

Abstract
Aim
The principal study objective was to investigate the pharmacokinetic characteristics of a new sublingual ketamine wafer and to establish its absolute bioavailability and local tolerability.
Methods
The study was of open label, two way randomized crossover design in eight healthy male volunteers. Each participant received either a single 10 mg intravenous dose as a constant rate 30 min infusion or a 25 mg sublingual dose of ketamine wafer in two treatment periods with a 7 day wash out. Pharmacokinetic blood sampling and local tolerability and safety assessments were carried out during 24 h following both dosing occasions. Plasma concentrations were analyzed by non-compartmental methods and local tolerability was assessed using modified Likert scales.
Results
The median (90% CI lower, upper limit) absolute bioavailability of sublingual ketamine was 29% (27, 31%). The first quantifiable plasma ketamine concentration was observed within 5 min for all eight participants for both routes of administration and the median (min–max) time of the peak plasma concentration was 0.75 h (0.25–1.0 h) after sublingual administration. The ketamine wafer had very good local tolerability.
Conclusion
Sublingual administration of the ketamine wafer resulted in rapid absorption. The ketamine wafer has comparable bioavailability with other oral transmucosal formulations of ketamine but with markedly reduced inter-subject variability, warranting further evaluation as an analgesic adjunct.
Keywords: bioavailability, ketamine, pharmacokinetics, sublingual
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Ketamine is used as an analgesic adjuvant. Non-injected formulations such as via the oral or sublingual routes have low and variable bioavailability which require care and titration in use.
WHAT THIS STUDY ADDS
A novel formulation of sublingual ketamine has been developed, which applies the drug in the sublingual space for a few minutes. Bioavailability was comparable with other non-injected formulations but with considerably lower inter-subject variability which makes it attractive for further clinical development.
Introduction
Ketamine is a general anaesthetic licensed for use by the intravenous (i.v.) route and has been in clinical practice for over four decades. In recent years there has been increasing interest in its use at non-anaesthetic low doses as an adjunct in acute and chronic pain management 1–5. Its pain modifying properties are attributed to its antagonism at N-methyl-D-aspartate (NMDA) receptors, binding non-competitively to the phencyclidine binding site 6,7. When administered at sub-anaesthetic doses ketamine is effective at producing analgesia and also demonstrates some opioid sparing activity, although the mechanisms behind this remain poorly understood 8. Ketamine's analgesic efficacy correlates well with its inhibition of NMDA receptor-mediated pain facilitation and a decrease in activity of brain structures that respond to noxious stimuli 9. Therefore its utility in the management of acute pain is of interest 10–12.
The licensed formulation of ketamine is a racemic mixture of two enantiomers R-(–) and S-(+) of which the S-(+) enantiomer is four times more potent than the R-(–) enantiomer in humans when administered via the parenteral route 13–16. Metabolism after parenteral administration is extensive and rapid, and is mediated by various isoforms of cytochrome P450, specifically CYP3A4 and CYP2B6 17,18. Norketamine, a major metabolite, also has NMDA antagonist properties, although due to differences in potency and pharmacokinetics, it plays a minor role in overall drug action when ketamine is administered by the i.v. route but not necessarily by the oral route 19,20.
Because of high hepatic first pass metabolism, oral formulations of ketamine have low bioavailability with higher norketamine/ketamine plasma area under the curve (AUC) ratios than after i.v. administration 20,21. When administered sublingually (SL) as a liquid formulation or as a tablet, the AUCs were comparable 21 or about 50% higher 20 than after oral administration, suggesting that the SL formulations were largely swallowed. Recently, a novel rapidly dissolving SL wafer formulation has been developed. By releasing the drug in a small volume immediately adjacent to the mucosal membranes, there is the possibility of significant direct SL absorption with higher bioavailability than other oral formulations. The primary aim of this study was to assess the absolute bioavailablity of a single 25 mg SL dose of racemic ketamine administered as a wafer formulation to healthy male volunteers. A formulation that does not require i.v. administration may be of use as an adjunct in both acute and chronic pain management. The pharmacokinetic characteristics and local tolerability of the novel wafer formulation were also assessed.
Methods
The study was approved by the Royal Adelaide Hospital Human Research Ethics Committee and was registered with the Australian Therapeutic Goods Administration under the Clinical Trial Notification scheme and with the Australian and New Zealand Clinical Trials Registry (Number: 2011/0292). The study was conducted in accordance with the principles of the Declaration of Helsinki 22 and Good Clinical Practice Guidelines 23.
Design
The study was of open label two way randomized, crossover design in eight healthy male volunteers who all gave written informed consent. Each participant received either a single 10 mg i.v. dose as a constant rate 30 min infusion or a 25 mg SL dose of ketamine in two treatment periods with a 7 day washout. Both the SL and i.v. doses, and the duration of the i.v. infusion were chosen to ensure adequate characterization of the plasma concentration–time profiles and good quality estimates of pharmacokinetic (PK) variables for both routes of administration. The i.v. dose of 10 mg has been used in similar studies and has been well tolerated. Bioavailability values of 24–32.2% have been reported in the literature for sublingually administered ketamine. Even if the bioavailability of the wafer formulation was higher, a 25 mg dose was not expected to show a systemic tolerability markedly different from that of the i.v. dose. The sequence of the two formulations was according to a computer-generated randomization code.
Clinical
The SL wafer formulation was a freeze dried solid dispersion of racemic ketamine hydrochloride in a porous matrix using lactose as a filling agent. Prior to administration of the wafer the sublingual space was rinsed with 3 ml of water after which the wafer was placed sublingually by a member of the study staff. The participants were instructed to avoid chewing or swallowing of the wafer within 5 min of its placement. For i.v. administration, commercially available ketamine (Ketalar®) was diluted to 30 ml in saline and administered over 30 min using a volumetrically controlled syringe driver. The infusion line was primed prior to start of the infusion.
Pharmacokinetic blood sampling and clinical assesment of local tolerability and safety were carried out for 24 h following both dosing occasions.
Key inclusion criteria were healthy adult males aged 18–65 years with a BMI 19–30 kg m−2 in good general health including mental health as assessed by the Symptom Checklist-90-R (SCL-90-R®), a screening instrument which evaluates a broad range of psychological problems and symptoms of psychopathology.
Pharmacokinetic blood samples (5 ml), were taken following both i.v. and SL administration at predose 5, 10, 15, 30, 35 and 45 min, and at 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 8.0, 12 and 24 h post-dose.
Whole blood was drawn into prechilled lithium heparin tubes and remained on ice post-sample collection until centrifugation. Samples were centrifuged at 1800 g for 10 min in a refrigerated centrifuge at 4°C. Plasma was decanted and frozen at −80°C.
To assess the local tolerability profile of the SL formulation, modified Likert scales (0–10) were recorded at 5, 10, 15, 30 and 45 min and 1 h post-dose administration at various time points for both the SL and i.v. formulation:
Mucosal irritation
Burning sensation
Bitterness
Nausea
Residual grittiness in the mouth
Safety assessments included scheduled adverse event probes, spontaneous adverse event (AE) reporting, physical examination, routine laboratory investigations, ECGs and vital sign evaluation.
Vital signs (including systolic and diastolic blood pressure, pulse, respiratory rate and body temperature) were performed predose and at hours 0.5, 1, 2, 4, 6, 8, 12 and 24 h post-dose. Pulse oximetry was recorded predose and continuously for the first 3 h post-dose administration.
Laboratory
Safety laboratory testing (biochemistry, haematology and urinalysis) was performed predose and at hour 24 post-dose administration in each period.
Quantification of the plasma concentrations of racemic ketamine was performed using a validated HPLC method with u.v. detection, a lower limit of quantification (LLOQ) of 2 ng ml−1 and <20% bias and imprecision 24.
Data analysis
Standard non-compartmental methods using the PK Solver plug-in for Microsoft Excel were used to derive pharmacokinetic variables, except for Cmax, tmax and tlast, which were taken as observations from the plasma concentration–time profile of each participant. Actual times were used when reporting tmax. The terminal rate constant (λz) was estimated by log-linear regression, of the slope of the natural log plasma concentration vs. time curve where λz = −1 x slope. The linear regression in the terminal phase used the last three to six data points. The terminal t1/2 was calculated as t1/2 = ln(2)/λz.
The area under the plasma concentration time curve from time zero to the last quantifiable concentration (AUC(0,tlast)) was obtained using the linear trapezoidal method and extrapolated to infinity to obtain the total area, AUC(0,∞), with Clast/λz, where Clast is the last quantifiable plasma concentration. The AUCextr (extrapolated portion of AUC(0,∞)) was calculated as (1 − AUC(0,tlast)/AUC(0,∞) x 100. For the i.v. dose, clearance (CL) was calculated as dose/AUC(0.∞) and Vz was calculated as CL/λz. The bioavailability (F) of ketamine was calculated as the ratio of the dose adjusted AUC(0,∞) following i.v. and SL dosing according to AUC(0,∞)(SL)/AUC(0,∞)(i.v.) x dosei.v./doseSL.
Results
Eight healthy male volunteers of mean (SD) 25 (7.6) years and BMI 26.1 (2.83) kg m−2 took part in the study.
The individual and mean plasma concentration profiles are shown graphically for i.v. and SL administration in Figures 1 and 2, respectively. The mean profiles for i.v. and SL are shown in Figure 3. The pharmacokinetic results are provided in Table 1. In all participants and for both administration routes, the first quantifiable ketamine plasma concentrations were observed at the first post-dose sample at 5 min. The SL plasma concentration profiles showed minor fluctuations in a few participants. In one participant three comparable peaks were observed during the first 1.5 h following SL administration, although no noticable difference in PK characteristics could be observed in comparison with the other participants. Following the Cmax, concentrations declined biphasically for both i.v. and SL with the trend being more prominent for i.v. Peak plasma concentrations following the i.v. infusion occurred at the end of the infusion in all but one participant, where the peak occurred 5 min after the end of the infusion. For the SL formulation, peak plasma concentrations were observed between 0.25 and 1 h, with a median tmax of 0.75 h. In one participant the dissolution time of the wafer was noticably longer, 6 min, than the 30–60 s noted in all other participants. The same participant showed among the highest scores for ‘residual grittiness’ during the first 30 min after dosing, but scores had returned to 1 at 45 min and to baseline values at 60 min post-dose. The longer dissolution time did not translate into generally differing PK or systemic tolerability characteristics of ketamine in this participant. The cause of the prolonged dissolution time is unknown. Plasma concentrations were below the LLOQ in six participants at 24 h and in one participant at 12 h following SL dosing. Following i.v. dosing, all participants had quantifiable levels at 12 h and four participants at 24 h. The median (min–max) terminal half-lives for i.v. and SL were comparable at 4.5 (2.5–7.0) h and 3.4 (1.8–5.5.) h, respectively. The extrapolated portion of the AUC(0,∞) was very small for both routes of administration with min–max of 3–7% for i.v. and 2–9% for SL dosing. The median (lower, upper 90% CI limit) for the bioavailability of the wafer was 29 (27, 31) % showing very low inter-subject variability. The participant who had the highest bioavailability, 38%, also had the highest clearance, 59.8 l h−1.
Figure 1.
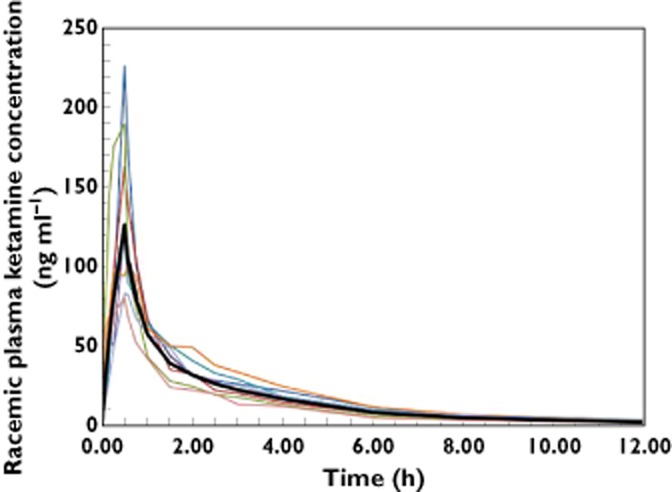
Individual racemic ketamine plasma concentration–time curves and geometric mean (bold line) during the first 12 h following a 10 mg dose given during a 30 min i.v. infusion to eight healthy volunteers
Figure 2.
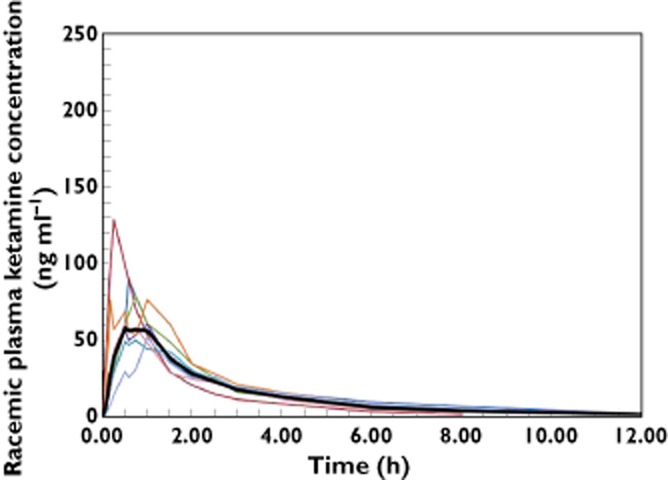
Individual racemic ketamine plasma concentration–time profiles and geometric mean (bold line) during the first 12 h following a 25 mg sublingual dose to eight healthy volunteers
Figure 3.
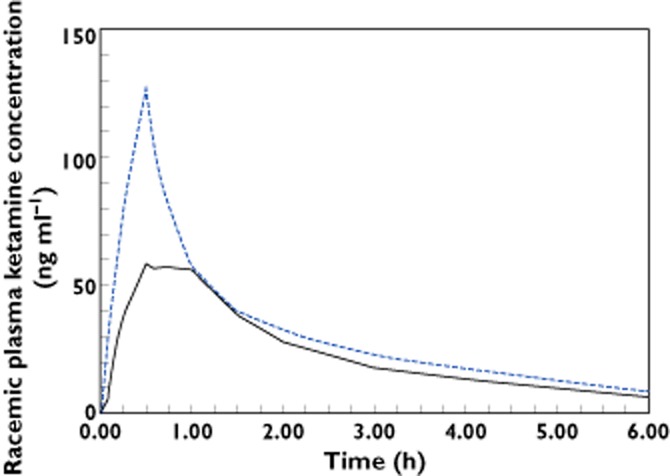
Geometric mean racemic ketamine plasma concentration–time profiles during the first 6 h following sublingual administration of 25 mg (continuous line) and 10 mg as a 30 min i.v. infusion (dashed line) to eight healthy volunteers
Table 1.
Individual pharmacokinetic variables and summary statistics of RS ketamine following administration of 10 mg as a 30 min i.v. infusion and 25 mg sublingually as a wafer to eight healthy volunteers
| Subject | Cmax,i.v. (ng ml−1) | Cmax,SL (ng ml−1) | tmax,SL (h) | AUC(0,∞)i.v. (ng ml−1 h) | AUC(0,∞)SL (ng ml−1 h) | CL (l h−1) | Vz (l) | t1/2,i.v. (h) | t1/2,SL (h) | F (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 226.7 | 88.8 | 0.58 | 282.7 | 202.9 | 35.4 | 126 | 2.5 | 2.9 | 28 |
| 2 | 163.3 | 128.3 | 0.25 | 243.2 | 162.5 | 41.1 | 158 | 2.7 | 1.8 | 27 |
| 3 | 190.3 | 78.7 | 0.75 | 254.6 | 184.3 | 39.3 | 283 | 5.0 | 3.2 | 29 |
| 4 | 124.2 | 60.2 | 1 | 270.0 | 203.5 | 37.0 | 253 | 4.7 | 5.5 | 30 |
| 5 | 120.4 | 50.0 | 0.75 | 289.2 | 171.9 | 34.6 | 300 | 6.0 | 3.5 | 23 |
| 6 | 101.9 | 76.1 | 1 | 299.4 | 211.3 | 33.4 | 164 | 3.4 | 2.3 | 29 |
| 7 | 83.2 | 51.8 | 1 | 261.0 | 186.1 | 38.3 | 385 | 7.0 | 4.6 | 29 |
| 8 | 81.1 | 61.2 | 0.5 | 167.2 | 161.6 | 59.8 | 375 | 4.3 | 5.1 | 38 |
| Gmean* | 128.1 | 71.1 | 0.75 | 255.0 | 184.6 | 39.2 | 237 | 4.5 | 3.4 | 29 |
| Min–max | 81.1–226.7 | 50.0–128.3 | 0.25-1 | 167.2–299.4 | 161.6–211.3 | 33.4–59.8 | 126–385 | 2.5–7.0 | 1.8–5.5 | 23–38 |
| CV (%) | 16 | 14 | 21 | 8 | 4 | 8 | 18 | 16 | 17 | 6 |
| 90% CI† | 27, 31 |
Cmax, peak plasma concentration; tmax, time of Cmax; AUC(0,∞), area under the plasma concentration–time curve from time zero to infinity; CL, clearance following i.v. administration; Vz, apparent volume of distribution following i.v. administration; t1/2, terminal half-life; F, bioavailability; NA, Not applicable; SL, sublingual.
Gmean is provided for all variables except for bioavailability, tmax and t1/2 where medians are shown.
90% confidence interval (lower, upper).
Nineteen adverse events thought to be related to treatment were reported. Most were expected CNS-type effects typical of ketamine: light headed (n = 1 for i.v. and n = 3 for SL), hazy (i.v. n = 2), numbness in mouth and/or face (i.v. n = 5, SL n = 1), and one each of body feels heavy, dry mouth and visual disturbance for i.v., and for SL one each of terrible taste in mouth, blurred vision, decreased sensation in arm and dizziness, respectively. The onset was comparable for the two routes of administration, being 6–22 min for i.v. and 5–18 min for SL dosing. All AEs were mild and had a short duration of less than 1 h with only three AEs ‘possibly’ or ‘probably’ related to treatment lasting over 30 min. There were no serious adverse events. Local tolerability of the SL formulation was excellent with transient bitterness the only effect of note.
Discussion
In this study the pharmacokinetic characteristics and absolute bioavailability of a novel SL wafer formulation of racemic ketamine were determined, and the local tolerability was assessed. A majority of the adverse events were typical CNS effects of ketamine, and were more frequently observed for the i.v. dose, which is likely due to the higher plasma concentrations achieved in comparison with the SL dose. However, all AEs were mild, resolved within 1 h and both the local and systemic tolerability was very good for both routes of administration. The extrapolated portion of the AUC(0,∞) was very small in all participants, indicating high quality in the estimates of AUC and hence bioavailability. The dissolution and subsequent absorption following SL administration was rapid, as shown by the early quantifiable plasma concentrations. The similar terminal half-lives across dosing routes confirmed that absorption was rapid and not rate limiting for the elimination. The early tmax was also indicative of fast absorption, in the light of the similar terminal half-life values across dosing routes. The tmax was comparable with previously reported values for SL administration of ketamine, with a median (min–max) tmax of 0.75 h (0.25–1 h) in the present study, a median (interquartile range) of 0.5 h (0.3–0.8 h) for a lozenge 21 and a mean (SD) of 40 (20) min for a tablet formulation 20. The median bioavailability at 29% was also very similar to that observed for the lozenge formulation, median of 24% 21 and tablet, mean of 32.2% 20. However what differed markedly with the novel wafer formulation compared with formulations presented in previous studies was that the between subject variability in bioavailability was noticeably lower. The 90% CI was over a very narrow range of 27–31%, in comparison with an interquartile range of 19–49% for the lozenge 21 and a standard deviation of 8.2% for the SL tablet 20. It should be noted that the variability estimates for all three formulations have been derived from a small number of subjects with three healthy volunteers for the SL tablet 20, 10 patients for the lozenge 21 and eight volunteers in the present trial. The low inter-subject variability in bioavailability of the novel wafer might be due to the formulation delivering a more controlled release of drug into the sublingual space than a SL lozenge 21 or tablet 20. The inter-variability estimate for the novel wafer formulation will require confirmation in future trials in a larger number of subjects. In the context of a narrow therapeutic index drug such as ketamine, reliable and consistent delivery is particularly important and hence the low variability in bioavailability makes the new wafer formulation especially attractive for further evaluation as an analgesic adjunct.
In conclusion the clinical safety and tolerability of ketamine and the adverse event profile was as expected for the dose levels used and prevailing clinical experience and mild and transient local effects were seen. The bioavailability of ketamine in the novel SL wafer formulation was comparable with previously reported SL formulations and in addition promises a very low inter-subject variability. In view of ketamine's relatively narrow therapeutic index, low variability is appealing as it signifies reproducible exposure and consequently clinical effect.
Acknowledgments
We would like to thank the staff of the Pain and Anaesthesia Research Clinic, University of Adelaide, for conducting the study.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare PR, SL, VS, YL and VM had support from iX Biopharma for the submitted work, PR and VS are directors of iX Biopharma in the previous 3 years and there are no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Niesters M, Martini C, Dahan A. Ketamine for chronic pain: risks and benefits. Br J Clin Pharmacol. 2014;77:357–367. doi: 10.1111/bcp.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elia N, Tramèr MR. Ketamine and postoperative pain – a quantitative systematic review of randomised trials. Pain. 2005;113:61–70. doi: 10.1016/j.pain.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 3.Bell RF, Dahl JB, Moore RA, Kalso EA. Perioperative ketamine for acute postoperative pain. Cochrane Database Syst Rev. 2006;(1) doi: 10.1002/14651858.CD004603.pub2. CD004603. doi: 10.1002/14651858.CD004603.pub2. [DOI] [PubMed] [Google Scholar]
- 4.Persson J. Ketamine in pain management. CNS Neurosci Ther. 2013;19:396–402. doi: 10.1111/cns.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visser E, Schug SA. The role of ketamine in pain management. Biomed Pharmacother. 2006;60:341–348. doi: 10.1016/j.biopha.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Martin D, Lodge D. Ketamine acts as a non-competitive N-methyl-D-aspartate antagonist on frog spinal cord in vitro. Neuropharmacology. 1985;24:999–1003. doi: 10.1016/0028-3908(85)90128-5. [DOI] [PubMed] [Google Scholar]
- 7.Petrenko AB, Yamakura T, Askalany AR, Kohno T, Sakimura K, Baba H. Effects of ketamine on acute somatic nociception in wild-type and N-methyl-D-aspartate (NMDA) receptor e1 subunit knockout mice. Neuropharmacology. 2006;50:741–747. doi: 10.1016/j.neuropharm.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Munoz M, Sanchez-Blazquez P, Vicente-Sanchez A, Berrocoso E, Garzon J. The mu-opioid receptor and the NMDA receptor associate in PAG neurons: implications in pain control. Neuropsychopharmacology. 2012;37:338–349. doi: 10.1038/npp.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simoni SD, Schwarz AJ, O'Daly OG, Marquand AF, Brittain C, Gonzales C, Stephenson S, Williams SCR, Mehta MA. Test–retest reliability of the BOLD pharmacological MRI response to ketamine in healthy volunteers. NeuroImage. 2013;64:75–90. doi: 10.1016/j.neuroimage.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 10.Jennings PA, Cameron P, Bernard S. Ketamine as an analgesic in the pre-hospital setting: a systematic review. Acta Anaesthesiol Scand. 2011;55:638–643. doi: 10.1111/j.1399-6576.2011.02446.x. [DOI] [PubMed] [Google Scholar]
- 11.Lui F, Ng KFJ. Adjuvant analgesics in acute pain. Expert Opin Pharmacother. 2011;12:363–385. doi: 10.1517/14656566.2011.521743. [DOI] [PubMed] [Google Scholar]
- 12.Carr DB, Goudas LC, Denman WT, Brookoff D, Staats PS, Brennen L, Green G, Albin R, Hamilton D, Rogers MC, Firestone L, Lavin PT, Mermelstein F. Safety and efficacy of intranasal ketamine for the treatment of breakthrough pain in patients with chronic pain: a randomized, double-blind, placebo-controlled, crossover study. Pain. 2004;108:17–27. doi: 10.1016/j.pain.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Ryder S, Way WL, Trevor AJ. Comparative pharmacology of the optical isomers of ketamine. Eur J Pharmacol. 1978;49:15–23. doi: 10.1016/0014-2999(78)90217-0. [DOI] [PubMed] [Google Scholar]
- 14.Henthorn TK, Krejcie TC, Niemann CU, Enders-Klein C, Shanks CA, Avram MJ. Ketamine distribution described by a recirculatory pharmacokinetic model is not stereoselective. Anesthesiology. 1999;91:1733–1743. doi: 10.1097/00000542-199912000-00027. [DOI] [PubMed] [Google Scholar]
- 15.Geisslinger G, Hering W, Thomann P, Knoll R, Kamp HD, Brune K. Pharmacokinetics and pharmacodynamics of ketamine enantiomers in surgical patients using a stereoselective analytical method. Br J Anaesth. 1993;70:666–671. doi: 10.1093/bja/70.6.666. [DOI] [PubMed] [Google Scholar]
- 16.Sigtermans M, Dahan A, Mooren R, Bauer M, Kest B, Sarton E. S(+)-ketamine effect on experimental pain and cardiac output. Anesthesiology. 2009;111:892–903. doi: 10.1097/ALN.0b013e3181b437b1. [DOI] [PubMed] [Google Scholar]
- 17.Kharasch ED, Labroo R. Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology. 1992;77:1201–1207. doi: 10.1097/00000542-199212000-00022. [DOI] [PubMed] [Google Scholar]
- 18.Hijazi Y, Boulieu R. Contribution of CYP3A4, CP2B6 and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–858. doi: 10.1124/dmd.30.7.853. [DOI] [PubMed] [Google Scholar]
- 19.Olofsen E, Noppers I, Niesters M, Kharasch E, Aarts L, Sarton E, Dahan A. Estimation of the contribution of norketamine to ketamine-induced acute pain relief and neurocognitive impairment in healthy volunteers. Anesthesiology. 2012;117:353–364. doi: 10.1097/ALN.0b013e31825b6c91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanagihara Y, Ohtani M Kariya S, Uchino K, Hiraishi T, Ashizawa N, Aoyama T, Yamamura Y, Yamada Y, Iga T. Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharm Drug Dispos. 2003;24:37–43. doi: 10.1002/bdd.336. [DOI] [PubMed] [Google Scholar]
- 21.Chong C, Schug SA, Page-Sharp M, Jenkins B, Illett KF. Development of a sublingual/oral formulation of ketamine for use in neuropathic pain. Clin Drug Investig. 2009;29:317–324. doi: 10.2165/00044011-200929050-00004. [DOI] [PubMed] [Google Scholar]
- 22.World Medical Association (WMA) Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. Adopted by the 18th WMA General Assembly, Helsinki, June 1964, and amended by the 52nd WMA General Assembly, Edinburgh October 7, 2000. Available at http://www.wma.net/en/30publications/10policies/b3/index.html (last accessed 20 September 2007)
- 23.European Agency for the Evaluation of Medicinal Products (EMEA) International Conference on Harmonisation–World Health Organization. Guideline for Good Clinical Practice. ICH topic E 6 (R1). Available at http://www.ema.europa.eu/pdfs/human/ich/013595en.pdf (last accessed 1 December 2009)
- 24.Menelaou A, Doverty M, Somogyi AA. Development of a sensitive assay for the quantification of (S)-ketamine in plasma using HPLC with UV detection. Proc Aust Soc Clin Exp Pharmacol Toxicol. 2001;9:82. [Google Scholar]