig. 6F). We also observed the same pattern of enhanced T cell recall and improved immune protection following CL-13 boosting relative to Armstrong boosting in mice that were primed with other Ad vectors (Ad5, Ad35, or Ad48) (data not shown). Overall, these results demonstrate that a highly replicating boosting antigen resulted in improved recall CD8 T cell responses with superior protective capacity following both bacterial and viral challenges.
Robust immunological control and survival following lethal Listeria challenge are associated with minimal expansion and activation of memory CD8 T cells.
The previous experiments demonstrated that Ad26-GP priming followed by LCMV CL-13 boosting afforded complete protection following lethal Listeria challenges. We next assessed the recall CD8 T cell response on day 2 postchallenge to determine the extent of expansion of memory CD8 T cells (Fig. 7A). Interestingly, Ad26-GP-primed, CL-13-boosted mice showed very limited proliferation of memory CD8 T cells in spleen and liver following Listeria challenge, as measured by Ki67 staining (Fig. 7B and C). Following the LMgp33 challenge, the expression of Ki67 per cell was also reduced in mice that had previously received a CL-13 boost than in mice that received Armstrong boost (Fig. 7D and E). This same pattern of Ki67+ expression was also evident longitudinally on memory CD8 T cells from blood (Fig. 7F).
FIG 7.

Rapid immune control is associated with minimal recall expansion and modest activation of memory CD8 T cells. (A) Experimental design. Mice were primed with Ad26-GP, and after 60 days, they received an LCMV Armstrong or LCMV CL-13 boost. After >30 days, mice were challenged with LMgp33, and early anamnestic CD8 T cell responses were analyzed on day 2. (B) Representative FACS plots showing the percentages of CD8 T cells that expressed the proliferation marker Ki67. (C) Summary of the percentages of Ki67-expressing CD8 T cells. (D) Representative histograms showing Ki67 expression by splenic CD8 T cells. (E) Summary of the MFI of Ki67-expressing CD8 T cells. (F) Percentage of Ki67-expressing CD8 T cells in blood before and after Listeria challenge. (G) Representative histograms showing the mean fluorescence intensity (MFI) for CD44 and PD-1 by CD8 T cells. (H) Summary of the MFI of CD44 and PD-1 expression in spleen. (I) Summary of the MFI of CD44 and PD-1 expression in liver. Data are gated from DbGP33-specific CD8 T cells from the indicated tissues. Error bars represent SEM. Data are from two experiments (four mice per group per experiment). Values that were statistically significant are indicated by asterisks as follows: *, P = 0.05; **, P = 0.02.
Moreover, memory CD8 T cells of mice that were boosted with LCMV CL-13 showed very limited immune activation following Listeria challenge as evidenced by modest CD44 and PD-1 (programmed cell death protein 1) expression (Fig. 7G to I). This same pattern was observed following VVgp33 challenge (data not shown). Taken together, these results suggested that rapid immune control is associated with a recall CD8 T cell response that did not require additional expansion or activation.
Note that although Ad26-GP priming followed by LCMV CL-13 boosting resulted in optimal CD8 T cell boosting, inversion of this prime-boost regimen resulted in impaired boosting capacity. Priming with LCMV CL-13 and boosting with Ad26-GP resulted in minimal or no expansion of CD8 T cells (Fig. 8), which could be due to induction of high levels of PD-1 upon priming with a highly replicating antigen. Note that whereas challenge of LCMV Armstrong-immune mice with LMgp33 results in robust immune protection, challenge of CL-13-infected mice with LMgp33 results in suboptimal immune protection (evidenced by bacterial plaque assays on day 2 after LMgp33 challenge [data not shown]). These observations parallel previous studies that show vigorous cytotoxicity by CD8 T cells from mice infected with LCMV Armstrong, but not from mice infected with LCMV CL-13 (20, 37). Importantly, PD-1 blockade has been shown to improve the boosting of exhausted CD8 T cells generated after chronic CL-13 infection (38), and thus, blockade of multiple coinhibitory pathways may represent a promising strategy to further improve the boosting capacity of exhausted CD8 T cells. Overall, these data suggest that the order of priming and boosting is critical and that the benefits of enhancing the replicative capacity of the boost may not apply to the prime.
FIG 8.
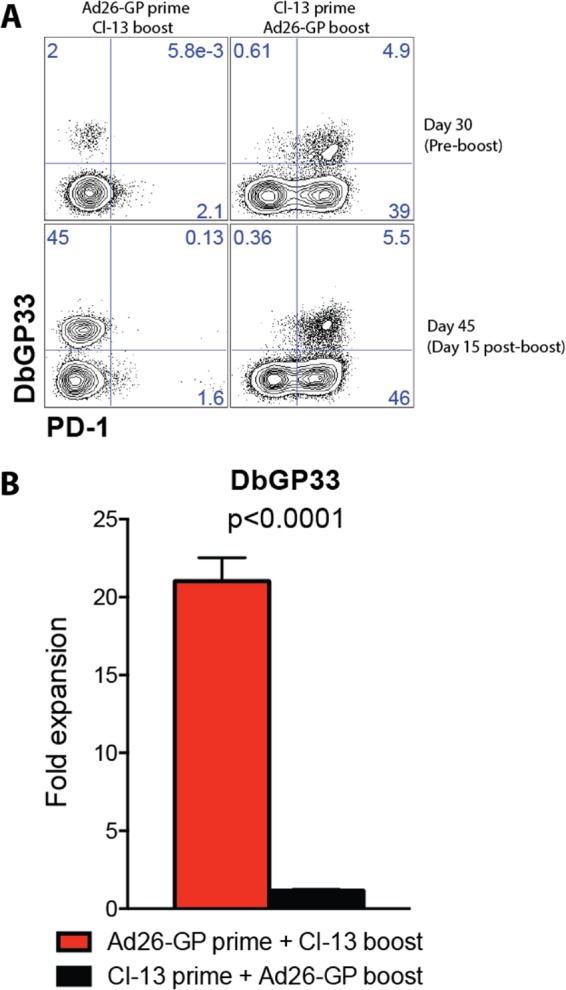
Boosting potential is influenced by the nature of the priming antigen. (A) Representative FACS plots showing the percentage of CD8 T cells in blood that were DbGP33+ PD-1+ following Ad26-GP prime and CL-13 boost in comparison to CL-13 prime and Ad26-GP boost. (B) Fold expansion of DbGP33 CD8 T cell responses in blood on day 15 postboost. Error bars represent SEM. Data are from two experiments (four mice per group per experiment).
DISCUSSION
In the present study, we have shown that the replicative capacity of the boosting antigen impacts the magnitude and protective efficacy of adaptive immune responses elicited by Ad/LCMV prime-boost vaccine regimens. In particular, boosting with LCMV CL-13 resulted in a prolonged and greater pulse of antigen that resulted in augmented cellular and humoral immune responses and improved protection against both Listeria and vaccinia virus challenges compared to boosting with LCMV Armstrong. These data demonstrate that the immunologic properties of the boosting antigen, likely reflecting the extent of antigen expression, substantially impact the efficacy of heterologous prime-boost vaccine regimens.
Strikingly, memory GP-specific CD8 T cells from mice that were primed with Ad26-GP and boosted with LCMV CL-13 underwent a substantial recall expansion, with several GP-specific CD8 T cell responses constituting more than 70% of total CD8 T cells in the spleen even after day 60 postboost (Fig. 3A). Of note, if the order of the prime-boost is inverted, T cell responses are quickly exhausted following priming with LCMV CL-13 and cannot be significantly boosted by Ad-GP vectors (Fig. 8). Thus, our data suggest that the order of a prime-boost immunization is critical.
Of note, the robust recall of GP-specific T cell responses in Ad26-GP-primed mice was associated with reduced generation of NP-specific T cell responses following LCMV boost (Fig. 4). This could be due to interclonal competition between memory and naive T cells for cytokines and access to antigen-presenting cells.
Anamnestic CD8 T cell responses elicited by a CL-13 boost also expressed higher levels of granzyme B compared to those induced by an Armstrong boost (Fig. 5). Although it is not completely clear why the CL-13 boost induced a greater number of granzyme B-expressing CD8 T cells relative to the Armstrong boost, it is possible that a strong level of immune stimulation following boosting with a highly replicating antigen may result in improved induction of granzyme B-expressing effector CD8 T cells that migrate to lymphoid sites. Note that migration of secondary effectors to the lymph nodes is necessary for immune control of CL-13 (17), and thus, it is plausible that a CL-13 boost induces effector CD8 T cells that are preferentially poised at lymphoid sites. This observation is potentially relevant, since localization of cytotoxic CD8 T cells to the lymph nodes has been reported to be associated with control of persistent infection and sustainment of CD8 T cell responses (17). In addition, functional CD8 T cell responses in lymph nodes have been associated with immune protection elicited by live attenuated SIV vaccines, as lymph nodes constitute a crucial site of pathogen intercept especially after systemic challenges (39, 40).
These data indicate that the CD8 T cells elicited by Ad26-GP prime and CL-13 boost were highly functional effector cells that did not require further expansion or overt activation to control high-dose pathogen challenges. Highly functional effector CD8 T cells elicited by cytomegalovirus (CMV) vectors have also been shown to provide rapid control against SIV in rhesus monkeys (41). The correlates of immune control in our murine studies are similar with those of previous vaccine studies in monkey models of SIV infection. Protective SIV-specific T cell responses elicited by attenuated SIV vaccines are associated with minimal anamnestic expansion and activation, whereas nonprotective T cell responses exhibit robust expansion and activation following SIV challenges (likely in response to uncontrolled viral replication) (39, 42). Thus, our observations with Listeria and vaccinia virus challenges may be generalizable to human immunodeficiency virus (HIV)/SIV and other infections, suggesting that recall proliferation and activation of vaccine-elicited CD8 T cells correlate inversely with pathogen control. We also observed very low PD-1 upregulation in anamnestic CD8 T cell responses of mice that exhibited 100% survival following Listeria challenge (Ad26-GP/CL-13 boost).
Our data suggest that the replicative capacity of the boost impacts immunogenicity and protective efficacy of prime-boost vaccine regimens. We hypothesize that this is due to the increased magnitude and duration of boosting antigen exposure, but we cannot exclude the possible contribution of other variables, such as differences in the tropism between LCMV Armstrong and LCMV CL-13.
Taken together, our data suggest that the replicative capacity of the boost influences the efficacy of prime-boost vaccine regimens. Following priming with replication-defective Ad vectors, boosting with highly replicating LCMV CL-13 was more effective than boosting with LCMV Armstrong for inducing protective immune responses. Protective efficacy in this model may have reflected differences in boosting antigen stimulation and early IFN responses, which impacted the overall magnitude and functionality of CD8 T cells that localized to multiple tissues, thus providing early cytotoxic control of the pathogen. These data suggest potential benefits of live, replicating vaccine vectors over replication-defective vaccine vectors in candidate booster immunizations in humans. We thus propose a novel model of “escalating stringency” in prime-boost vaccination regimens, which consists of a mild prime (e.g., replication-incompetent vectors), followed by a more stringent boost (e.g., live, replication-competent vectors) to maximize immune responses.
ACKNOWLEDGMENTS
We thank R. Larocca and K. Stephenson for general assistance. We also acknowledge Dave Masopust for discussions.
This work was supported by grants from the NIH (AI007245 and AI07387 to P.P.M.; AI078526 and AI096040 to D.H.B.), the Bill and Melinda Gates Foundation (OPP1033091), and the Ragon Institute of MGH, MIT, and Harvard.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 19 March 2014
REFERENCES
- 1.Kaufman DR, Bivas-Benita M, Simmons NL, Miller D, Barouch DH. 2010. Route of adenovirus-based HIV-1 vaccine delivery impacts the phenotype and trafficking of vaccine-elicited CD8+ T lymphocytes. J. Virol. 84:5986–5996. 10.1128/JVI.02563-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pulendran B, Ahmed R. 2006. Translating innate immunity into immunological memory: implications for vaccine development. Cell 124:849–863. 10.1016/j.cell.2006.02.019 [DOI] [PubMed] [Google Scholar]
- 3.Teigler JE, Iampietro MJ, Barouch DH. 2012. Vaccination with adenovirus serotypes 35, 26, and 48 elicits higher levels of innate cytokine responses than adenovirus serotype 5 in rhesus monkeys. J. Virol. 86:9590–9598. 10.1128/JVI.00740-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. 2006. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 203:413–424. 10.1084/jem.20051720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penaloza-MacMaster P, Ur Rasheed A, Iyer SS, Yagita H, Blazar BR, Ahmed R. 2011. Opposing effects of CD70 costimulation during acute and chronic lymphocytic choriomeningitis virus infection of mice. J. Virol. 85:6168–6174. 10.1128/JVI.02205-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerder S, Carding SR, Flavell RA. 1995. B7 costimulation is necessary for the activation of the lytic function in cytotoxic T lymphocyte precursors. J. Immunol. 155:5167–5174 [PubMed] [Google Scholar]
- 7.Zhang B, Maris CH, Foell J, Whitmire J, Niu L, Song J, Kwon BS, Vella AT, Ahmed R, Jacob J, Mittler RS. 2007. Immune suppression or enhancement by CD137 T cell costimulation during acute viral infection is time dependent. J. Clin. Invest. 117:3029–3041. 10.1172/JCI32426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watts TH. 2005. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 23:23–68. 10.1146/annurev.immunol.23.021704.115839 [DOI] [PubMed] [Google Scholar]
- 9.Sharpe AH, Abbas AK. 2006. T-cell costimulation-biology, therapeutic potential, and challenges. N. Engl. J. Med. 355:973–975. 10.1056/NEJMp068087 [DOI] [PubMed] [Google Scholar]
- 10.Vezys V, Penaloza-MacMaster P, Barber DL, Ha SJ, Konieczny B, Freeman GJ, Mittler RS, Ahmed R. 2011. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. J. Immunol. 187:1634–1642. 10.4049/jimmunol.1100077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan WG, Jin HT, West EE, Penaloza-MacMaster P, Wieland A, Zilliox MJ, McElrath MJ, Barouch DH, Ahmed R. 2013. Comparative analysis of simian immunodeficiency virus Gag-specific effector and memory CD8+ T cells induced by different adenovirus vectors. J. Virol. 87:1359–1372. 10.1128/JVI.02055-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234. 10.1038/ni889 [DOI] [PubMed] [Google Scholar]
- 13.Qureshi H, Ma ZM, Huang Y, Hodge G, Thomas MA, DiPasquale J, DeSilva V, Fritts L, Bett AJ, Casimiro DR, Shiver JW, Robert-Guroff M, Robertson MN, McChesney MB, Gilbert PB, Miller CJ. 2012. Low-dose penile SIVmac251 exposure of rhesus macaques infected with adenovirus type 5 (Ad5) and then immunized with a replication-defective Ad5-based SIV gag/pol/nef vaccine recapitulates the results of the phase IIb step trial of a similar HIV-1 vaccine. J. Virol. 86:2239–2250. 10.1128/JVI.06175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geisbert TW, Bailey M, Hensley L, Asiedu C, Geisbert J, Stanley D, Honko A, Johnson J, Mulangu S, Pau MG, Custers J, Vellinga J, Hendriks J, Jahrling P, Roederer M, Goudsmit J, Koup R, Sullivan NJ. 2011. Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against Ebolavirus challenge. J. Virol. 85:4222–4233. 10.1128/JVI.02407-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, Buchbinder SP, Robertson MN, Mehrotra DV, Self SG, Corey L, Shiver JW, Casimiro DR. 2008. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet 372:1894–1905. 10.1016/S0140-6736(08)61592-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraser KA, Schenkel JM, Jameson SC, Vezys V, Masopust D. 2013. Preexisting high frequencies of memory CD8+ T cells favor rapid memory differentiation and preservation of proliferative potential upon boosting. Immunity 39:171–183. 10.1016/j.immuni.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolz JC, Harty JT. 2011. Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity 34:781–793. 10.1016/j.immuni.2011.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.West EE, Youngblood B, Tan WG, Jin HT, Araki K, Alexe G, Konieczny BT, Calpe S, Freeman GJ, Terhorst C, Haining WN, Ahmed R. 2011. Tight regulation of memory CD8+ T cells limits their effectiveness during sustained high viral load. Immunity 35:285–298. 10.1016/j.immuni.2011.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. 2007. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J. Immunol. 179:6704–6714 [DOI] [PubMed] [Google Scholar]
- 20.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927. 10.1128/JVI.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. 2012. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J. Virol. 86:8161–8170. 10.1128/JVI.00889-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. 2012. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37:1130–1144. 10.1016/j.immuni.2012.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wherry EJ, Ahmed R. 2004. Memory CD8 T-cell differentiation during viral infection. J. Virol. 78:5535–5545. 10.1128/JVI.78.11.5535-5545.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. 2004. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc. Natl. Acad. Sci. U. S. A. 101:16004–16009. 10.1073/pnas.0407192101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. 2007. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684. 10.1016/j.immuni.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 26.Wherry EJ. 2011. T cell exhaustion. Nat. Immunol. 12:492–499 [DOI] [PubMed] [Google Scholar]
- 27.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. 2009. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10:29–37. 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penaloza-MacMaster P, Provine NM, Ra J, Borducchi EN, McNally A, Simmons NL, Iampietro MJ, Barouch DH. 2013. Alternative serotype adenovirus vaccine vectors elicit memory T cells with enhanced anamnestic capacity compared to Ad5 vectors. J. Virol. 87:1373–1384. 10.1128/JVI.02058-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barouch DH, Liu J, Li H, Maxfield LF, Abbink P, Lynch DM, Iampietro MJ, SanMiguel A, Seaman MS, Ferrari G, Forthal DN, Ourmanov I, Hirsch VM, Carville A, Mansfield KG, Stablein D, Pau MG, Schuitemaker H, Sadoff JC, Billings EA, Rao M, Robb ML, Kim JH, Marovich MA, Goudsmit J, Michael NL. 2012. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature 482:89–93. 10.1038/nature10766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, O'Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH. 2009. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 457:87–91. 10.1038/nature07469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salvato M, Borrow P, Shimomaye E, Oldstone MB. 1991. Molecular basis of viral persistence: a single amino acid change in the glycoprotein of lymphocytic choriomeningitis virus is associated with suppression of the antiviral cytotoxic T-lymphocyte response and establishment of persistence. J. Virol. 65:1863–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvato M, Shimomaye E, Southern P, Oldstone MB. 1988. Virus-lymphocyte interactions. IV. Molecular characterization of LCMV Armstrong (CTL+) small genomic segment and that of its variant, clone 13 (CTL-). Virology 164:517–522 [DOI] [PubMed] [Google Scholar]
- 33.Abbink P, Lemckert AA, Ewald BA, Lynch DM, Denholtz M, Smits S, Holterman L, Damen I, Vogels R, Thorner AR, O'Brien KL, Carville A, Mansfield KG, Goudsmit J, Havenga MJ, Barouch DH. 2007. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 81:4654–4663. 10.1128/JVI.02696-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. 2001. Preparation of cell cultures and vaccinia virus stocks. Curr. Protoc. Protein Sci. Chapter 5:Unit 5.12. 10.1002/0471140864.ps0512s13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Busch DH, Vijh S, Pamer EG. 2001. Animal model for infection with Listeria monocytogenes. Curr. Protoc. Immunol.. Chapter 19:Unit 19.9. 10.1002/0471142735.im1909s36 [DOI] [PubMed] [Google Scholar]
- 36.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8:177–187. 10.1016/S1074-7613(00)80470-7 [DOI] [PubMed] [Google Scholar]
- 37.Barber DL, Wherry EJ, Ahmed R. 2003. Rapid in vivo killing by memory CD8 T cells. J. Immunol. 171:27–31 [DOI] [PubMed] [Google Scholar]
- 38.Ha SJ, Mueller SN, Wherry EJ, Barber DL, Aubert RD, Sharpe AH, Freeman GJ, Ahmed R. 2008. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. J. Exp. Med. 205:543–555. 10.1084/jem.20071949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukazawa Y, Park H, Cameron MJ, Lefebvre F, Lum R, Coombes N, Mahyari E, Hagen SI, Bae JY, Reyes MD, III, Swanson T, Legasse AW, Sylwester A, Hansen SG, Smith AT, Stafova P, Shoemaker R, Li Y, Oswald K, Axthelm MK, McDermott A, Ferrari G, Montefiori DC, Edlefsen PT, Piatak M, Jr, Lifson JD, Sekaly RP, Picker LJ. 2012. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat. Med. 18:1673–1681. 10.1038/nm.2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Picker LJ, Hansen SG, Lifson JD. 2012. New paradigms for HIV/AIDS vaccine development. Annu. Rev. Med. 63:95–111. 10.1146/annurev-med-042010-085643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genesca M, Skinner PJ, Hong JJ, Li J, Lu D, McChesney MB, Miller CJ. 2008. With minimal systemic T-cell expansion, CD8+ T cells mediate protection of rhesus macaques immunized with attenuated simian-human immunodeficiency virus SHIV89.6 from vaginal challenge with simian immunodeficiency virus. J. Virol. 82:11181–11196. 10.1128/JVI.01433-08 [DOI] [PMC free article] [PubMed] [Google Scholar]