

Abstract
Current European pharmaceutical legislation is not adequate to meet advances in science and technologies that will lead to rapid development of custom-made medicines. Using existing legislation for custom-made medical devices as a template and anti-sense oligonucleotides as model medicinal products, we propose new European pharmaceutical legislation to permit timely access to custom-made anti-sense oligonucleotide medicinal products. The proposals may be more widely applicable to other medicinal products.
Keywords: anti-sense oligonucleotide, custom-made medicine, Europe, personalized medicine, pharmaceutical regulation, RNA
Introduction
Medicines may only be placed on the European marketplace for use in patients if they have been given a licence either through the national regulatory body (the Medicines and Healthcare products Regulatory Agency for the UK) or the European Medicines Agency. In order to obtain such a licence, a pharmaceutical company must ensure acceptable quality of the final product, obtain supportive pre-clinical data and conduct clinical studies to demonstrate that the product bears a positive benefit : risk ratio. The company is required to formulate a risk management plan and to carry out post-marketing surveillance in order to detect adverse events arising from treatment after the licence is granted. Regulatory agencies may also request construct of a registry of patients and post-approval efficacy and safety studies 1.
Personalized medicine, described by a European Commission workshop as ‘a medical model using molecular profiling technologies for tailoring the right therapeutic strategy for the right person at the right time …’ 2 is an emerging field that promises to bring radical changes in healthcare. Knowledge of human health and disease is rapidly expanding as a result of the development and implementation of high throughput technologies such as those associated with determining the sequence of the human genome 3 and, in support of these advances, the European Commission has so far provided about 1 billion euros in funding to develop personalized medicine through the EU Framework Programme for Research and Technological Development 4.
We have previously described challenges to the current European pharmaceutical regulatory system that are likely to arise from development of anti-sense oligonucleotides 5. For instance, about 5000 different mutations of the dystrophin gene have been described and that lead to Duchenne muscular dystrophy. In theory, exon skipping will benefit about 80% of boys with Duchenne muscular dystrophy. Phase III clinical trials are currently underway to assess the effect of an anti-sense oligonucleotide that causes skipping of exon 51 of the dystrophin gene (the dystrophin gene has 79 exons, exon 51 is at the most commonly affected area of the gene) and yet, even if successful, skipping exon 51 will only benefit about 15% of boys with Duchenne muscular dystrophy 6. Duchenne muscular dystrophy affects only about 1 in 3000 boys. The remaining 65% of boys who may benefit from exon skipping and whose genetic abnormalities are scattered along the 2.4 million bases of the dystrophin gene (and away from exon 51) form subsets with very small numbers. In practical terms, how will companies gather together enough participants to carry out clinical development programmes of exon skippers other than that for exon 51? Could custom-made anti-sense oligonucleotide medicinal products provide the answer and, if so, how might such medicines be made available?
At present, European pharmaceutical legislation (European Directive 2001/83/EC, as amended) permits two routes to gain access to custom-made medicines: (i) the hospital exemption scheme for advanced therapy medicinal products and (ii) the ‘specials’ scheme 1.
According to EC regulation 1394/2007, as amended, advanced therapy medicinal products encompass gene therapies, somatic cell therapies and tissue engineered products 7. Anti-sense oligonucleotides do not fall into these categories and so the hospital exemption scheme would not be applicable for these substances.
Under the ‘specials’ scheme, a medicine may be supplied in response to an unsolicited order and in order to satisfy the need of an individual patient. The ‘special’ is an unlicensed medicine formulated according to the specification of the prescriber. The quality of the product may be variable, patient information texts are not required, there is not an obligation to carry out pharmacovigilance and professional responsibility must be taken by the prescribing person (unless it can be shown that the product was faulty). In practice, the ‘specials’ scheme is mostly used to reformulate licensed medicines. ‘Specials’ may be made by pharmacists in their dispensaries or by companies that hold a ‘specials’ manufacturing licence. There are about 800 000 prescriptions per year for ‘specials’ in the UK alone.
With the exception of ‘specials’ that are reformulations of existing licences, we are of the opinion that the ‘specials’ scheme does not provide incentive to companies to make novel custom-made medicines and that the scheme is not adequate to ensure safe use of such products because there is not an obligation to provide patient information texts or carry out pharmacovigilance. We believe that there is a need to develop new pharmaceutical regulation for custom-made medicinal products that reflects advances in science and technology and that also protects patients (and prescribers and manufacturers) and encourages innovation 8,9. We now propose a scheme for custom-made anti-sense oligonucleotide medicinal products that draws on experience from legislation for custom-made medical devices.
The Medical Devices Directive 93/42/EEC, as amended 10, describes custom-made medical devices as follows:
‘custom-made’ means, in relation to a device:
that it is manufactured specifically in accordance with a written prescription of a duly qualified medical practitioner or a professional user which gives, under his responsibility, specific characteristics as to its design and
that it is intended for the sole use of a particular patient.
The company must be registered with the regulatory agency as a manufacturer of custom-made medical devices. The company will hold the intellectual property for a medical device that can be tailored to the requirements of the individual. The company must maintain documentation allowing an understanding of the design, manufacture and performances of the medical device and also review and document experience gained in the post-production phase. A post-marketing vigilance system of reporting to regulatory agencies must be set up in order to report any accidents resulting from the constituents or design of the device that pose a potential serious risk to public health. The company must make available a ‘statement’ to the named patient for whom the device has been manufactured and the ‘statement’ must contain the following information: the name and address of the manufacturer, data allowing identification of the device in question, confirmation that the device is intended for exclusive use by a named patient, the name of the medical practitioner or the authorized person who made out the prescription and, where applicable, the name of the clinic concerned and the specific characteristics of the product as indicated by the prescription.
Proposal for new European pharmaceutical regulation for custom-made anti-sense oligonucleotide medicinal products
By drawing on regulatory experience of custom-made medical devices, we make the following proposals for new European pharmaceutical regulation for custom-made anti-sense oligonucleotide medicinal products:
-
A custom-made medicinal product means that the product is manufactured in accordance with a written prescription of a qualified medical practitioner and is intended for the sole use of a named patient.
There must be appropriate safeguards on release and distribution of the new product in order to ensure that it is delivered to the named patient.
-
The custom-made anti-sense oligonucleotide medicinal product is tailor-made based on intellectual property of a master anti-sense oligonucleotide medicinal product. The company must hold the licence for the master anti-sense oligonucleotide medicinal product and be registered with the regulatory agency to produce custom-made medicinal products.
We believe that this will ensure an acceptable, high quality for the custom-made medicinal product.
-
The master anti-sense oligonucleotide medicinal product will have obtained its market licence through a conventional development programme that will have included in vitro studies of biological activity of the master anti-sense oligonucleotide medicinal product and phase III clinical studies.
[The in vitro assay is used at this point for research purposes only and so does not need to be CE marked]
The company will construct and justify a model that describes the link between in vitro biological effect of the master anti-sense oligonucleotide medicinal product and clinical benefit. The model will be plausible.
The custom-made anti-sense oligonucleotide will have the same sugar-phosphate backbone as the master anti-sense oligonucleotide.
A custom-made anti-sense oligonucleotide medicinal product will bear a new form of licence that is derived from and dependent upon that for the master anti-sense oligonucleotide medicinal product. The licence for the custom-made anti-sense oligonucleotide medicinal product will be conjoined to the licence for the master anti-sense oligonucleotide medicinal product. The benefit : risk profile of the master product will be positive at the time of a grant being issued (and will remain constantly so unless evidence of harm accumulates). If the benefit : risk profile of the master product becomes negative then all custom-made medicinal products derived from it will acquire a negative benefit : risk profile.
-
The company will provide acceptable quality data for the custom-made anti-sense oligonucleotide medicinal product. If pre-clinical data are not available for the custom-made anti-sense oligonucleotide medicinal product then this should be justified by the company. In vitro assay results (the same in vitro assays that supported the master medicinal product) on tissue from the intended recipient of the custom-made anti-sense oligonucleotide medicine must confirm that the custom-made anti-sense oligonucleotide product has an acceptable biological effect before a licence may be granted for the custom-made anti-sense oligonucleotide medicinal product. In other respects, clinical studies of patient exposure will not be required before the licence is granted.
[The in vitro assay is now intended to be used for medical purpose and so will need to be CE marked in accord with the In Vitro Diagnostic Device Directive 98/79/EC 11. At present, most companion diagnostic devices are regulated under the lowest risk category. It is understood that forthcoming revision of the In Vitro Diagnostic Device Directive and associated standards and guidelines will result in such devices being regarded as medium-risk and so subject to review by a notified body]
Based on in vitro studies of the custom-made anti-sense oligonucleotide medicinal product and with reference to the master anti-sense oligonucleotide medicinal product, the company will construct and justify a model that describes the link between in vitro biological effect of the custom-made anti-sense oligonucleotide medicinal product and proposed clinical benefit. An intellectual bridge from the custom-made anti-sense oligonucleotide medicinal product to clinical data supporting the master anti-sense oligonucleotide medicinal product is thereby formed but the intellectual bridge is considered to be imperfect.
Consent must be obtained from the named patient (or guardian) before administration of a custom-made anti-sense oligonucleotide medicinal product.
A ‘statement’ (similar to that described by the Medical Devices Directive 93/42/EEC, as amended, for a custom-made device) should be given to the named patient and the prescribing physician at the time of receipt from the hospital pharmacist.
-
The patient is given a patient information leaflet that makes use of the imperfect intellectual bridge to the master anti-sense oligonucleotide medicinal product by listing the adverse events that may be expected with the master anti-sense oligonucleotide medicinal product and that draws attention to the unknowns and risks associated with a custom-made anti-sense oligonucleotide medicinal product.
All patient information texts will display appropriate and prominent warning information including an easily recognized symbol (symbol to be decided).
-
The benefit : risk profile of the custom-made anti-sense oligonucleotide medicinal product will be considered to oscillate. Thus, the custom-made anti-sense oligonucleotide medicinal product is presumed to convey benefit immediately prior to administration to the named patient. During administration and until the next administration, the same custom-made anti-sense oligonucleotide medicinal product is presumed to convey harm or futility to the named patient. The onus will therefore lie with the manufacturer and prescribing physician to collect data in order to refute harm and futility (in that order of assessment). This will be done by conducting an open-labelled continuous evaluation study with analysis of change from baseline for each patient 12,13 in order to collect data on efficacy and safety during the life of exposure to the custom-made anti-sense oligonucleotide medicinal product. The nature of the data collected during the continuous evaluation study will be the same as that collected for the clinical studies that supported the application for the master product. Data from the study will be compared with baseline data that have been obtained over an acceptable time period prior to administration of the custom-made medicine.
A description of the continuous evaluation study for each custom-made anti-sense oligonucleotide medicinal product will be included in its risk management plan. Records must be kept and be available for inspection.
Initial administration of a custom-made anti-sense oligonucleotide medicinal product to each named patient will be considered to be a first-in-human undertaking. The company must justify place of administration, initiating dose, route and rate of administration, dose escalation decisions, maximum permissible dosage and the handling of safety and tolerability issues. Stopping rules should be clearly stated 14,15.
-
Serious and unexpected adverse drug reactions to the custom-made anti-sense oligonucleotide medicinal product must be reported immediately to the regulatory agency: non-serious adverse drug reactions should be reported by the manufacturer annually to the regulatory agency.
We anticipate that close medical supervision during the continuous evaluation study will enhance the clinical safety of the recipient.
The benefit : risk profile of the custom-made anti-sense oligonucleotide medicinal product will be judged for each named patient. If the benefit : risk profile becomes negative for the named patient then the custom-made anti-sense oligonucleotide medicinal product will no longer be administered to the named patient.
The regulatory agency will maintain surveillance of custom-made anti-sense oligonucleotide medicinal products by the established mechanisms of pharmacovigilance.
Professional responsibility will be shared between the prescribing medical practitioner and the manufacturer.
The time taken from application for a conjoined licence to granting will be 60 days.
Figure 1 summarizes the route to obtaining a licence for a custom-made medicinal product.
Figure 1.
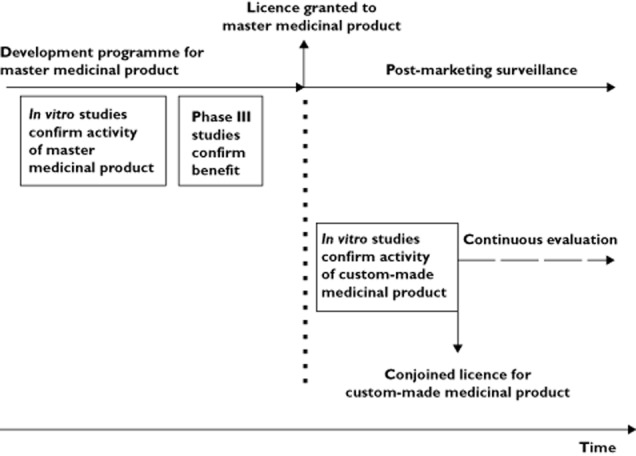
Timeline of development programmes for master and conjoined medicinal products. The development programme of the master anti-sense oligonucleotide medicinal product includes in vitro data showing that tissue biopsies respond to exposure. A phase III study confirms a positive benefit : risk balance. A market licence is granted for the master medicinal product that then enters into post-marketing surveillance.Once a licence has been obtained for the master medicinal product, a licence for a custom-made anti-sense oligonucleotide medicinal product may be obtained by submission of the same type of in vitro data that supported the master medicinal product. An imperfect bridge is constructed to data that supported the master anti-sense oligonucleotide medicinal product. A conjoined licence is issued for the custom-made product. Each patient who will be given the custom-made medicinal product is entered into a continuous evaluation study. Collection of data in the continuous evaluation study is used to refute or confirm the presumptions of harm and futility and benefit
Scenario for a custom-made anti-sense oligonucleotide medicinal product
We use the following scenario to illustrate how we envisage a custom-made medicine may be produced and licensed based on the genomic model of personalised medicine.
Syndrome X is a rare condition that affects children from age 5 years leading to death at 20 years. Syndrome X has obtained ‘orphan status’. The protein defect leading to syndrome X has been identified. The protein is known to have five domains. The active site of syndrome X protein lies in domain 3. Figure 2A represents the healthy state where the five exons of pre-mRNA are processed through to a five domain protein required for health. 95% of patients with syndrome X have a disruption of exon 2 of the mRNA that codes for syndrome X protein and the disruption results in a non-functioning protein leading to disease.
Figure 2.
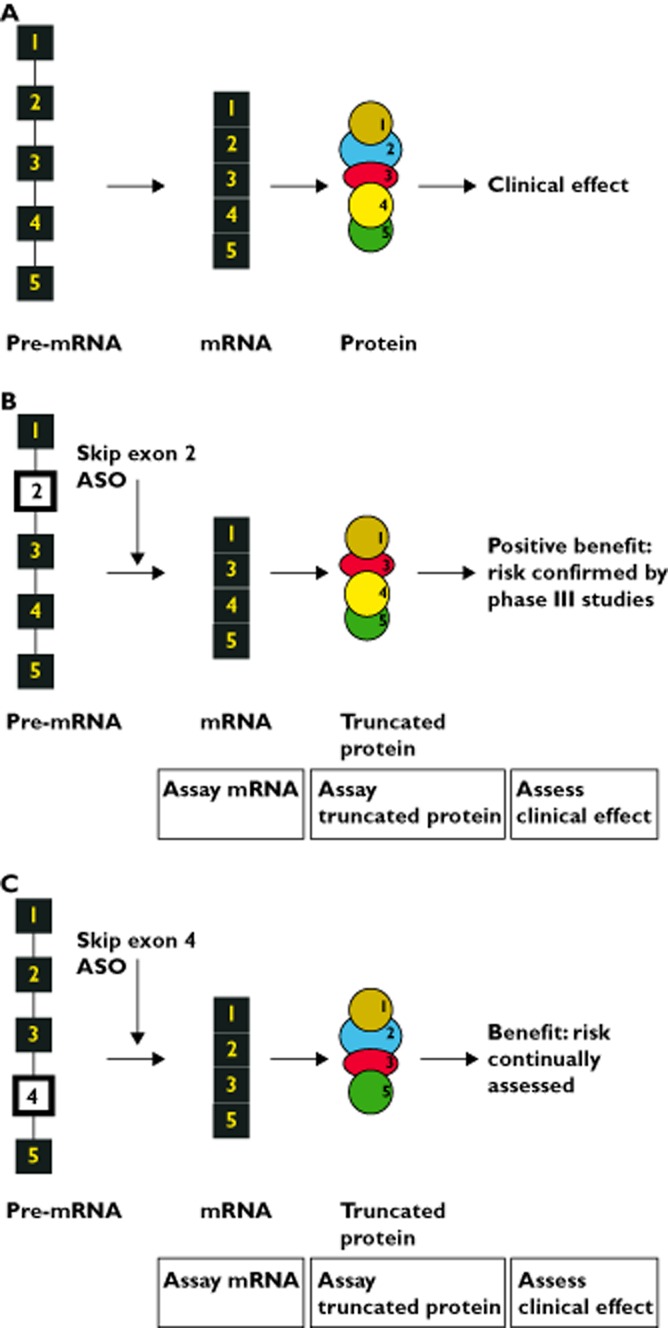
(A) Processing of pre-mRNA for syndrome X protein in health. The pre-mRNA that codes for the syndrome X protein has 5 exons, shown here as numbered boxes, the connecting lines between boxes represent introns. Pre-mRNA is spliced to form mRNA, all 5 exons are retained. In this model, each exon in mRNA is transcribed to a separate domain (numbered 1 to 5 to correspond to the exons) of the syndrome X protein. The active site of the protein resides in domain 3. (B) Processing of pre-mRNA for syndrome X protein in patient with exon 2 abnormality and who is exposed to exon 2 skipper (the master medicinal product). 95% of patients with syndrome X have a defect in exon 2, shown as an open box in the pre-mRNA. This pre-mRNA leads to production of a non-functioning syndrome X protein and so to the development of syndrome X in the patient. Company A has conducted conventional phase III trials of an anti-sense oligonucleotide (ASO) that causes ‘skipping’ of exon 2. Exposure to this ‘master’ anti-sense oligonucleotide leads to an mRNA with sequence of exons 1-3-4-5, confirmed by quantification of mRNA on tissue biopsy. The mRNA is transcribed to a truncated protein with domains 1-3-4-5 and assay of this protein in tissue biopsy confirms 70% of the activity of the native protein. A positive clinical benefit : risk ratio is confirmed for patients who enter the phase III trial of the exon 2 skipper. Company A obtains a licence for the exon 2 skipper. (C) Processing of pre-mRNA for syndrome X protein in patient with exon 4 abnormality and who is exposed to exon 4 skipper (the conjoined medicinal product). 5% of patients with syndrome X have a defect in exon 4, shown as an open box in the pre-mRNA, that leads to production of a non-functioning syndrome X protein and so to the development of syndrome X. Patient 1 has the defect in exon 4 of the pre-mRNA. Patient 1 presents to Company A. Company A makes an anti-sense oligonucleotide (ASO) that will cause ‘skipping’ of exon 4. There are too few patients with a defect in exon 4 to conduct a phase III trial. Tissue biopsies are taken from patient 1 and exposed to the exon 4 skipper. Production of mRNA with sequence 1-2-3-5 is confirmed. Assay of truncated protein with domains 1-2-3-5 confirms 50% activity of the native protein. Company A obtains a licence for the exon 4 skipper (the custom-made anti-sense oligonucleotide medicinal product) that is conjoined to the licence for the exon 2 skipper (the master anti-sense oligonucleotide medicinal product). Patient 1 is entered into a continuous evaluation study and is administered the exon 4 skipper. Clinical measurements that reflect those that were taken in the phase III trial of the master medicinal product are recorded
Company A has conducted a clinical development programme (including a conventional phase III study) for an anti-sense oligonucleotide that skips exon 2 resulting in truncated syndrome X protein with an intact domain 3 that retains function (Figure 2B). The development programme had included in vitro studies on tissue biopsy samples (from patients with exon 2 disruption) that were exposed to the exon 2 skipper. Results of the in vitro assays confirm that a truncated mRNA (sequence 1-3-4-5) and truncated protein are produced and that the protein has 70% activity of the protein found in health. Only those patients who returned positive results for the in vitro assays are permitted to go forward to the phase III study. Clinical data collected during the phase III study confirm a positive benefit : risk balance for the exon 2 skipper.
Company A constructs a model that links the in vitro results of the exon 2 skipper to clinical benefit. The model is constructed in silico.
Company A applies for a licence for the exon 2 skipper. The regulatory agency assesses the application. The model presented by the company that describes the links between exposure to anti-sense oligonucleotide, mRNA production and increase in activity of syndrome X protein is accepted. Company A is granted a licence for exon 2 skipper with an indication for patients who are shown to have disruption of exon 2.
Company A registers itself with the regulatory agency to produce custom-made anti-sense oligonucleotide medicinal products based on the exon 2 skipper (the master anti-sense oligonucleotide medicinal product).
5% of patients with syndrome X have a disruption of exon 4 of the mRNA that codes for syndrome X protein and this disruption also results in a non-functioning protein leading to disease. There are too few patients with disruption of exon 4 to carry out conventional phase III studies.
Patient 1 presents to his physician with syndrome X. Patient 1 is shown to have a disruption of exon 4. The physician contacts Company A because Company A has a licence to make custom-made medicines based on the licence for exon 2 skipper. Company A makes an anti-sense oligonucleotide that will cause ‘skipping’ of exon 4, as shown in Figure 2C. Tissue biopsies are taken from patient 1 and exposed to the exon 4 skipper. Quantification of mRNA confirms that mRNA with sequence 1-2-3-5 is produced and assay of the resulting truncated protein with an intact domain 3 confirms 50% activity compared with health.
Company A justifies use of the exon 4 skipper in patient 1 with reference to data held on the exon 2 skipper and to the in silico model of exon skipping.
Company A informs the regulatory agency that it has made an exon 4 skipper and that it intends that this product will be administered to patient 1.
Company A submits quality data for exon 4 skipper, a consent form, a patient information leaflet and a ‘statement’. Company A justifies the absence of pre-clinical data on the grounds that an animal equivalent for exon 4 disruption is not known.
A protocol for a continuous evaluation study for patient 1 is submitted in which Company A confirms and justifies the place of administration, the rate and route of administration, the starting dose and dose escalation criteria and how safety and tolerability information will be handled. Stopping rules are clearly stated. The continuous evaluation study will collect clinical measurements that are of the same type as those used in the phase III trial of the exon 2 skipper.
Company A confirms to the regulatory agency that it will immediately report any serious or unexpected reaction to the exon 4 skipper and that it will submit an annual report on all in-house custom-made medicines issued for syndrome X.
The regulatory agency assesses the application. The model presented by the company that links exposure to the exon 4 skipper and increase in activity of syndrome X protein is accepted. The regulatory agency issues a licence for the exon 4 skipper that is conjoined to the licence for the exon 2 skipper. From submission of an application for a licence for the exon 4 skipper to issuing of the licence for the exon 4 skipper has taken 60 days.
Discussion
Personalized medicine represents a new approach to classifying, understanding, treating and preventing disease by using information on individual biological differences 1–3. It may be anticipated that personalized medicine will place new demands upon the regulatory frameworks that control the licensing of medicinal products. Current European pharmaceutical legislation describes the ‘specials’ scheme to gain access to custom-made medicines 1. ‘Specials’ products have variable quality, do not have evidence of clinical efficacy and safety, are not obliged to be accompanied by patient information texts and are not subject to pharmacovigilance. We are concerned that the current regulatory process is not adequate for custom-made medicinal products that will undoubtedly arise from advances in technology and medicine. We believe that change is needed and that such change needs to be bold whilst building upon current practice and retaining emphasis on protection of public health. We have used the example of anti-sense oligonucleotide medicinal products to illustrate the innovations that we propose.
At present, about 50 chemically-modified single-stranded anti-sense oligonucleotides between 20 and 30 bases long are being investigated as potential medicinal agents in clinical trials for a variety of diseases including cancers, genetic disorders and viral infections. In general, for cancers and viral infections, anti-sense oligonucleotides are used to block production of a protein that is important in pathogenesis whilst for genetic disorders, anti-sense oligonucleotides are used to ‘skip’ the sequence of pre-mRNA that contains the genetic abnormality and so lead to production of a (potentially) functioning truncated protein.
For genetic disorders, anti-sense oligonucleotides are being developed to address the most frequent mutation of the affected target gene but the percentage of subjects with the most frequent mutation may be small in conditions that already qualify for orphan status. For instance, the most commonly affected area of the dystrophin gene leading to Duchenne muscular dystrophy is in the region of exon 51 but is found in only about 15% of affected boys. The most commonly affected mutation leading to Leber Congenital Amaurosis is a mutation in the intron between exons 26 and 27 of CEP290 but is to be found in only 10% of cases. In practical terms, companies face great difficulties gathering together enough participants to carry out conventional clinical development programmes of anti-sense oligonucleotides other than for the most common mutation of an affected gene. We therefore propose new pharmaceutical legislation for custom-made medicines to bridge this gap.
Our proposals for custom-made anti-sense oligonucleotide medicinal products are based on current European legislation for custom-made medical devices 10 produced by a registered company that holds intellectual property in the form of a master design. In our proposals for custom-made anti-sense oligonucleotide medicinal products, the company holds the licence for the master anti-sense oligonucleotide medicinal product and has created a model that links in vitro results of the master product to clinical benefit. This model then acts as a platform from which to justify (by creation of an ‘imperfect intellectual bridge’) a custom-made anti-sense oligonucleotide medicinal product that is tailored to the needs of an individual patient. Conventional phase III clinical studies of the custom-made anti-sense oligonucleotide medicinal product are not required but continued use of the custom-made medicine in any one patient will rely on collection of data that matches the nature of the data collected during the phase III clinical studies for the master medicinal product.
We acknowledge that the custom-made medicinal product carries with it a higher (or different) risk compared with the master medicinal product. This higher risk is most obviously conveyed by a first-in-man exposure done with a licensed medicine as is the case with the custom-made medicinal product. Nevertheless, we have taken steps to mitigate risk (i) by use of a conjoined licence, (ii) by the first exposure for each patient being done in compliance with requirements for a first-in-man exposure 14,15, (iii) by the presumptions of harm and futility (in that order of assessment) consequential to administration of the custom-made product and that these presumptions must be refuted to permit long term exposure of the individual, (iv) by providing a ‘statement’ and hybrid patient information leaflet that emphasize the unknowns and risks associated with a custom-made medicinal product and (v) by carrying out continuous evaluation of each patient to monitor for adverse effects (and futility and efficacy). We also advocate that clinicians engage patients in a shared decision-making throughout the period of exposure by discussing progress and evaluating results 16.
We believe that shared professional responsibility between the prescribing medical practitioner and the manufacturer reflects the commitment of the manufacturer to produce a product of acceptable, high quality and the decision of the physician to prescribe it. We acknowledge that there are complex legal issues around sharing of responsibilities and liabilities and that the obligations of the prescribing medical practitioner and the producer of the product as regards breach of duty in negligence and in statute (product liability/consumer protection) will need be satisfied. We consider that the key legal issues will be informed consent and causation.
We advise that approval for treatment be obtained from the local ethics committee and that informed consent be obtained from the patient or guardian by discussion and by written confirmation before administration of the product 17. Consent to treatment will have to obtained when safety and efficacy data are generated at a much earlier stage of development than for products licensed under the current regulatory regime and when the benefit : risk profile of the custom-made product has not yet been established. It is acknowledged that patients will be exposed to higher risk than with conventional medicines and so the process of consent must emphasize the unknowns associated with the custom-made product. The consent should also clearly state that data obtained during continuous evaluation after exposure will be made available to the company monitor, the local ethics committee and the regulatory agency.
As with conventional medicines, causation will also be a key component in any dispute over injury associated with a custom-made product. Proof of causation of injury will be a particularly complex problem in both negligence and product liability because of the limited data available for a custom-made medicinal product. In this regard, however, custom-made medicinal products do not differ from unlicensed ‘specials’.
We consider that the adoption of the proposed framework for custom-made anti-sense oligonucleotide medicinal products will enable regulatory agencies to maintain guard on the safety of public health and cope with the demand for such medicines. Finally, as experience is gained, and if that experience is encouraging, then we would hope that the concepts described for anti-sense oligonucleotide medicinal products may be more broadly applicable to other medicines.
Competing Interests
Both authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Contributors
Both JDJ and PF contributed and wrote the review. Although the authors work for regulatory authorities, the views expressed are their own and do not reflect those of any agency or institution.
References
- 1. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. 2012. Available at http://ec.europa.eu/health/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf (last accessed July 2013)
- 2. Workshop on personalised medicine. Published by the European Commission, DG Research. 2010. Available at http://ec.europa.eu/research/health/pdf/summary-report-omics-for-personalised-medicine-workshop_en.pdf (last accessed July 2013)
- 3.Gibson IR, Jiang R, Yu F. The 1000 Genomes Project: paving the way for personalized genomic medicine. Pers Med. 2013;10:321–324. doi: 10.2217/pme.13.22. [DOI] [PubMed] [Google Scholar]
- 4.Draghia-Akli R. Enabling personalized medicine in Europe: a look at the European Commission's funding activities in the field of personalized medicine research. Pers Med. 2012;9:152–155. doi: 10.2217/pme.11.91. [DOI] [PubMed] [Google Scholar]
- 5.Johnston JD, Feldschreiber P. Challenges posed to the European pharmaceutical regulatory system by highly-personalized medicines. Br J Clin Pharmacol. 2014;77:421–426. doi: 10.1111/bcp.12173. Available at http://onlinelibrary.wiley.com/doi/10.1111/bcp.12173/pdf (last accessed October 2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, Takeda S, Wilton SD, Wolff JA, Wooddell CI, Xiao X, Tremblay JP. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19:830–840. doi: 10.1038/mt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004. 2010. Available at http://ec.europa.eu/health/files/eudralex/vol-1/reg_2007_1394_cons_2012-07/reg_2007_1394_cons_2012-07_en.pdf (last accessed July 2013)
- 8. Personalised medicine for the European citizen. Published by the European Science Foundation. 2012. Available at http://www.esf.org/uploads/media/Personalised_Medicine.pdf (last accessed July 2013)
- 9.Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363:301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 10. Council Directive 93/42/EEC of 14 June 1993 concerning medical devices. 2007. Available at http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:1993L0042:20071011:en:PDF (last accessed July 2013)
- 11. Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. 1998. Available at http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:1998:331:0001:0037:EN:PDF (last accessed July 2013)
- 12. Guideline on clinical trials in small populations. Published by the Committee for Medicinal Products for Human Use of the European Medicines Agency. 2006. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000366.jsp&mid=WC0b01ac0580032ec4 (last accessed July 2013)
- 13. Points to consider on adjustment for baseline covariates. Published by the Committee for Medicinal Products for Human Use of the European Medicines Agency. 2006. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000366.jsp&mid=WC0b01ac0580032ec4 (last accessed July 2013)
- 14. Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products. Published by the Committee for Medicinal Products for Human Use of the European Medicines Agency. 2007. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf (last accessed July 2013)
- 15. First in human studies: points to consider in study placement, design and conduct. Published by the Association of the British Pharmaceutical Industry. 2011. Available at http://www.abpi.org.uk/our-work/library/guidelines/Pages/first-human-studies.aspx (last accessed July 2013)
- 16.Coulter A, Parsons S, Askham J. Where are the patients in decision-making about their own care? Published by the World Health Organisation 2008. Available at http://www.who.int/management/general/decisionmaking/WhereArePatientsinDecisionMaking.pdf (last accessed July 2013)
- 17. Consent: patients and doctors making decisions together. Published by the General Medical Council. 2008. Available at http://www.gmc-uk.org/static/documents/content/GMC_Consent_0513_Revised.pdf (last accessed July 2013)