

Abstract
Development of anxiety-like behaviors during ethanol withdrawal has been correlated with increased histone deacetylase (HDAC) activity and decreased brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton-associated protein (Arc) gene expression in the amygdala. Furthermore, HDAC-mediated histone modifications play a role in synaptic plasticity. In this study we used the HDAC inhibitor trichostatin A (TSA) to determine whether HDAC inhibition could prevent ethanol withdrawal-induced deficits in dendritic spine density (DSD), BDNF or Arc expression in the amygdala of rats. It was found that decreased BDNF and Arc expression in the central (CeA) and medial nucleus of amygdala (MeA), observed during withdrawal after chronic ethanol exposure, were normalized following acute TSA treatment. TSA treatment was also able to attenuate anxiety-like behaviors during ethanol withdrawal and correct the observed decrease in DSD in the CeA and MeA of ethanol-withdrawn rats. Taken together, these findings demonstrate that correcting the deficits in histone acetylation through TSA treatment also amends downstream synaptic plasticity-related deficits such as BDNF and Arc expression, and DSD in the CeA and MeA as well as attenuates anxiety-like behaviors in rats during withdrawal after chronic ethanol exposure.
Keywords: Alcoholism and Anxiety, HDAC inhibitor, BDNF, Arc, Dendritic spines
Introduction
The development of alcoholism occurs following repeated ethanol exposures, and is mediated by both positive and negative effects of ethanol (Koob, 2003a; Pandey, 2004; Gilpin and Koob, 2008). It is believed that the anticipation of the euphoric effect of alcohol promotes the positive reinforcement of alcohol intake, while relief from alcohol withdrawal symptoms fosters the negative reinforcing properties of alcohol drinking (Koob, 2003a; Pandey, 2004; Grüsser et al., 2006). Chronic ethanol exposure-induced neuroadaptations within specific neurocircuitry produces withdrawal symptoms upon the cessation of alcohol consumption (Pandey, 2004; Gilpin and Koob, 2008). For example, anxiety, one of the major alcohol withdrawal symptoms, is a key element driving continuous alcohol consumption, thus playing a critical role in the maintenance of alcohol addiction (Kushner et al., 2000; Koob, 2003a; Pandey, 2003).
The amygdala is a structure located in the rostromedial part of the temporal lobe (Koob, 2003a; 2003b) which functions as the modulator of the autonomic (Sah et al., 2003) and the emotional responses to fear and anxiety (Pessoa and Adolphs, 2010). The central and medial nucleus of amygdala (CeA and MeA) serve as an output station for distribution of sensory information (Pitkanen et al., 1997) throughout other limbic regions and play a crucial role in alcohol addiction (Koob 2003a; Pandey, 2004). In addition, the basolateral amygdala (BLA) which receives innervations from cortical area and sends projections to the CeA and other regions of extended amygdala, has been shown to be involved in associative memory, anxiety, and alcoholism (LeDoux and Muller, 1997; McCool et al., 2010; Kryger and Wilce, 2010; Tye et al., 2011). It is therefore important to investigate the molecular mechanisms underlying the neuroadaptive changes in these specific amygdaloid nuclei in response to chronic ethanol exposure to better understand the pathophysiology of alcoholism.
Chromatin remodeling, either due to histone modifications or changes in DNA methylation, plays an important role in the regulation of synaptic plasticity associated with brain disorders (Tsankova et al., 2007; Lubin et al., 2008; Moonat and Pandey, 2012; Moonat et al., 2013). Histone modifications occur on the amino (N)-terminal tail of histones and play a crucial role in switching between open and closed forms of chromatin structure changing gene promoter accessibility for transcriptional machinery, thus altering gene transcription (Tsankova et al., 2007; Moonat and Pandey, 2012). We have shown that development of anxiety-like behaviors during alcohol withdrawal is correlated with increased histone deacetylase (HDAC) activity, decreased histone acetylation (H3-K9 & H4-K8), and decreased neuropeptide Y(NPY) expression in amygdala, while treatment with the HDAC inhibitor trichostatin A (TSA) during ethanol withdrawal was able to reverse these deficits (Pandey et al., 2008a). In addition, decreased brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton-associated (Arc) protein expression and dendritic spines in the CeA and MeA have been shown to be involved in ethanol withdrawal-related anxiety and alcohol-drinking behaviors in rats (Pandey et al., 2008b; Moonat et al., 2011; 2013). However, it is unknown if the correction of deficits in histone acetylation by TSA treatment can also correct the deficits of synaptic plasticity related events produced during ethanol withdrawal after chronic ethanol exposure. We, therefore, examined the effects of HDAC inhibition on amygdaloid BDNF, Arc expression and dendritic spines during alcohol dependence.
Methods
Chronic ethanol exposure and TSA treatment
All experiments were approved by the Institutional Animal Care and Use Committee and adhered to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals. Adult male Sprague-Dawley rats were housed individually in a temperature-controlled room with a 12/12-hr light/dark cycle, with food and water provided ad libitum. Rats were randomly separated into five groups: (1) Control+ Vehicle, (2) Ethanol+ Vehicle, (3) Withdrawal + Vehicle, (4) Withdrawal + TSA, (5) Control+ TSA. As described previously by us (Pandey et al., 2008a; 2008b), rats were offered 80 ml/day of the nutritionally complete Lieber-DeCarli liquid control diet (Lieber-DeCarli Diet 82; Bio-Serv, Frenchtown, NJ) for 3 days. Control groups continued with control diet for 16 days, while ethanol groups were gradually introduced to ethanol (1.8% through 8.1% within 7 days), and then maintained on 9% v/v ethanol diet for 15 or 16 days. Rats were pair fed and their liquid diet intake and body weights were closely monitored. One group of ethanol-diet fed rats were withdrawn from ethanol for 24 h (Withdrawal group) and given control diet. Another group of ethanol-diet fed rats were withdrawn for 0 h (Ethanol group) and continued to be fed with ethanol diet. Two hours before measuring anxiety-like behaviors, all rats were intraperitoneally (i.p.) injected with either vehicle [DMSO, 1:5 dilution with phosphate buffered saline (PBS)], or TSA [2 mg/kg; dissolved in DMSO and then diluted (1:5 dilution) with PBS] (Pandey et al., 2008a). Anxiety-like behaviors were measured by elevated plus maze (EPM) or light/dark-box (LDB) exploration tests. Immediately after behavioral measurements, rats were anesthetized using pentobarbital (50 mg/kg) and brains were collected for gold immunolabeling, in situ RT-PCR, or Golgi-Cox staining, as described below. Same rats were used for the behavioral and biochemical studies. Blood was also collected to measure blood ethanol levels using an Analox Alcohol Analyzer (Analox Instruments, Lunenburg, MA).
Elevated plus maze test (EPM)
The EPM procedure has been previously described by our lab (Pandey et al., 2008a; Pandey et al., 2008b). The EPM apparatus consists of two open arms and two closed arms arranged perpendicular to each other and connected by a central platform. Following a 5-minute habituation period in the procedure room, rats were placed on the central platform facing an open arm and exploratory behaviors in both open and closed arms were observed for 5 min. The number of entries and the time spent in open or closed arms were recorded and expressed as the percent of open-arm entries and the mean percent of time spent on the open arms (open-arm activity). The general activity of the each rat was represented by total number of closed arm entries.
Light/dark-box (LDB) exploration test
The LDB exploration test procedure was followed, as published by us previously (Pandey et al., 2008a; Sakharkar et al., 2012). The LDB was located in a dark room and consisted of a dark compartment without illumination and a light compartment with illumination connected through an opening. Rats were allowed a 5 minute pretest habituation period in the procedure room prior to testing. Rats were then placed in the dark compartment facing away from the opening and behavior was recorded for 5 minutes. The time spent in each compartment of the LDB was recorded through the use of an infrared beam connected to the LDB compartments and exploratory behaviors of rats were monitored by a computer. The percentage of time spent in either the dark compartment or light compartment was calculated for each animal. The total ambulations for each rat were also calculated as a measure of general activity.
Gold-Immunolabeling procedure for BDNF and Arc in rat brain
Rats were anesthetized and perfused with 200 ml of n-saline, followed by 300 ml of 4% ice-cold paraformaldehyde (PFA) fixative prepared in 0.1M phosphate buffer (PB; pH 7.4). Following perfusion, brains were removed, post-fixed overnight in PFA at 4°C and cryoprotected using a sucrose gradient (10%, 20%, and 30%) prepared in 0.1M PB. Brains were then frozen until gold-immunolabeling histochemical processing to measure protein levels of BDNF and Arc, as previously described by us (Pandey et al., 2008b; Moonat et al., 2011). The coronal brain sections (20 μm) were washed (twice for 10 min) with 0.01 M PBS and then blocked with RPMI 1640 medium with L-glutamine (Life Technologies, Grand Island, NY) for 30 min, followed by 10% normal goat serum diluted in PBS containing 0.25% Triton X-100 (PBST) for 30 min and then 1% BSA (prepared in PBST) for 30 min at room temperature. Sections were then incubated with either anti-BDNF antibody (H-117, Santa Cruz Biotechnology, Santa Cruz, CA, 1:200 dilution; antibody only binds to BDNF, not pro-BDNF) or anti-Arc antibody (H-300, Santa Cruz Biotechnology, Santa Cruz, CA, 1:200 dilution) in 1% BSA (prepared in PBST) for 18 h at room temperature. After two 10 min PBS washes and two 10 min 1% BSA in PBS washes, sections were incubated with gold particle (1.4 nm) conjugated anti-rabbit secondary antibody (Nanoprobes, Yaphank, NY; 1:200 dilution in 1% BSA in PBS) for 1 hr at room temperature. Sections were then rinsed several times in 1% BSA in PBS followed by distilled water. The gold immunolabeling was developed using silver enhancement solution (Ted Pella, Redding, CA) for 15–30 min and sections underwent a final rinsing with tap water and then mounted on slides. Gold-immunolabeled proteins were quantified at high magnification (100x) using an image analysis system connected to a light microscope. The threshold of each image was set up to ensure that an area without staining should give zero counts. Under this condition, gold particles in the defined areas (3 fields in each section) of 3 adjacent brain sections (bregma: 2.12–2.56 mm) of each rat were counted and values averaged for each rat. The results were represented as the number of immunogold particles/100 μm2 area of a defined amygdaloid (CeA, MeA, or BLA) structure.
In situ RT-PCR for Arc mRNA Measurement
In order to measure Arc mRNA levels, we used the in situ RT-PCR procedure described previously (Pandey et al., 2008b; Moonat et al., 2011). Briefly, 40 μm free-floating brain sections were treated with proteinase K (1 μg/ml in 1X PBS containing 0.05% Triton X-100 and prepared in DEPC treated water) for 15 minutes at 37°C, followed by washing with 1X PBS and DNase digestion. After a subsequent 1X PBS wash, sections were transferred to PCR tubes containing 100 μl of reverse transcription reaction mixture (Applied Biosystems, Foster City, CA) containing reverse transcriptase enzyme in the presence of oligo d(t)16. For negative sections, no reverse transcriptase enzyme was added. PCR was performed with Taq DNA polymerase enzyme and Arc primers (IDT DNA, Coralville, IA). The primer sequences we used are based on previous studies and do not span introns (Pandey et al., 2008b; Moonat et al., 2011): 5′-ACAGAGGATGAGACTGAGGCAC-3′ and 5′-TATTCAGGCTGGGTCCTGTCAC-3′. PCR conditions for Arc mRNA were: 94 °C for 5 min; 30 cycles of 95 °C for 15 sec; 55 °C for 30 sec; 72 °C for 30 sec; and a final elongation step of 72 °C for 3 min. After PCR, sections were mounted on slides. Arc-positive cell bodies were detected using an alkaline phosphatase-conjugated anti-DIG antibody and subsequent staining of the complex with the specific substrate, nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indoylphosphate (Roche Diagnostics, Indianapolis, IN). The optical density (OD) of Arc-positive cell bodies was calculated by the image analysis system. The OD of negative brain sections was subtracted from the OD of positive brain sections. The mean OD of Arc-positive cell bodies in the areas of CeA, MeA, and BLA were calculated and values were averaged for each rat. The results were represented as mean OD/100 pixels of area.
Golgi-Cox staining procedure
The labeling of dendritic spines in the amygdaloid brain regions was performed using the Golgi-Cox staining procedure (FD NeuroTechnologies, Baltimore, MD). As we described before (Pandey et al., 2008b; Moonat el al., 2011), brains were rapidly immersed in impregnation solution for at least 1 week. Then 200 μm brain sections were cut, mounted and stained according to the protocol provided by FD NeuroTechnologies. After staining, sections were dehydrated and cleared in xylene solution and then cover slipped using mounting medium. Sections were observed under a light microscope at high magnification (100x), and the dendritic spines were counted using the Neurolucida Neuroexplorer program (MicroBrightField, Williston, VT). The spines displaying complete impregnation as observed by the labeling of dendrites connected to the soma were counted. A total of nine dendrites were counted within each amygdala nuclei (CeA, MeA and BLA) for each rat. Values are depicted as dendritic spine density (DSD) per 10 μm of dendritic length, averaged for each rat.
Statistical analysis
The differences between the various groups for anxiety-like behaviors and neurochemical data were evaluated by a one-way ANOVA test. Post hoc comparisons were performed using Tukey’s test and p<0.05 was considered to be significant.
Results
Effect of TSA treatment on anxiety-like behaviors during ethanol withdrawal in rats
The mean ± SEM blood ethanol level in ethanol diet-fed rats (n=13) was 172 ± 8.6 mg%. The mean ± SEM consumption of ethanol diet (ml/day) by three groups of ethanol-fed rats did not significantly differ from each other (Ethanol+ Vehicle=53.7± 0.69; Withdrawal+ Vehicle=53.2± 0.48; Withdrawal+ TSA=53.2± 0.53). There were no significant differences in the body weights (mean ± SEM) among the various rat groups [Control+ Vehicle, 330± 2.5 g (n=12); Control+ TSA, 331± 2.5 g (n=12); Ethanol+ Vehicle, 330± 2.7 g (n=13); Withdrawal+ Vehicle, 328± 2.0 g (n=12); Withdrawal+ TSA, 325± 2.9 g (n=14)]. In accordance with our earlier findings, (Pandey et al., 2008a), we found that TSA treatment was able to attenuate anxiety-like behaviors in rats withdrawn from chronic ethanol exposure. Two well established behavioral tests, the EPM and LDB, were used to determine anxiety-like behaviors in rats with or without ethanol exposure and/or TSA treatment. In the EPM test (Fig 1), we found that rats withdrawn from chronic ethanol exposure display both decreased percentage of open-arm entries (p<0.001) and percentage of time (p<0.001) spent on open arms, compared with control-diet and ethanol-diet fed rats indicating increased anxiety-like behaviors. TSA treatment during ethanol withdrawal rescued anxiety-like behaviors by increasing both the percentage of open-arm entries and percentage of time spent on open arms in ethanol-withdrawn rats indicating an anxiolytic-like effect of HDAC inhibition in rats undergoing ethanol withdrawal. Control-diet fed rats did not differ in anxiety measures regardless of whether they received vehicle or TSA injections. There were no significant differences in the numbers of total arm entries or closed arm entries among the various groups (Fig 1).
Figure 1.

Measurement of anxiety-like behaviors using the elevated plus maze (EPM) shows increased anxiety levels in ethanol-withdrawn rats and attenuation of anxiety-like behaviors following TSA treatment. The anxiety-like behaviors are represented by open arm and closed arm activity. Values are the mean ± SEM of 6–8 rats in each group. * Significantly different (p < 0.001; one-way ANOVA followed by post hoc analysis by Tukey’s test; percentage of open arm entries [F4, 27=21.25, p<.001] and the percentage of time spent in the open arms [F4, 27=17.141, p<.001]) from all other groups (Control+ Vehicle, Ethanol+ Vehicle, Withdrawal+ TSA, Control+ TSA injected rats).
During the LDB exploration test (Fig 2), withdrawal rats treated with vehicle spent significantly (p<0.001) more time in the dark compartment and significantly (p<0.001) less time in the light compartment indicating increased anxiety-like behaviors. On the contrary, withdrawal rats treated with TSA spent significantly (p<0.001) more time in the light and significantly (p<0.001) less time in the dark, compared to control-diet fed and ethanol-withdrawn rats injected with vehicle. The anxiety measures of chronic ethanol-diet fed rats not undergoing withdrawal or rats fed with control-diet treated with TSA did not differ from their respective control groups. There were no significant differences in total ambulations among the various groups (Fig 2), indicating normal general activities of the rats. Both behavioral tests demonstrated the development of anxiety-like behaviors during ethanol withdrawal in rats, an effect which was blocked by treatment with TSA.
Figure 2.

Measurement of anxiety-like behaviors in rats using light/dark box (LDB) shows increased anxiety levels in ethanol-withdrawn rats. TSA treatment prevents the withdrawal-induced anxiogenic effect. The data are represented as percentage of time spent in light and dark compartment of LDB. The general activities of the rats are measured by total ambulations during the exploration of LDB. Values are the mean ± SEM of 6–7 rats in each group. * Significantly different (p < 0.001; one-way ANOVA followed by post hoc analysis by Tukey’s test; percentage of time spent in the light and dark compartments [F4, 26=43.71, p<.001]) from all other groups (Control+ Vehicle, Ethanol+ Vehicle, Withdrawal+ TSA, Control+ TSA injected rats). # Groups significantly different (p<0.001) from each other.
Effect of TSA treatment on BDNF and Arc expression in the amygdala during ethanol withdrawal
In order to examine the effect of TSA treatment on BDNF and Arc levels during ethanol withdrawal, we measured BDNF protein, Arc protein and Arc mRNA levels in amygdaloid brain regions of rats withdrawn from chronic ethanol exposure with or without TSA treatment. We observed significant (p<0.001) decreases (Fig 3A and 3B) in protein levels of BDNF and Arc and decreases in Arc mRNA levels (p<0.001) in the CeA and MeA, but not BLA of ethanol withdrawn rats compared with control-diet fed or ethanol-fed rats. Interestingly, these BDNF and Arc deficits were rescued by TSA treatment. Control-diet fed rats treated with TSA showed no significant differences in BDNF and Arc expression in the amygdala. These data suggest that TSA treatment was able to prevent the withdrawal-induced decreases in BDNF and Arc expression in the CeA and MeA of rats.
Figure 3.
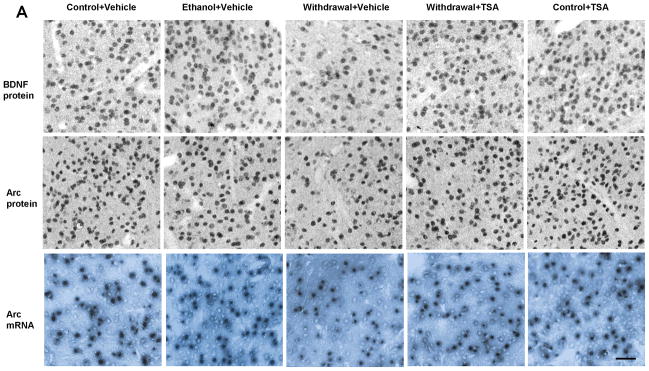

A, Representative photomicrographs of brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton-associated (Arc) protein gold-immunolabeling and Arc mRNA in situ RT-PCR in the central nucleus of amygdala (CeA) of various rat groups (Scale bar = 40 μm). B, Bar diagram showing the effects of chronic ethanol treatment and its withdrawal (with or without TSA treatment) on BDNF protein, Arc protein and mRNA levels in CeA, medial nucleus of amygdala (MeA) and basolateral amygdala (BLA) of rats. Values are mean ± SEM of 6 rats in each group. * Significantly different (p <0.001; one-way ANOVA followed by post hoc analysis by Tukey’s test; [BDNF protein: CeA, F4,25=109.11 , p<.001; MeA, F4,25=100.51 , p<.001; Arc protein: CeA, F4,25=60.23, p<.001; MeA, F4,25=29.93, p<.001; Arc mRNA: CeA, F4,25=25.59, p<.001; MeA, F4,25=16.24, p<.001]) from all other groups (Control+ Vehicle, Ethanol+ Vehicle, Withdrawal+ TSA, Control+ TSA injected rats).
Effects of TSA treatment on dendritic spine density (DSD) in the amygdala during ethanol withdrawal
We next examined whether TSA treatment-induced reversal of deficits in BDNF and Arc expression might also have an effect on the observed deficits in DSD in the CeA and MeA during withdrawal after chronic ethanol exposure. Golgi-Cox staining was used to investigate changes in the DSD in amygdaloid brain regions. The representative photomicrographs of Golgi-Cox staining in the CeA and BLA regions of various groups are shown in Figure 4A and quantification of dendritic spines are shown in Figure 4B. In agreement with our earlier findings (Pandey et al., 2008b), we confirmed a significant decrease (p<0.001) in DSD in the CeA and MeA, but not BLA of ethanol-withdrawn rats as compared with control-diet fed or ethanol-diet fed rats. We extended these findings and found that reductions in dendritic spines in the CeA and MeA were attenuated by TSA treatment (p <0.001). Similar to BDNF and Arc, DSD in the amygdaloid brain regions of control-diet fed rats was not altered by TSA treatment. These results indicate that TSA treatment not only attenuated anxiety-like behaviors but also corrected the deficits in BDNF, Arc, and DSD in the CeA and MeA of rats during withdrawal after chronic ethanol exposure.
Figure 4.

A, Representative photomicrographs of dendritic spines in the central nucleus of amygdala (CeA) and basolateral amygdala (BLA) of various rat groups as measured by Golgi-Cox staining (scale bar = 10 μm). B, Effect of chronic ethanol treatment and its withdrawal (with or without TSA treatment) on dendritic spine density in the amygdaloid brain regions of rats. Values are mean ± SEM of 6 rats in each group. *Significantly different [p <0.001; one-way ANOVA followed by post hoc analysis by Tukey’s test; CeA, F4, 25=14.89, p<.001; Medial nucleus of amygdala (MeA), F4, 25=13.51, p<.001] from all other groups (Control+ Vehicle, Ethanol+ Vehicle, Withdrawal+ TSA, Control+ TSA injected rats).
Discussion
The current study demonstrates a possible role of HDAC-induced histone modifications in the regulation of synaptic plasticity related events in the CeA and MeA during withdrawal from chronic ethanol exposure in rats. We have previously reported that the anxiogenic effects of ethanol withdrawal are attenuated by HDAC inhibition, which in turn is associated with correcting the deficits in histone H3-K9 and H4-K8 acetylation in the CeA and MeA in rats (Pandey et al., 2008a). Additionally, BDNF and Arc gene expression and DSD were found to be reduced in the CeA and MeA of ethanol-withdrawn rats and BDNF signaling along with dendritic spines in the CeA have been shown to be involved in anxiety-like behaviors during ethanol withdrawal in rats (Pandey et al., 2008b). Here, we extended these studies and found that TSA treatment is able to rescue the deficits in BDNF, Arc, and DSD, indicating an association between HDAC-induced histone modifications and synaptic plasticity in the development of anxiety-like behaviors during withdrawal from chronic ethanol exposure in rats.
Histone acetylation appears to play an important role in the adaptive changes produced by ethanol exposure. For example, an in vitro study revealed an ethanol-induced dose-dependent increase of HDAC2 expression, which was corrected by TSA treatment (Agudelo et al., 2011). In rats, acute ethanol exposure produced anxiolytic-like effects, decreased HDAC activity and increased histone H3-K9 and H4-K8 acetylation in the amygdala (Pandey et al., 2008a), whereas withdrawal from chronic ethanol exposure decreased histone H3-K9 and H4-K8 acetylation and increased HDAC activity (Pandey et al., 2008a). Inhibition of HDAC through TSA not only attenuated development of anxiety-like behaviors but also corrected the reduction of histone H3-K9 and H4-K8 acetylation in the CeA and MeA (Pandey et al., 2008a). It is important to point out that similar to our previous findings (Pandey et al., 2008a) TSA treatment not only attenuated anxiety-like behaviors in ethanol-withdrawn rats but also produced anxiolytic-like effects compared with control rats (Fig 2). It has been shown that cancer patients with high HDAC2 levels in their peripheral blood mononuclear cells respond well clinically with the HDAC inhibitor Vorinostat treatment (Munster et al., 2011). There is an upregulation of HDAC activity in the amygdala of rats undergoing ethanol withdrawal (Pandey et al., 2008a) and this is possibly the reason why TSA is effective in producing anxiolytic-like effects in ethanol-withdrawn rats but not in control rats.
The involvement of histone acetylation in alcohol intake and alcohol tolerance has also been investigated in several studies. It was found that inhalation of benzyl alcohol regulates potassium channels at the transcriptional level by histone H4 acetylation leading to alcohol tolerance (Wang et al., 2007). Further evidence suggests epigenetic mechanisms play a role in the development of rapid ethanol tolerance in rats (Sakharkar et al., 2012) and in the expression of ethanol-induced behavioral sensitization in mice (Botia et al., 2012). Acute ethanol exposure inhibits amygdaloid HDAC activity and increases histone acetylation in the CeA and MeA of rats, a phenomenon that could not be repeated by a second administration of the same dose of ethanol but could be replicated with a higher dose of ethanol, indicating the development of tolerance (Sakharkar et al., 2012). Recently, we demonstrated that higher expression of the HDAC2 isoform in the CeA regulates anxiety-like and alcohol drinking behaviors of alcohol preferring (P) rats (Moonat et al., 2013). Also, it has been shown that various HDAC inhibitors (TSA and Vorinostat) were able to attenuate alcohol intake in animal models (Warnault et al., 2013). HDAC inhibitors are well tolerated in cancer patients (Khan and La Thangue, 2012). However, some dose dependent side effects such as fatigue, hepatotoxicity, and thrombocytopenia have been reported (Gräff and Tsai, 2013). The dose of TSA (2mg/kg) used here appears to be in the non-toxic range since higher dose of TSA (7.5 mg/kg) has been shown to produce non-toxic effects in animal model and was effective in ameliorating autoimmune encephalomyelitis (Camelo et al., 2005). Because of the multiple roles that HDACs play in different stages of alcoholism, the development of HDAC isoform specific inhibitors with reduced side effects may have therapeutic implications in the treatment of alcoholism. Here, we have not performed experiments to check persistent effects of TSA. It is possible that acute TSA treatment effects may be short lived and longer chronic treatments are needed for persistent effects. These possibilities will be investigated in future studies.
Involvement of the BDNF system in anxiety and alcoholism is well established. Deficits in BDNF levels and BDNF gene polymorphism promote anxiety-like and alcohol drinking behaviors (Pandey et al., 2004; Jeanblac et al., 2009; Colzato et al., 2011; Moonat et al., 2011). It has been shown that BDNF and Arc expression in the CeA and MeA was decreased during withdrawal after chronic ethanol exposure but increased by acute ethanol exposure (Pandey et al., 2008b). Rats selectively bred to prefer alcohol have been found to exhibit innate anxiety-like behaviors as well as lower Arc and BDNF expression in the CeA and MeA (Moonat et al., 2011). BDNF anti-sense oligodeoxynucleotides (ODNs) infusion into the CeA and MeA or Arc anti-sense ODNs infusion into CeA provoked anxiety-like behaviors and increased alcohol intake in an unselected stock of rats (Pandey et al., 2006; 2008b). Conversely, BDNF infusion into CeA was able to rescue withdrawal-induced anxiety-like behaviors and normalize decreased Arc expression (Pandey et al., 2008b). We have previously shown that decreased histone H3-K9 and H4-K8 acetylation and NPY expression in amygdaloid brain regions as a result of increased HDAC activity after ethanol withdrawal, and inhibition of HDAC reversed the withdrawal-induced deficits in histone acetylation and gene expression (Pandey et al., 2008a). Our current data indicate that BDNF and Arc are most likely regulated by HDAC, suggesting the possible involvement of histone acetylation in the regulation of BDNF and Arc expression during ethanol withdrawal-induced anxiety-like behaviors. Several studies in the field indicate that histone acetylation regulates the expression of BDNF in the hippocampus during depression-like behaviors and learning and memory (Tsankova et al., 2006; Bredy et al., 2007; Lubin et al., 2008; Takei et al., 2011). However, further study is needed to determine the direct role of histone acetylation in the regulation of various BDNF exons and Arc expression in the amygdala or other brain regions during alcohol dependence.
In the current study, we demonstrated a close association between BDNF and Arc expression with dendritic spine density (DSD) in the CeA and MeA during alcohol dependence. HDAC inhibition not only reversed deficits in BDNF and Arc expression, but also normalized the DSD in the CeA and MeA indicating a mechanism by which histone modifications underlie synaptic plasticity during alcohol dependence. These results are consistent with several recent studies that identify critical roles of histone acetylation in synaptic plasticity. For example, over-expression of HDAC2 decreased DSD and impaired long-term potentiation (LTP) in the hippocampus of mice; conversely, HDAC2 knock out mice were found to have increased DSD in the hippocampus and increased LTP (Guan et al., 2009). Histone acetylation was increased specifically within the BDNF promoter II in HDAC2 knock out mice and decreased in mice over-expressing HDAC2 (Guan et al., 2009). Chronic treatment with the HDAC inhibitor Vorinostat reversed the decreased DSD observed in over-expressing HDAC2 mice and ameliorated learning impairment (Guan et al., 2009). Furthermore, higher expression of HDAC2 in the CeA and MeA is associated with decreases in dendritic spines in P rats as compared with alcohol non-preferring (NP) rats and knock down of HDAC2 by siRNA in the CeA significantly increased dendritic spines, produced anxiolytic-like effects, and attenuated alcohol intake in P rats (Moonat et al., 2011; 2013). Interestingly, aging-related cognitive declines as well as a coupled loss of synaptic plasticity and dendritic spines in the hippocampus have been associated with increased HDAC2 and decreased histone acetylation at specific promoters of BDNF; treatment with TSA was able to restore BDNF expression and reverse age-dependent reductions in dendritic spines (Zeng et al., 2011). However, it has been shown that neuronal morphological changes in the cortical area of Tg2527 amyloid precursor protein mutant mice compared with wild-type mice does not produce corresponding functional changes at electrophysiological levels (Rocher et al., 2008). Nonetheless, epigenetic studies possibly suggest the involvement of HDAC2-induced chromatin remodeling in synaptic plasticity-associated events in the brain.
Based on present and our previous findings (Pandey et al., 2008a), we propose that ethanol withdrawal-induced adaptive changes in epigenetic processes such as HDAC hyper-activity condenses chromatin structure which, in turn, decreases downstream gene expression. BDNF and Arc gene expression would therefore be decreased, resulting in decreased dendritic spine density and possibly an overall regulation of synapse function. These deficits in the CeA and MeA as well as anxiety-like behaviors were attenuated by treatment with TSA during ethanol withdrawal. We also reported earlier that TSA treatment during ethanol withdrawal normalized deficits in H3-K9 and H4-K8 acetylation due to inhibition of HDAC activity in the amygdala (Pandey et al., 2008a). These results collectively provide an important epigenetic mechanism that possibly contributes to the pathogenesis of alcoholism.
Acknowledgments
This work was supported by grants from the National Institute on Alcohol Abuse and Alcoholism Grants AA-016690, AA-019971(NADIA project), AA-013341, and AA-010005 and by the Department of Veterans Affairs (Merit Review Grant-l01BX000143; Research Career Scientist award) to SCP.
Footnotes
Conflict of Interest
SCP reports that a US patent application on a related topic (serial number 60/848237 filed on September 29th, 2006) is currently pending. All other authors reported no biomedical financial interests or potential conflicts of interest.
The data of the manuscript has been presented at the Research Society on Alcoholism scientific meeting and is a part of the PhD dissertation of CY.
References
- Agudelo M, Gandhi N, Saiyed Z, Pichili V, Thangavel S, Khatavkar P, Yndart-Arias A, Nair M. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin A (TSA) Alcohol Clin Exp Res. 2011;35(8):1550–1556. doi: 10.1111/j.1530-0277.2011.01492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botia B, Legastelois R, Alaux-Cantin S, Naassila M. Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in mice. PLoS One. 2012;7(10):e47527. doi: 10.1371/journal.pone.0047527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14(4):268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164(1–2):10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Colzato LS, Van der Does AJ, Kouwenhoven C, Elzinga BM, Hommel B. BDNF Val66Met polymorphism is associated with higher anticipatory cortisol stress response, anxiety, and alcohol consumption in healthy adults. Psychoneuroendocrinology. 2011;36(10):1562–1569. doi: 10.1016/j.psyneuen.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Koob GF. Neurobiology of Alcohol Dependence: Focus on Motivational Mechanisms. Alcohol Res Health. 2008;31(3):185–195. [PMC free article] [PubMed] [Google Scholar]
- Gräff J, Tsai LH. The potential of HDAC inhibitors as congnitive enhancers. Annu Rev Pharmacol Toxicol. 2013;53:311–330. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- Grüsser SM, Mörsen CP, Flor H. Alcohol craving in problem and occasional alcohol drinkers. Alcohol Alcohol. 2006;41(4):421–425. doi: 10.1093/alcalc/agl035. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanblac J, He DY, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci. 2009;29(43):13494–13502. doi: 10.1523/JNEUROSCI.2243-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90 (1):85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Expt Res. 2003a;27(2):232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neuroadaptive mechanisms of addiction: studies on the extended amygdala. Euro Neuropsychopharmacol. 2003b;13(6):442–452. doi: 10.1016/j.euroneuro.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Kushner MG, Abrams K, Borchardt C. The relationship between anxiety disorders and alcohol use disorders: a review of major perspectives and findings. Clin Psychol Rev. 2000;20(2):149–171. doi: 10.1016/s0272-7358(99)00027-6. [DOI] [PubMed] [Google Scholar]
- Kryger R, Wilce PA. The effects of alcoholism on the human basolateral amygdala. Neuroscience. 2010;167(2):361–371. doi: 10.1016/j.neuroscience.2010.01.061. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28(42):10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Muller J. Emotional memory and psychopharmacology. Philos Trans R Soc Lond B Biol Sci. 1997;352(1362):1719–1726. doi: 10.1098/rstb.1997.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Christian DT, Diaz MR, Läck AK. Glutamate plasticity in the drunken amygdala: the making of an anxious synapse. Int Rev Neurobiol. 2010;91:205–233. doi: 10.1016/S0074-7742(10)91007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Pandey SC. Stress, epigenetics, and alcoholism. Alcohol Res. 2012;34(4):495–505. [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Pandey SC. The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addict Biol. 2011;16(2):238–250. doi: 10.1111/j.1369-1600.2010.00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC. Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry. 2013;73(8):763–773. doi: 10.1016/j.biopsych.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the tratment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104(12):1828–1835. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol Sci. 2003;24(9):456–460. doi: 10.1016/S0165-6147(03)00226-8. [DOI] [PubMed] [Google Scholar]
- Pandey SC. The gene transcription factor cyclic AMP-responsive element binding protein: role in positive and negative affective states of alcohol addiction. Pharmacol Ther. 2004;104(1):47–58. doi: 10.1016/j.pharmthera.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H, Xu T. Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci. 2004;24(21):5022–5030. doi: 10.1523/JNEUROSCI.5557-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. 2008a;28(14):3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006;26(32):8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K. Effector immediate-early gene arc in the amygdala plays a critical role in alcoholism. J Neurosci. 2008b;28(10):2589–2600. doi: 10.1523/JNEUROSCI.4752-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessoa L, Adolphs R. Emotion processing and the amygdala: from a ‘low road’ to ‘many roads’ of evaluating biological significance. Nat Rev Neurosci. 2010;11(11):773–783. doi: 10.1038/nrn2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20(11):517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- Rocher AB, Kinson MS, Luebke JI. Significant structural but not physiological changes in cortical neurons of 12-month-old Tg2576 mice. Neurobiol Dis. 2008;32(2):309–318. doi: 10.1016/j.nbd.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83(3):803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res. 2012;36(1):61–71. doi: 10.1111/j.1530-0277.2011.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei S, Morinobu S, Yamamoto S, Fuchikami M, Matsumoto T, Yamawaki S. Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J Psychiatr Res. 2011;45(4):460–468. doi: 10.1016/j.jpsychires.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8(5):355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9(4):519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471(7338):358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krishnan HR, Ghezzi A, Yin JC, Atkinson NS. Drug-induced epigenetic changes produce drug tolerance. PLoS Biol. 2007;5(10):e265. doi: 10.1371/journal.pbio.0050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnault V, Darcq E, Levine A, Barak S, Ron D. Chromatin remodeling-anovel strategy to control excessive alcohol drinking. Transl Psychiatry. 2013;3:e231. doi: 10.1038/tp.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Tan M, Kohyama J, Sneddon M, Watson JB, Sun YE, Xie CW. Epigenetic enhancement of BDNF signaling rescues synaptic plasticity in aging. J Neurosci. 2011;31(49):17800–17810. doi: 10.1523/JNEUROSCI.3878-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]