

Abstract
Early detection of drug-induced kidney injury is vital in drug development. Generally accepted biomarkers such as creatinine and blood urea nitrogen (BUN) lack sensitivity and early injury responses are missed. Many new biomarkers to detect nephrotoxicity for pre-clinical utilization have been described and their use is adopted in regulatory guidelines. However, guidance on appropriate biomarkers for clinical trials is minimal. We provide an overview of potentially useful kidney biomarkers that can be used in clinical trials. This includes guidance to select biomarkers suitable to capture specific characteristics of the (expected) kidney injury. We conclude that measurement of urinary kidney injury marker-1 (KIM-1) serves many purposes and is often an appropriate choice. Cystatin C captures effects on glomerular filtration rate (GFR), but this marker should preferably be combined with more specific markers to localize the origin of the observed effect. Untoward effects on tubules can be captured relatively well with several markers. Direct detection of glomerular injury is currently impossible since specific biomarkers are lacking. Indirect assessment of toxic effects on glomeruli is possible by using carefully selected panels of other injury markers. We conclude that it is possible to obtain appropriate information on nephrotoxicity in clinical drug development by using carefully selected panels of injury markers and suggest that identification and validation of specific glomerular biomarkers could be of great value.
Keywords: biomarkers, early clinical trials, nephrotoxicity
Introduction
Case: Administration of a low dose locked nucleic acid antisense oligonucleotide caused toxic acute tubular necrosis in a 56-year-old otherwise healthy female volunteer. She received three weekly subcutaneous doses of experimental drug SPC5001, a PCSK9 inhibiting antisense oligonucleotide (developed to lower LDL-cholesterol). Five days after the last dose serum creatinine reached 2.67 mg dl–1 (a 238% change from baseline). Urine microscopy revealed red blood cells, white blood cells and granular casts. Renal biopsy showed multifocal tubular necrosis. Kidney biomarkers were measured retrospectively and were already increased after the first dose, clearly preceding changes in serum creatinine, urea, dipstick or urine microscopy (see Figure 1). Serum creatinine peaked 1 week after the last antisense dose and was 3.81 mg dl–1 at that time. Upon supportive treatment, serum creatinine decreased gradually. Baseline levels were observed from 44 days after last antisense administration onwards. She recovered completely without any persisting abnormalities. Which biomarkers for kidney injury would have prevented this? (van Poelgeest et al. [1]).
As illustrated by the above described case, early detection of drug-induced nephrotoxicity and prevention of clinical manifestations such as tubular necrosis are vital in early drug development in humans. The problem of drug-related renal toxicity is important as renal drug toxicities in animal studies account for more than 30% of the attrition of compounds from drug development 2. Despite this pre-selection, prevalence of (acute) kidney injury due to drug toxicity in clinical practice is as high as 18–27% of all episodes of acute kidney injury 3. In addition, efficacious interventions to reverse kidney damage are non-existent and clinicians can only apply supportive therapies while awaiting recovery of renal function. Animal models have shown that intervention directly after early changes have been noted is preferable as the window of opportunity to apply treatment appears to be limited to a few hours 4. Unfortunately, signals of early injury responses and the underlying mechanism of damage are often missed by traditional methods for monitoring renal function, such as serum creatinine, serum blood urea nitrogen (BUN), estimated glomerular filtration rate (GFR) and actual GFR. Serum creatinine and BUN are easy to obtain and the assays are part of standard care measurements rendering the results readily available. However, as markers for kidney injury, both have several shortcomings. Creatinine concentration in serum depends on multiple other factors than decline in renal function that differ intra- and inter-individually, such as age, gender, muscle mass, protein intake and certain drugs 4,5. Also, BUN concentrations are influenced by variable factors, such as diet, dehydration, liver function and tissue breakdown 6. This lack of specificity complicates the interpretation of these markers. The problems can be partially overcome by calculating GFR. Based upon data from large groups of patients different equations have been derived to estimate the actual eGFR using the serum creatinine value 7. The eGFR is commonly used in clinical practice as an overall index for kidney function 8. The Cockcroft–Gault formula, described in 1976 is based on a cohort of 249 patients and takes into account the factors age, weight and gender 9. The eGFR Modification of Diet in Renal Disease (MDRD) was derived from a study with renal disease patients and includes age, race, gender, serum BUN and serum albumin as input parameters 10. In addition, the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) was derived from two study cohorts, one consisting of patients with known renal disease (mGFR < 90 ml min−1) and a second cohort made up of kidney transplant donors all of whom had an mGFR > 90 ml min−1 11. The CDK-EPI formula includes the factors gender, age and race. Thus all formulae correct for certain variables and offer a tool to estimate GFR and monitor kidney function in chronic kidney disease patients. However, they still lack accuracy 12,13 and miss early and relatively small changes in GFR 14. This is particularly true when monitoring healthy subjects with normal renal function and normal baseline serum creatinine values. In this situation very small changes in plasma creatinine, which are in the noise of the assay, may still reflect considerable loss of renal function, as illustrated in Figure 2. Another disadvantage of serum creatinine (and eGFRs derived from this marker) and BUN is that a considerable time delay exists between the onset of kidney injury and the moment that the signal is observed. For creatinine this is, for example, illustrated in Figure 1. BUN concentrations correlate closely with histopathological changes in the kidney 15. However, the signal is delayed and by the time the damage is revealed by this marker 70–80% of renal epithelial mass is already lost 16.
Figure 2.
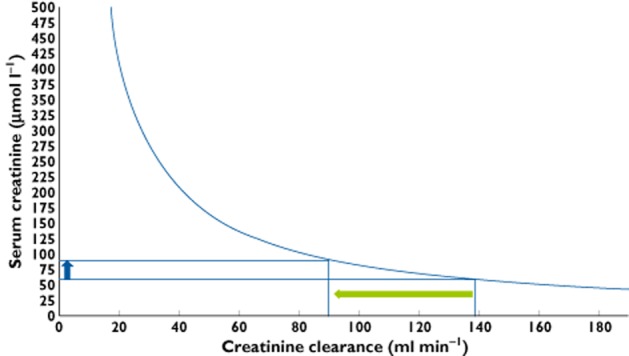
Theoretical relationship between serum creatinine and glomerular filtration rate (GFR) calculated with creatinine clearance for a subject with normal muscle mass (12 mmol creatinine 24 h–1). In the range of creatinine for healthy subjects large declines in GFR are associated with relatively small increases in serum creatinine. For example, if serum creatinine increases from 60 to 90 μmol l−1 (blue arrow), which is a 50% increase (a cut off value for safety often used in clinical trials), GFR decreases to 50 ml min−1, which reflects a considerable functional loss of over 35%. When the GFR decreases below 60 ml min−1, further decrements are associated with larger increases in serum creatinine, which makes the marker more sensitive to change
Figure 1.
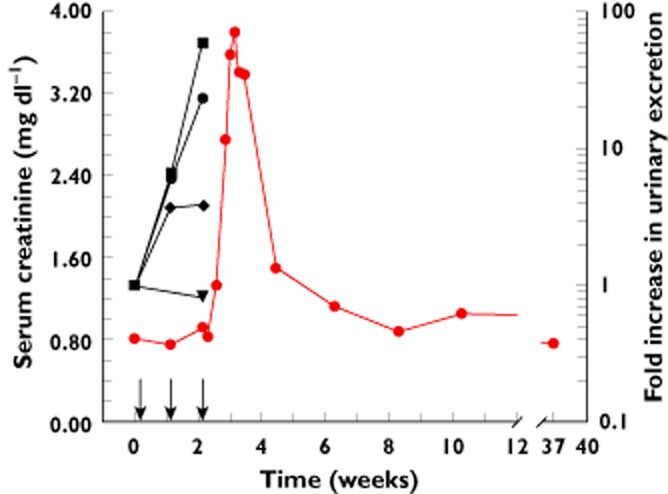
Time course of serum creatinine and urinary kidney damage markers. Arrows denote drug administration on study days 1, 8 and 15.  serum creatinine,
serum creatinine,  urinary B2M,
urinary B2M,  urinary αGST,
urinary αGST,  urinary KIM-1,
urinary KIM-1,  urinary NAG
urinary NAG
It has been advocated to not estimate but measure GFR for a more reliable assessment of kidney function. This can be achieved by measuring clearance after intravenous administration of exogenous markers that are solely removed by glomerular filtration. This approach also avoids GFR estimates influenced by extra-glomerular clearance such as tubular secretion. Different markers are used among which are inulin 17 and radio-isotopic agents, such as 99mTc-diethylenetriaminepentaacetic acid (DPTA), 169Yb-DTPA, 125I-iothalamate and 51Cr-ethylenediaminetetra-acetic acid (51Cr-EDTA). These markers have a high accuracy and can detect subtle changes in GFR 17,18. These methods are time consuming and therefore the results may not be readily available 19,20. More importantly, however, GFR is only one measure of kidney function while many different mechanisms of drug induced kidney injury exist, such as reduction in renal perfusion, direct tubular toxicity, intratubular obstruction, allergic interstitial nephritis and haemolytic-uraemic syndrome 21. All these mechanisms will lead directly or indirectly to reduction in the GFR, for instance by tubuloglomerular feedback after tubular toxicity 22. However, these changes in GFR may be detected too late for adaptations in dose or schedule of dosing, and do not provide information on the mechanism of toxicity. In this review we focus on measures that may provide earlier and more specific warning signals of renal damage than a decreased GFR.
In preclinical research detection of early signs of kidney injury is done by histological examination, which is considered to be the gold standard. Histopathology provides accurate anatomical information on the kidney injury as well as its severity and leads to a diagnosis. However, performing a kidney biopsy in humans is rarely an option and is an inappropriate tool to be used in drug development trials.
These difficulties regarding early detection of kidney injury may substantially influence the path of drug development. For instance, development of compounds for which renal toxicity is observed during preclinical experiments may be ceased in order to avoid safety issues during the clinical development. However, it is known that only 40–60% of animal findings are predictive of toxicities in humans 23,24. This results in the undesirable situation in which potentially efficacious compounds are abandoned for the wrong reason. Moreover, as these compounds do not reach the clinical phase, no insight is gained regarding the (potentially avoidable) nephrotoxic mechanisms of injury. The development of immortalized human proximal tubule cell lines expressing functional influx and efflux transporters 25, which can be challenged with toxins 26, appears to be a promising approach that can be used preclinically. However, it is not yet confirmed that these cell lines and the subsequent incubations sufficiently mimic the in vivo situation. Therefore translating findings from these cell-lines directly to the clinical situation seems premature. Adequate use of renal biomarkers could thus be of great value during early drug development.
For preclinical assessment, many new potential biomarkers of toxicity have been identified and their possible benefit has been evaluated by comparing their performance with the traditional markers. This extensive research has resulted in an EMEA/FDA guideline (published in 2009), that identified acceptable biomarkers that can be used to detect drug-induced nephrotoxicity in preclinical research 27. This panel includes the urinary excreted biomarkers kidney injury molecule-1 (KIM-1), albumin, total protein, beta-2-microglobulin (B2M), clusterin, trefoil factor 3 (TFF3) and cystatin C (CysC). These markers can be used to capture acute drug-induced nephrotoxicity of tubular or glomerular (with associated tubular involvement) origin 27. These markers were shown to provide additional and complementary information to BUN and serum creatinine and correlate with histopathological alterations. However, it was recognized that for the clinical setting these markers were insufficiently qualified to justify their general use. It was suggested to explore further their potential as clinical biomarkers for acute drug-induced kidney injury and recovery/reversibility. The guideline also advised to consider the biomarkers in clinical trials on a case-by-case basis to gather further data on their usefulness to monitor drug-induced renal toxicity in man.
With this review, we aim to offer guidance to select the biomarkers that suit the specific characteristics of the (expected) kidney injury. We provide an overview of promising biomarkers for nephrotoxicity, focusing on their possible use and limitations in clinical trials during early drug development. Certain properties of biomarkers of kidney injury are considered to be critical. First, the origin of the biomarker and the mechanism and/or site of injury should be clarified as much as possible. Furthermore the biomarker should be sensitive to early injury in order to outperform the traditional biomarkers and enable early intervention. High specificity and correlation with established outcome measures, such as histopathology, are also vital to avoid confusion on the value of the observed signal. As the majority of data on kidney injury biomarkers originates from preclinical research, the translational step to humans is of great importance. This could be achieved if supportive clinical evidence on biomarker profiles reflecting kidney injury in humans is available. Finally, reliable assays must be readily available.
The biomarkers were selected using the previously mentioned EMEA/FDA guidelines on preclinical markers, and on the condition that the pertaining assay is validated for human use and commercially available. The selection was expanded with the biomarkers for which promising data have been reported. These markers are neutrophil gelatinase-associated lipocalin (NGAL), alpha glutathione S-transferase (αGST), N-acetyl-beta-glucosaminidase (NAG) and interleukin-18 (IL18). Established markers such as serum creatinine, serum BUN, urinary albumin and protein were not considered in detail, although these are used as reference. Based on the literature, several considerations and recommendations regarding selection of kidney biomarkers in clinical drug development are given.
Summary on selected markers
CysC is a small molecule cysteine proteinase inhibitor synthesized by all nucleated cells and filtered freely by the glomerulus. After filtration it is not secreted nor reabsorbed by the tubules, but catabolized completely and thus reflects true GFR when measured in blood. Preclinically, CysC appears to be the most sensitive marker for early proximal tubular damage in animals, although a consistent dose response relation is lacking 28. CysC is suitable to assess kidney function in general, regardless of specific lesion site, as this marker is devoid of extra-glomerular clearance, variability in production and limited sensitivity that apply for BUN and serum creatinine 29. It has been suggested that CysC measured in blood could be a suitable translational biomarker as it avoids laborious urine collection in animals 29. Possible disadvantages of CysC are its dependency on factors other than decline of renal function alone, such as age, gender, weight and height, smoking and high serum C-reactive protein concentrations 5. In the clinic, CysC has been shown to be a sensitive marker of early renal dysfunction following ischaemic injury 30.
NGAL is an acute phase protein secreted as a response to acute injury of proximal and distal tubular epithelial cells. It is freely filtered by the glomerulus after which rapid clearance occurs via receptor binding and endocytosis 4. NGAL has been reported to be the most sensitive marker for proximal tubular damage in the preclinical setting 28. After gentamicin exposure in rats a clear signal is detected as early as 24 h after exposure. However, specificity to the location of injury must be questioned, since in a rodent glomerular damage model increased NGAL concentrations were also observed 31. Clinical research has demonstrated that urinary NGAL is increased in several forms of chronic kidney injury 32,33 and in patients with urinary tract infections 34.
IL18 is a pro-inflammatory cytokine produced by leukocytes and renal parenchymal cells such as tubular epithelial cells, podocytes and mesangial cells. It plays an important role in the exacerbation of acute tubular necrosis 35 and the inflammatory pathways involved are partly clarified 36. The IL18 receptor (IL-18R) is expressed on these cells in cisplatin-induced acute kidney injury and urinary IL18 excretion has proven to be an early diagnostic marker for acute kidney injury in humans, particularly in critically ill patients 37. However, at present it is unclear if IL18 reflects location-specific injury and therefore its potential for preclinical and clinical use in drug development is unclear. An obvious disadvantage is that increased IL18 concentrations can also be observed upon many forms of inflammation not limited to the kidney.
NAG is a lysosomal enzyme which is contained abundantly in the renal tubular epithelia and involved in the degradation of mucopolysaccharides and glycoproteins. Its size precludes glomerular filtration and elevated urinary concentrations are considered to reflect tubular dysfunction. In preclinical research the sensitivity of NAG is higher compared with serum creatinine and comparable with BUN 38. The NAG response profile is dependent on the toxin causing proximal tubule injury 39. For example, gentamicin triggers an early response that lasts for 8 h after dosing, whereas chromium triggers a response after 8 h and mercury does not trigger a significant NAG increase at all 39. Nevertheless, clinical evidence supports the usefulness of NAG as an early marker of mild tubular injury 40 and demonstrates that it has predictive properties regarding the development of acute tubular necrosis 41.
αGST is a detoxification enzyme that is produced in numerous tissues. Urinary αGST concentrations are very low under physiological conditions, but substantial amounts are excreted in various manifestations of tubular injury, including cisplatin- 42 and gentamicin-induced 43 toxicity and in acute tubular necrosis after mercuric chloride and potassium dichromate exposure 44. αGST appears to be an adequate preclinical early toxicity biomarker to detect onset of epithelial necrosis, but is less suitable to monitor reversibility 29. In drug-induced injury of proximal tubular cells with cisplatin and gentamicin, αGST correlated more closely with histopathological confirmed injury compared with NAG, BUN and serum creatinine 45. In a preclinical model of tubular injury limited to the pars recta of proximal tubule cells, it was demonstrated that αGST excretion reflects injury at low grade toxicity, outperforming numerous other markers, and with equal sensitivity to KIM-1 46. However, injury to the collecting duct was associated with a decreased αGST, which is not well understood yet. As a consequence it has been suggested that qualification of this biomarker has to await further results 45. Although limited information is available, clinical evidence suggests that αGST is informative for tubular dysfunction or injury 47,48.
KIM-1 is a transmembrane protein expressed by proximal tubular epithelial cells. KIM-1 functions as a phosphatidylserine receptor and has phagocytic capacity 28. Expression is markedly upregulated in response to injury 49. Urinary KIM-1 concentration provides a more sensitive predictor of histopathological confirmed injury in 11 well-established rat models of acute kidney injury when compared with BUN, serum creatinine or urinary NAG, even in cases of low grade toxicity 38. One study reported that urinary KIM-1 concentrations also correlated with different grades of kidney tubular histopathologies. This was supported by the finding of dose-dependent upregulation of the KIM-1 gene in segment-specific toxicity models 50. Whereas αGST appears to be a good early toxicity biomarker for epithelial necrosis, KIM-1 and clusterin concentrations persist during regeneration and appear to reflect the triggering and continuation of the repair process 29,51. KIM-1 responses seem to depend on toxin, for example Sasaki et al. 52 report that after cisplatin exposure KIM-1 increases (together with clusterin and aGST) after 3 days (confirmed by Vinken et al. 15), whereas in a model for papillary necrosis using 2-bromoethylamine hydrobromide KIM-1 concentrations (together with clusterin, albumin and osteopontin) are convincingly increased as early as day 1 after exposure. Interestingly, measurement of urinary KIM-1 also enables detection of subchronic and chronic injury and correlates closely with histopathology 28. This study also showed that CysC and NGAL are the most sensitive markers for early kidney damage of proximal tubular damage, but subchronic or chronic injury was best reflected by KIM-1 concentrations. In keeping with the notion that urinary KIM-1 may be an early marker for chronic nephrotoxicity in animals, increased levels of urinary KIM-1 concentrations were reported in an experiment using cadmium 53. Clinical evidence that the results in animals translate to humans is currently limited. KIM-1 did indeed show a significant signal following acute kidney injury, although sampling for KIM-1 in this study was rather late, precluding assessment of its suitability as an early marker 54.
B2M is produced by all cells expressing major histocompatibility complex (MHC) class I antigen. Under normal conditions the main source is activated lymphocytes from which shedding from cell surface of the MHC occurs through proteolysis. Synthesis is stimulated in various conditions characterized by proliferation of lymphoid cells that occurs in various disease states, such as neoplasms, (auto-) immune disorders or infections 55–57. B2M is filtered freely across the glomerulus and complete reabsorption occurs by proximal tubular cells 28. Impaired tubular uptake results in increased B2M urinary excretion. Glomerular protein loss may also increase urinary B2M excretion as B2M shares a common rate-limited tubular reabsorption pathway with other proteins. In the preclinical setting B2M has a better diagnostic performance than BUN and serum creatinine to detect glomerular injury (together with urinary total protein and CysC) 58. As tubular dysfunction without glomerular impairment also increases B2M 59,60 specificity regarding the location of damage using B2M alone is questionable. Interestingly, B2M might be excreted via other pathways as compared with other markers, as can be concluded from the findings on a model for papillary necrosis. This massive injury gave rise to impressive increases in KIM-1, clusterin, albumin and osteopontin, but concentrations of B2M remained close to normal 52. In the clinical setting, B2M has proven to be a marker for disease severity in autosomal polycystic kidney disease 61 and for renal damage by fumaric acid esters 62.
Clusterin is a glycosated protein associated with apoptosis and clearance of cellular debris and can be found in several tissues. Within kidney cells, clusterin has been suggested to possess anti-apoptotic properties and facilitates cell protection, lipid recycling and cell attachment, and aggregation 63. Clusterin cannot be filtered by glomeruli due to its size and therefore urinary concentrations are specific for kidney injury. Clusterin performed better to detect proximal tubular injury than CysC, B2M and total protein 58 and evidence suggests it can be used as an early marker with a similar profile to KIM-1 [15, 52]. The clusterin response correlates well with tubular injury regardless of the location, particularly when regeneration is present 45. Elevated clusterin concentrations persist during regeneration and appear to reflect the triggering and continuation of the repair process 29. Clinical data on urinary clusterin in relation to kidney injury is not extensively available. However, it has been shown that clusterin expression is increased in renal injury and cystic diseases 64.
TFF3 is a small peptide hormone secreted by mucus-producing and other epithelial cells 65. In the kidney it is produced/secreted by cells of the collecting ducts 66. TFF3 is involved in many functions including restoration of intestinal epithelium 67, but its physiological function within the kidney is still elusive. Because in ageing rats decreasing amount of kidney TFF3 are found, it has been suggested that TFF3 may have a general protective function 68. Significantly decreased concentrations of TFF3 have been observed in different rat models of proximal tubular injury. Combining TFF3 with urinary albumin increases sensitivity to early injury compared with traditional markers 69. However, studies comparing TFF3 with other novel biomarkers are lacking. In humans, it has been reported that certain populations (African descent, diabetes and antihypertensive medication use) have higher baseline urinary TFF3 concentrations, and that increase in urinary levels might indicate ongoing repair of chronic damage in the kidney 70.
Comparison and considerations
Concerns regarding nephrotoxicity are often encountered during the development of novel drugs. Particularly when the compound is about to be tested in humans for the first time, all indications of possible kidney injury are weighed. This may include compounds belonging to drug classes that are notoriously associated with renal injury, drugs specifically targeted to the kidney, but also drugs that are considered ‘suspicious’ because of their mechanism of action or pharmacokinetic properties. Preclinical suggestions for nephrotoxicity can be present. In case of clearly dose-related adverse renal effects at the high end of the tested dose range, it is usually possible to estimate a safe range for dosing in humans provided that adequate monitoring is possible. However, it is more difficult if preclinical data point towards an incidentally occurring event. This might lead to a more variable risk for susceptible human subjects, which is difficult to catch. Whatever the findings in animals, close monitoring of the kidney in clinical trials to detect untoward effects as soon as possible is necessary in all cases.
Every drug has its specific features and other factors such as dosing schedule, cumulative dose, patient and/or disease characteristics may play a role. Thus, each drug requires the selection of an appropriate biomarker or most likely a panel of biomarkers to provide comprehensive information. As subject safety is a primary goal in first into human trials, sensitivity for the event is crucial. A strategy for selecting biomarkers in humans could be to first focus on the localization of the site of injury and the mechanism by which the compound causes injury. This may be achieved by using histopathological information on the compound and comparing this with histopathological and biomarker profiles of known nephrotoxic agents that target similar sites (Table 1).
Table 1.
Established nephrotoxic agents and accompanying biomarker signal
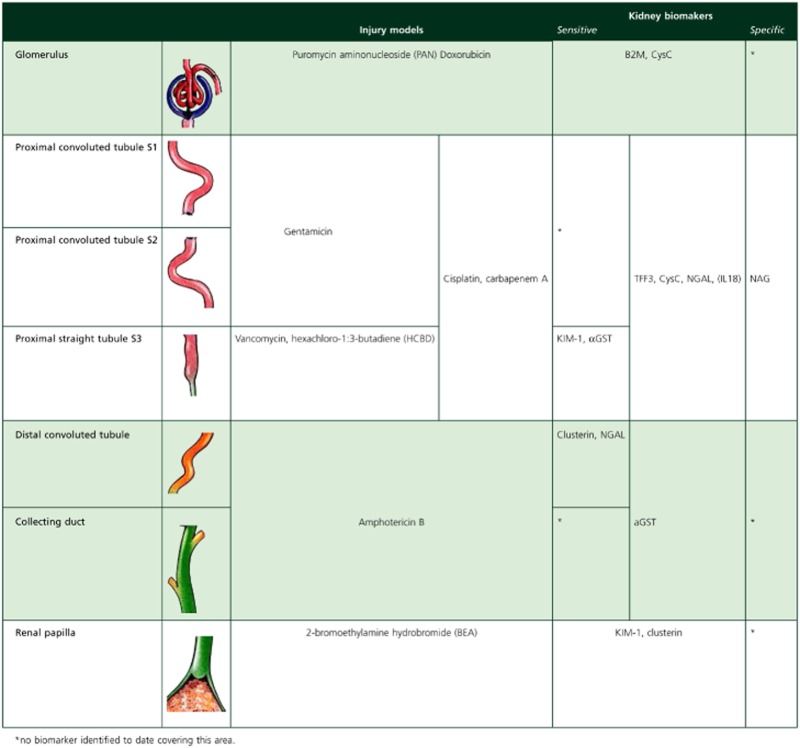 |
The selection of a panel of biomarkers should obviously be based on the possibilities and limitations of individual markers and the aim should be to compose a panel of which the combination of biomarkers provides complimentary information (Table 2).
Table 2.
Aims of research and appropriate biomarkers
| Aim: | Suitable marker |
|---|---|
| Monitoring general kidney function (GFR) | CysC |
| Differentiate between glomerular and tubular damage | KIM-1, clusterin, αGST |
| Monitor toxicity with suspected glomerular localization | B2M |
| Monitor toxicity with suspected non-glomerular localization | CysC, NGAL, |
| Detect early kidney injury with suspected proximal tubular localization | αGST, KIM-1 |
| Monitor reversibility/regeneration with suspected proximal tubular localization | KIM-1 |
| Monitor toxicity with suspected distal tubular localization | Clusterin |
| Monitor reversibility/regeneration with suspected distal tubular localization | Clusterin |
| Elucidate pathophysiological mechanism with known proximal tubular injury site | NAG |
| High risk of acute tubular necrosis (Phase II/III trials) | IL18 |
We first point out that currently a specific marker for glomerular injury is unavailable and thus the nature of renal damage is often defined as glomerular by excluding other causes. Identification of a biomarker specific for glomerular injury would be of great value. CysC can be used to monitor general kidney function as it reflects GFR. Further, CysC and NGAL are the most sensitive markers for early kidney damage of proximal tubular damage 28. However, it should be realized that the organ specificity of CysC is poor and for differentiation between tubular and glomerular damage other biomarkers are recommended. The dependency of CysC concentrations on other factors than renal function is important in diagnostic work-ups, but not so much in clinical drug research as these circumstances can be taken into account by choice of population and by focusing on the time course and relative changes from baseline. If this is unfeasible or undesired, CycC appears to be a less suitable biomarker.
Although NGAL seems more tubular injury-specific compared with CysC, this has been challenged by the finding that NGAL also increases in a model of glomerular injury 31. This could reflect generalized (glomerular and tubular) injury or could indicate that the biomarker is not injury-site specific. NGAL is not recommended as a monitoring tool, as it appears to possess equal sensitivity to serum BUN. αGST and KIM-1 exhibit similar sensitivity to detect tubular damage to the pars recta of the proximal tubule 46. Nevertheless, KIM-1 is preferred as there is more evidence regarding its performance compared with other biomarkers 38. Moreover, the close correlation of KIM-1 with histopathology 38,50 and the finding that it can be used also to monitor reversibility 29 suggest KIM-1 to be the biomarker of choice. Furthermore, KIM-1 can be used to detect subchronic or chronic injury 28,71.The limited information available on TFF3 suggests that this biomarker does not have a clear advantage over other biomarkers for proximal tubular injury. We suggest to not use this marker unless more information is available on its profile and its performance compared with other biomarkers in animals and humans.
B2M is generally considered to be a glomerular marker, but increased urinary excretion could also reflect impaired reabsorption by proximal tubular cells. It appears that B2M as single measure does not have great value 58. However, when included in a panel of biomarkers it allows distinguishing between glomerular and tubular dysfunction. The choice to incorporate clusterin depends largely on the expected toxicity. Clusterin is a sensitive marker and its urinary excretion increases as a response to tubular injury irrespectively of the site. This feature of non-specificity can be of value, when tubular injury to distal tubules is suspected or localization is unclear. It is advised to use clusterin only in a panel of biomarkers. The information on IL-18 as a suitable marker is too limited to justify its use in early clinical drug development. However, the finding that IL18 can be used to detect acute kidney injury very early in hospitalized patients 37 and its role in the pathophysiology of kidney injury 36 suggests that IL18 is potentially a useful marker but this should be explored further.
It is important to take into account that biomarkers might increase for reasons other than kidney injury (alone), which potentially results in an inaccurate conclusion. For instance, increased concentrations of acute-phase proteins such as NGAL, but also B2M and IL18, may be observed upon any inflammatory condition regardless of the site of inflammation. Also the population that is studied is important as illustrated by the finding that patients with an impaired glucose tolerance show a higher urinary excretion of NAG 72. These problems may be minimized by assessment of (changes in) renal function while taking all available information into account. In practice, it is advised to consider carefully the population that is studied, to have good information on the biomarkers at baseline, and to always measure chemistry and haematology at similar time points as the panel of renal biomarkers. This comprehensive approach prevents erroneous attribution of findings to the kidney.
Recommendation
We conclude that measurement of urinary KIM-1 suits many purposes and is therefore often an appropriate choice. It allows for early detection of proximal tubule injury, differentiation between glomerular and tubular damage and assessment of reversibility and regeneration provided this occurs. KIM-1 can also be used to detect sub-chronic and chronic kidney injury. NGAL is useful if tubular involvement is suspected, but no clear notion of tubular localization is present. Clusterin may add value in cases of suspected injury in the distal tubule. If general kidney function and thus a measure of clearance is required, it is useful to include CysC, preferably combined with more specific markers to localize the origin of the observed effect. IL-18 is considered less suitable for early phase trials, but might be helpful in phase II/III trials to be able to detect acute tubular necrosis at a very early stage. It may be useful to include B2M and αGST in the panel of biomarkers as this makes it possible to relate findings on new compounds to existing knowledge of known nephrotoxic agents.
Conflict of Interest Statement
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.van Poelgeest EP, Swart RM, Betjes MG, Moerland M, Weening JJ, Tessier Y, Hodges MR, Levin AA, Burggraaf J. Acute kidney injury during therapy with an antisense oligonucleotide directed against PCSK9. Am J Kidney Dis. 2013;62:796–800. doi: 10.1053/j.ajkd.2013.02.359. [DOI] [PubMed] [Google Scholar]
- 2.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates. Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 3.Loghman-Adham M, Kiu Weber CI, Ciorciaro C, Mann J, Meier M. Detection and management of nephrotoxicity during drug development. Expert Opin Drug Saf. 2012;11:581–596. doi: 10.1517/14740338.2012.691964. [DOI] [PubMed] [Google Scholar]
- 4.McIlroy DR, Wagener G, Lee HT. Biomarkers of acute kidney injury: an evolving domain. Anesthesiology. 2010;112:998–1004. doi: 10.1097/ALN.0b013e3181cded3f. [DOI] [PubMed] [Google Scholar]
- 5.Knight EL, Verhave JC, Spiegelman D, Hillege HL, De ZD, Curhan GC, de Jong PE. Factors influencing serum cystatin C levels other than renal function and the impact on renal function measurement. Kidney Int. 2004;65:1416–1421. doi: 10.1111/j.1523-1755.2004.00517.x. [DOI] [PubMed] [Google Scholar]
- 6.Traynor J, Mactier R, Geddes CC, Fox JG. How to measure renal function in clinical practice. BMJ. 2006;333:733–737. doi: 10.1136/bmj.38975.390370.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herget-Rosenthal S, Bokenkamp A, Hofmann W. How to estimate GFR-serum creatinine, serum cystatin C or equations. Clin Biochem. 2007;40:153–161. doi: 10.1016/j.clinbiochem.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 8.Stevens PE, Levin A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158:825–830. doi: 10.7326/0003-4819-158-11-201306040-00007. [DOI] [PubMed] [Google Scholar]
- 9.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 10.Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–470. doi: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 11.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, III, Feldman HI, Kusek JW, Eggers P, Van LF, Greene T, Coresh J. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Botev R, Mallie JP, Wetzels JF, Couchoud C, Schuck O. The clinician and estimation of glomerular filtration rate by creatinine-based formulas: current limitations and quo vadis. Clin J Am Soc Nephrol. 2011;6:937–950. doi: 10.2215/CJN.09241010. [DOI] [PubMed] [Google Scholar]
- 13.Stevens LA, Coresh J, Greene T, Levey AS. Assessing kidney function–measured and estimated glomerular filtration rate. N Engl J Med. 2006;354:2473–2483. doi: 10.1056/NEJMra054415. [DOI] [PubMed] [Google Scholar]
- 14.Murty MS, Sharma UK, Pandey VB, Kankare SB. Serum cystatin C as a marker of renal function in detection of early acute kidney injury. Indian J Nephrol. 2013;23:180–183. doi: 10.4103/0971-4065.111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinken P, Starckx S, Barale-Thomas E, Looszova A, Sonee M, Goeminne N, Vermissen L, Buyens K, Lampo A. Tissue Kim-1 and urinary clusterin as early indicators of cisplatin-induced acute kidney injury in rats. Toxicol Pathol. 2012;40:1049–1062. doi: 10.1177/0192623312444765. [DOI] [PubMed] [Google Scholar]
- 16.Sieber M, Hoffmann D, Adler M, Vaidya VS, Clement M, Bonventre JV, Zidek N, Rached E, Amberg A, Callanan JJ, Dekant W, Mally A. Comparative analysis of novel noninvasive renal biomarkers and metabonomic changes in a rat model of gentamicin nephrotoxicity. Toxicol Sci. 2009;109:336–349. doi: 10.1093/toxsci/kfp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahn KH, Heidenreich S, Bruckner D. How to assess glomerular function and damage in humans. J Hypertens. 1999;17:309–317. doi: 10.1097/00004872-199917030-00002. [DOI] [PubMed] [Google Scholar]
- 18.Soares AA, Eyff TF, Campani RB, Ritter L, Camargo JL, Silveiro SP. Glomerular filtration rate measurement and prediction equations. Clin Chem Lab Med. 2009;47:1023–1032. doi: 10.1515/CCLM.2009.263. [DOI] [PubMed] [Google Scholar]
- 19.Prigent A. [Measurement of renal function in clinical practice: principles and limitations] J Radiol. 2011;92:274–279. doi: 10.1016/j.jradio.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Stevens LA, Levey AS. Measured GFR as a confirmatory test for estimated GFR. J Am Soc Nephrol. 2009;20:2305–2313. doi: 10.1681/ASN.2009020171. [DOI] [PubMed] [Google Scholar]
- 21.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 22.Blantz RC, Thomson SC, Peterson OW, Gabbai FB. Physiologic adaptations of the tubuloglomerular feedback system. Kidney Int. 1990;38:577–583. doi: 10.1038/ki.1990.245. [DOI] [PubMed] [Google Scholar]
- 23.Olson H, Betton G, Stritar J, Robinson D. The predictivity of the toxicity of pharmaceuticals in humans from animal data–an interim assessment. Toxicol Lett. 1998;102–103:535–538. doi: 10.1016/s0378-4274(98)00261-6. [DOI] [PubMed] [Google Scholar]
- 24.Knight A. Systematic reviews of animal experiments demonstrate poor human clinical and toxicological utility. Altern Lab Anim. 2007;35:641–659. doi: 10.1177/026119290703500610. [DOI] [PubMed] [Google Scholar]
- 25.Wilmer MJ, Saleem MA, Masereeuw R, Ni L, van der Velden TJ, Russel FG, Mathieson PW, Monnens LA, van den Heuvel LP, Levtchenko EN. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010;339:449–457. doi: 10.1007/s00441-009-0882-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moghadasali R, Mutsaers HA, Azarnia M, Aghdami N, Baharvand H, Torensma R, Wilmer MJ, Masereeuw R. Mesenchymal stem cell-conditioned medium accelerates regeneration of human renal proximal tubule epithelial cells after gentamicin toxicity. Exp Toxicol Pathol. 2013;65:595–600. doi: 10.1016/j.etp.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 27. EMEA/FDA guideline on detection of drug-induced nephrotoxicity. 2009. Ref Type: Internet Communication.
- 28.Hoffmann D, Fuchs TC, Henzler T, Matheis KA, Herget T, Dekant W, Hewitt P, Mally A. Evaluation of a urinary kidney biomarker panel in rat models of acute and subchronic nephrotoxicity. Toxicology. 2010;277:49–58. doi: 10.1016/j.tox.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 29.Ozer JS, Dieterle F, Troth S, Perentes E, Cordier A, Verdes P, Staedtler F, Mahl A, Grenet O, Roth DR, Wahl D, Legay F, Holder D, Erdos Z, Vlasakova K, Jin H, Yu Y, Muniappa N, Forest T, Clouse HK, Reynolds S, Bailey WJ, Thudium DT, Topper MJ, Skopek TR, Sina JF, Glaab WE, Vonderscher J, Maurer G, Chibout SD, Sistare FD, Gerhold DL. A panel of urinary biomarkers to monitor reversibility of renal injury and a serum marker with improved potential to assess renal function. Nat Biotechnol. 2010;28:486–494. doi: 10.1038/nbt.1627. [DOI] [PubMed] [Google Scholar]
- 30.Vassalos A, Young D, MacArthur K, Pollock J, Lyall F, Danton MH. Cystatin C: influence of perfusion and myocardial injury on early (<24 h) renal function after pediatric cardiac surgery. Paediatr Anaesth. 2011;21:1185–1191. doi: 10.1111/j.1460-9592.2011.03654.x. [DOI] [PubMed] [Google Scholar]
- 31.Tonomura Y, Tsuchiya N, Torii M, Uehara T. Evaluation of the usefulness of urinary biomarkers for nephrotoxicity in rats. Toxicology. 2010;273:53–59. doi: 10.1016/j.tox.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 32.Ding H, He Y, Li K, Yang J, Li X, Lu R, Gao W. Urinary neutrophil gelatinase-associated lipocalin (NGAL) is an early biomarker for renal tubulointerstitial injury in IgA nephropathy. Clin Immunol. 2007;123:227–234. doi: 10.1016/j.clim.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Brunner HI, Mueller M, Rutherford C, Passo MH, Witte D, Grom A, Mishra J, Devarajan P. Urinary neutrophil gelatinase-associated lipocalin as a biomarker of nephritis in childhood-onset systemic lupus erythematosus. Arthritis Rheum. 2006;54:2577–2584. doi: 10.1002/art.22008. [DOI] [PubMed] [Google Scholar]
- 34.Yilmaz A, Sevketoglu E, Gedikbasi A, Karyagar S, Kiyak A, Mulazimoglu M, Aydogan G, Ozpacaci T, Hatipoglu S. Early prediction of urinary tract infection with urinary neutrophil gelatinase associated lipocalin. Pediatr Nephrol. 2009;24:2387–2392. doi: 10.1007/s00467-009-1279-6. [DOI] [PubMed] [Google Scholar]
- 35.Bani-Hani AH, Leslie JA, Asanuma H, Dinarello CA, Campbell MT, Meldrum DR, Zhang H, Hile K, Meldrum KK. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009;76:500–511. doi: 10.1038/ki.2009.216. [DOI] [PubMed] [Google Scholar]
- 36.Nozaki Y, Kinoshita K, Yano T, Asato K, Shiga T, Hino S, Niki K, Nagare Y, Kishimoto K, Shimazu H, Funauchi M, Matsumura I. Signaling through the interleukin-18 receptor alpha attenuates inflammation in cisplatin-induced acute kidney injury. Kidney Int. 2012;82:892–902. doi: 10.1038/ki.2012.226. [DOI] [PubMed] [Google Scholar]
- 37.Parikh CR, Jani A, Melnikov VY, Faubel S, Edelstein CL. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am J Kidney Dis. 2004;43:405–414. doi: 10.1053/j.ajkd.2003.10.040. [DOI] [PubMed] [Google Scholar]
- 38.Vaidya VS, Ozer JS, Dieterle F, Collings FB, Ramirez V, Troth S, Muniappa N, Thudium D, Gerhold D, Holder DJ, Bobadilla NA, Marrer E, Perentes E, Cordier A, Vonderscher J, Maurer G, Goering PL, Sistare FD, Bonventre JV. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28:478–485. doi: 10.1038/nbt.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Y, Vaidya VS, Brown RP, Zhang J, Rosenzweig BA, Thompson KL, Miller TJ, Bonventre JV, Goering PL. Comparison of kidney injury molecule-1 and other nephrotoxicity biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol Sci. 2008;101:159–170. doi: 10.1093/toxsci/kfm260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouchi M, Suzuki T, Hashimoto M, Motoyama M, Ohara M, Suzuki K, Igari Y, Watanabe K, Nakano H, Urinary OK. N-acetyl-beta-d-glucosaminidase levels are positively correlated with 2-hr plasma glucose levels during oral glucose tolerance testing in prediabetes. J Clin Lab Anal. 2012;26:473–480. doi: 10.1002/jcla.21549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herget-Rosenthal S, Poppen D, Husing J, Marggraf G, Pietruck F, Jakob HG, Philipp T, Kribben A. Prognostic value of tubular proteinuria and enzymuria in nonoliguric acute tubular necrosis. Clin Chem. 2004;50:552–558. doi: 10.1373/clinchem.2003.027763. [DOI] [PubMed] [Google Scholar]
- 42.Feinfeld DA, Fuh VL, Safirstein R. Urinary glutathione-S-transferase in cisplatin nephrotoxicity in the rat. J Clin Chem.Clin Biochem. 1986;24:529–532. doi: 10.1515/cclm.1986.24.8.529. [DOI] [PubMed] [Google Scholar]
- 43.Feinfeld DA, Fleischner GM, Arias IM. Urinary ligandin and glutathione-S-transferase in gentamicin-induced nephrotoxicity in the rat. Clin Sci (Lond) 1981;61:123–125. doi: 10.1042/cs0610123. [DOI] [PubMed] [Google Scholar]
- 44.Feinfeld DA, Bourgoignie JJ, Fleischner G, Goldstein EJ, Biempica L, Arias IM. Ligandinuria in nephrotoxic acute tubular necrosis. Kidney Int. 1977;12:387–392. doi: 10.1038/ki.1977.129. [DOI] [PubMed] [Google Scholar]
- 45.Harpur E, Ennulat D, Hoffman D, Betton G, Gautier JC, Riefke B, Bounous D, Schuster K, Beushausen S, Guffroy M, Shaw M, Lock E, Pettit S. Biological qualification of biomarkers of chemical-induced renal toxicity in two strains of male rat. Toxicol Sci. 2011;122:235–252. doi: 10.1093/toxsci/kfr112. [DOI] [PubMed] [Google Scholar]
- 46.Swain A, Turton J, Scudamore C, Maguire D, Pereira I, Freitas S, Smyth R, Munday M, Stamp C, Gandhi M, Sondh S, Ashall H, Francis I, Woodfine J, Bowles J, York M. Nephrotoxicity of hexachloro-1:3-butadiene in the male Hanover Wistar rat; correlation of minimal histopathological changes with biomarkers of renal injury. J Appl Toxicol. 2012;32:417–428. doi: 10.1002/jat.1727. [DOI] [PubMed] [Google Scholar]
- 47.Bruning T, Sundberg AG, Birner G, Lammert M, Bolt HM, Appelkvist EL, Nilsson R, Dallner G. Glutathione transferase alpha as a marker for tubular damage after trichloroethylene exposure. Arch Toxicol. 1999;73:246–254. doi: 10.1007/s002040050613. [DOI] [PubMed] [Google Scholar]
- 48.Sundberg A, Appelkvist EL, Dallner G, Nilsson R. Glutathione transferases in the urine: sensitive methods for detection of kidney damage induced by nephrotoxic agents in humans. Environ Health Perspect. 1994;102(Suppl 3):293–296. doi: 10.1289/ehp.94102s3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amin RP, Vickers AE, Sistare F, Thompson KL, Roman RJ, Lawton M, Kramer J, Hamadeh HK, Collins J, Grissom S, Bennett L, Tucker CJ, Wild S, Kind C, Oreffo V, Davis JW, Curtiss S, Naciff JM, Cunningham M, Tennant R, Stevens J, Car B, Bertram TA, Afshari CA. Identification of putative gene based markers of renal toxicity. Environ Health Perspect. 2004;112:465–479. doi: 10.1289/ehp.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiusolo A, Defazio R, Zanetti E, Mongillo M, Mori N, Cristofori P, Trevisan A. Kidney injury molecule-1 expression in rat proximal tubule after treatment with segment-specific nephrotoxicants: a tool for early screening of potential kidney toxicity. Toxicol Pathol. 2010;38:338–345. doi: 10.1177/0192623310362244. [DOI] [PubMed] [Google Scholar]
- 51.Rouse RL, Zhang J, Stewart SR, Rosenzweig BA, Espandiari P, Sadrieh NK. Comparative profile of commercially available urinary biomarkers in preclinical drug-induced kidney injury and recovery in rats. Kidney Int. 2011;79:1186–1197. doi: 10.1038/ki.2010.463. [DOI] [PubMed] [Google Scholar]
- 52.Sasaki D, Yamada A, Umeno H, Kurihara H, Nakatsuji S, Fujihira S, Tsubota K, Ono M, Moriguchi A, Watanabe K, Seki J. Comparison of the course of biomarker changes and kidney injury in a rat model of drug-induced acute kidney injury. Biomarkers. 2011;16:553–566. doi: 10.3109/1354750X.2011.613123. [DOI] [PubMed] [Google Scholar]
- 53.Prozialeck WC, Vaidya VS, Liu J, Waalkes MP, Edwards JR, Lamar PC, Bernard AM, Dumont X, Bonventre JV. Kidney injury molecule-1 is an early biomarker of cadmium nephrotoxicity. Kidney Int. 2007;72:985–993. doi: 10.1038/sj.ki.5002467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaidya VS, Waikar SS, Ferguson MA, Collings FB, Sunderland K, Gioules C, Bradwin G, Matsouaka R, Betensky RA, Curhan GC, Bonventre JV. Urinary biomarkers for sensitive and specific detection of acute kidney injury in humans. Clin Transl Sci. 2008;1:200–208. doi: 10.1111/j.1752-8062.2008.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsoukas CM, Bernard NF. Markers predicting progression of human immunodeficiency virus-related disease. Clin Microbiol Rev. 1994;7:14–28. doi: 10.1128/cmr.7.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bethea M, Forman DT. Beta 2-microglobulin: its significance and clinical usefulness. Ann Clin Lab Sci. 1990;20:163–168. [PubMed] [Google Scholar]
- 57.Grundy JE. Role of beta 2-microglobulin in cytomegalovirus infection. Scand J Rheumatol Suppl. 1990;87:98–101. doi: 10.3109/03009749009097066. [DOI] [PubMed] [Google Scholar]
- 58.Dieterle F, Perentes E, Cordier A, Roth DR, Verdes P, Grenet O, Pantano S, Moulin P, Wahl D, Mahl A, End P, Staedtler F, Legay F, Carl K, Laurie D, Chibout SD, Vonderscher J, Maurer G. Urinary clusterin, cystatin C, beta2-microglobulin and total protein as markers to detect drug-induced kidney injury. Nat Biotechnol. 2010;28:463–469. doi: 10.1038/nbt.1622. [DOI] [PubMed] [Google Scholar]
- 59.Gatanaga H, Tachikawa N, Kikuchi Y, Teruya K, Genka I, Honda M, Tanuma J, Yazaki H, Ueda A, Kimura S, Oka S. Urinary beta2-microglobulin as a possible sensitive marker for renal injury caused by tenofovir disoproxil fumarate. AIDS Res Hum Retroviruses. 2006;22:744–748. doi: 10.1089/aid.2006.22.744. [DOI] [PubMed] [Google Scholar]
- 60.Del PM, Romero S, Casado JL. Proximal tubular renal dysfunction or damage in HIV-infected patients. AIDS Rev. 2012;14:179–187. [PubMed] [Google Scholar]
- 61.Meijer E, Boertien WE, Nauta FL, Bakker SJ, van Oeveren W, Rook M, van der Jagt EJ, van Goor H, Peters DJ, Navis G, de Jong PE, Gansevoort RT. Association of urinary biomarkers with disease severity in patients with autosomal dominant polycystic kidney disease: a cross-sectional analysis. Am J Kidney Dis. 2010;56:883–895. doi: 10.1053/j.ajkd.2010.06.023. [DOI] [PubMed] [Google Scholar]
- 62.Haring N, Mahr HS, Mundle M, Strohal R, Lhotta K. Early detection of renal damage caused by fumaric acid ester therapy by determination of urinary beta2-microglobulin. Br J Dermatol. 2011;164:648–651. doi: 10.1111/j.1365-2133.2010.10171.x. [DOI] [PubMed] [Google Scholar]
- 63.Rosenberg ME, Silkensen J. Clusterin: physiologic and pathophysiologic considerations. Int J Biochem Cell Biol. 1995;27:633–645. doi: 10.1016/1357-2725(95)00027-m. [DOI] [PubMed] [Google Scholar]
- 64.Dvergsten J, Manivel JC, Correa-Rotter R, Rosenberg ME. Expression of clusterin in human renal diseases. Kidney Int. 1994;45:828–835. doi: 10.1038/ki.1994.109. [DOI] [PubMed] [Google Scholar]
- 65.Madsen J, Nielsen O, Tornoe I, Thim L, Holmskov U. Tissue localization of human trefoil factors 1, 2, and 3. J Histochem Cytochem. 2007;55:505–513. doi: 10.1369/jhc.6A7100.2007. [DOI] [PubMed] [Google Scholar]
- 66.Chinery R, Poulsom R, Elia G, Hanby AM, Wright NA. Expression and purification of a trefoil peptide motif in a beta-galactosidase fusion protein and its use to search for trefoil-binding sites. Eur J Biochem. 1993;212:557–563. doi: 10.1111/j.1432-1033.1993.tb17693.x. [DOI] [PubMed] [Google Scholar]
- 67.Kinoshita K, Taupin DR, Itoh H, Podolsky DK. Distinct pathways of cell migration and antiapoptotic response to epithelial injury: structure-function analysis of human intestinal trefoil factor. Mol Cell Biol. 2000;20:4680–4690. doi: 10.1128/mcb.20.13.4680-4690.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Debata PR, Panda H, Supakar PC. Altered expression of trefoil factor 3 and cathepsin L gene in rat kidney during aging. Biogerontology. 2007;8:25–30. doi: 10.1007/s10522-006-9032-z. [DOI] [PubMed] [Google Scholar]
- 69.Yu Y, Jin H, Holder D, Ozer JS, Villarreal S, Shughrue P, Shi S, Figueroa DJ, Clouse H, Su M, Muniappa N, Troth SP, Bailey W, Seng J, Aslamkhan AG, Thudium D, Sistare FD, Gerhold DL. Urinary biomarkers trefoil factor 3 and albumin enable early detection of kidney tubular injury. Nat Biotechnol. 2010;28:470–477. doi: 10.1038/nbt.1624. [DOI] [PubMed] [Google Scholar]
- 70.Astor BC, Kottgen A, Hwang SJ, Bhavsar N, Fox CS, Coresh J. Trefoil factor 3 predicts incident chronic kidney disease: a case-control study nested within the Atherosclerosis Risk in Communities (ARIC) study. Am J Nephrol. 2011;34:291–297. doi: 10.1159/000330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prozialeck WC, Edwards JR. Early biomarkers of cadmium exposure and nephrotoxicity. Biometals. 2010;23:793–809. doi: 10.1007/s10534-010-9288-2. [DOI] [PubMed] [Google Scholar]
- 72.Fujita H, Narita T, Morii T, Shimotomai T, Yoshioka N, Kakei M, Ito S. Increased urinary excretion of N-acetylglucosaminidase in subjects with impaired glucose tolerance. Ren Fail. 2002;24:69–75. doi: 10.1081/jdi-120002662. [DOI] [PubMed] [Google Scholar]