

Abstract
Structural biology provides essential information for elucidating molecular mechanisms that underlie biological function. Advances in hardware, sample preparation, experimental methods, and computational approaches now enable structural analysis of protein complexes with increasing complexity that more closely represent biologically entities in the cellular environment. Integrated multidisciplinary approaches are required to overcome limitations of individual methods and take advantage of complementary aspects provided by different structural biology techniques. Although X-ray crystallography remains the method of choice for structural analysis of large complexes, crystallization of flexible systems is often difficult and does typically not provide insights into conformational dynamics present in solution. Nuclear magnetic resonance spectroscopy (NMR) is well-suited to study dynamics at picosecond to second time scales, and to map binding interfaces even of large systems at residue resolution but suffers from poor sensitivity with increasing molecular weight. Small angle scattering (SAS) methods provide low resolution information in solution and can characterize dynamics and conformational equilibria complementary to crystallography and NMR. The combination of NMR, crystallography, and SAS is, thus, very useful for analysis of the structure and conformational dynamics of (large) protein complexes in solution. In high molecular weight systems, where NMR data are often sparse, SAS provides additional structural information and can differentiate between NMR-derived models. Scattering data can also validate the solution conformation of a crystal structure and indicate the presence of conformational equilibria. Here, we review current state-of-the-art approaches for combining NMR, crystallography, and SAS data to characterize protein complexes in solution.
Keywords: nuclear magnetic resonance spectroscopy, small angle X-ray scattering, small angle neutron scattering, structural biology, multidomain proteins, protein complexes
Introduction
Recent technological advances in structural biology, for example, intense synchrotron radiation, improved detectors, ultrahigh-field nuclear magnetic resonance spectroscopy (NMR) magnets, cryogenic NMR probes, direct electron detectors, and improved computational tools have been driven by the challenges imposed by understanding ever more complex systems that mediate cellular biological activity. To understand how these protein complexes fulfill their critical roles in cellular pathways and their relation with disease high-resolution structural information is required. In eukaryotic systems, proteins are often comprised of several (structurally independent) domains connected by flexible linkers that ensure conformational flexibility and adaptability for molecular interactions.1 The inherent conformational dynamics associated with such proteins poses a challenge for structural analysis using X-ray crystallography. Crystallography is powerful in providing precise structural information for protein assemblies of stunning complexity, such as the ribosome,2,3 exosome,4 fatty acid synthase,5 RNA polymerase and its interactions,6–9 Argonaute proteins,10–12 the proteasome13–15 to name a few. However, the pictures obtained are static snapshots and crystallography can often not unravel mechanistic features relating to the intrinsic dynamics of these complexes, which is intimately linked with their functional activity (e.g., 7). Structures of large systems, where the presence of flexible and intrinsically disordered regions (IDRs) renders crystallization difficult, can be efficiently studied using cryo-electron microscopy (EM). Recent advances in electron technology, that is, direct electron detectors, now increase the resolution of EM reconstructions to 3–4 Å in favorable cases.16–19 Moreover, large domain or subunit conformational variability and/or dynamics can be detected.20 Nevertheless, flexible and disordered regions and conformational flexibility at the level of secondary structure elements and smaller domains escapes EM detection.
NMR has contributed greatly to understanding dynamics of large systems21–23 due to recent methodological advances for optimized isotope labeling and deuteration24–26 and NMR methods.27–33 With increasing flexibility and specifically when studying intrinsically disordered proteins (IDPs), solution NMR is the only method available to obtain high-resolution information on transient structures, conformational dynamics at various time scales, and weak molecular interactions, for example, 34–40, and see later.
With increasing molecular weight, NMR data are more and more difficult to obtain. Here, small angle scattering (SAS) can provide additional restraints for NMR-based structure calculations or for the validation of structural models derived from sparse (NMR) data. The combination of SAS and NMR is especially fruitful for studying large protein complexes in solution. In this review, we aim to outline and illustrate the power of integrated structural biology approaches specifically the combination of NMR spectroscopy and SAS experiments in the study of complex biological systems.
Structural Information Provided from NMR Spectroscopy and SAS
A comprehensive review of NMR methodology and SAS techniques is beyond the scope of this review. Therefore, recent advances will be briefly summarized focusing on the complementarity of the two methods, while for more detailed overviews on NMR and SAS we refer to recent reviews.41–48 Advances in NMR to study large systems have been tremendous in the last decade. The intrinsic low sensitivity of NMR has several reasons. With increasing molecular weight of a biomacromolecule, its molecular tumbling in solution slows down. As a result transverse relaxation rates, which are inverse proportional to the NMR line widths increase and, thus, the signal-to-noise at the maximum signal intensity is reduced. This sensitivity problem is addressed by several means. (i) Optimized isotope labeling and deuteration strategies reduce the amount of protons and, thus, relaxation pathways thus reducing the efficiency of relaxation (and thus the signal-to-noise).25,26 (ii) Transverse relaxation optimized pulse sequences reduce relaxation especially for large deuterated proteins.27,31,33,49 (iii) The introduction of residual dipolar couplings (RDCs) to determine structure and dynamics of proteins, their larger complexes and IDPs.50–52 (iv) The use of paramagnetic effects from spin labeling or lanthanide binding tags (LBTs) to obtain long-range distance restraints.53–58 (v) Developments in computational methods for structure calculation improves reliability of structural models of proteins and complexes, especially if experimental data are sparse.51,57,59–61 Efficient tools are available for data-driven docking of proteins and nucleic acids (e.g., HADDOCK62), and for de novo structure calculation of proteins with sparse NMR data, for example, CS-ROSETTA.63,64 In short, biological NMR spectroscopy has seen dramatic methodological advances and now also contributes to structural biology of large complexes.
In the last 2 decades, SAS has become a major method in structural biology due to great advances in hardware, methods, and efficient software tools for data analysis and modeling. Provided monodispersity of a sample measured SAS can provide information for the overall shape of a macromolecule or assembly in solution, independent of size, and composition. Small angle neutron scattering (SANS)65,66 especially in combination with contrast variation and perdeuteration of selected components of a complex can reveal the position of each subunit within a complex as has been demonstrated already 25 years ago with the ribosome of Escherichia coli.67,68 Allosteric conformational changes can be studied in detail and the complementarity of SAS data to crystallographic structures is well established.44 SAS data acquisition and analysis has been made accessible to users due to continuous improvement of analysis software69 and the availability of small angle X-ray scattering (SAXS) at synchrotron beam lines and home sources. Technical advances enabled high-throughput measurements70 and lead to on-line size-exclusion-chromatography-multiple-angle-laser-light-scattering-SAXS set-ups at synchrotron beamlines, for example, at ESRF (Grenoble, France) and EMBL (Hamburg, Germany). As data processing and interpretation is critical and structural information derived from SAXS data is highly ambiguous, guidelines for the presentation, and validation of SAS data have been established, similar to what has been implemented by crystallography and NMR communities.71–73 Furthermore, efforts have been made to reduce the user-bias during SAXS data processing and SAXS-based modeling.74,75
Combining NMR and SAS to Study Protein Complexes
The complementarity of NMR and SAS to study (large) protein complexes in solution is well-recognized. Where NMR faces size limitations and data become sparse with increasing molecular weight it can still provide high-resolution information from paramagnetic and orientational restraints to define domain and subunit arrangements. SAS has no such size limitations and yields low resolution structural information and subunit arrangements (Fig. 1). Observables that are readily accessible by NMR also for high molecular weight complexes are chemical shifts for binding interface mapping, RDCs to define domain/subunit orientations, and long-range distance restraints from spin labels covalently attached to a cysteine (e.g., a nitroxyl spin label can attenuate NMR signals for spins up to 20–25 Å away from the paramagnetic center53) or from paramagnetic cosolutes to identify binding interfaces shielded from solvent55,76,77 (Fig. 1).
Figure 1.
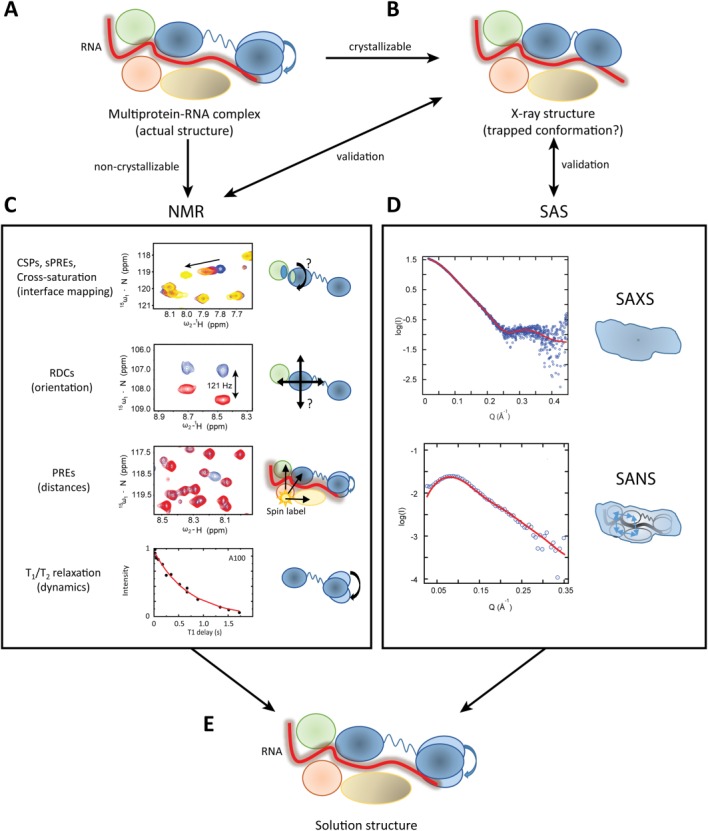
Complementarity of NMR and SAS illustrated with a multiprotein RNA complex. (A) The example shown assumes a complex comprised of five macromolecules, namely monomeric proteins (green, pink, and brown), a protein with two domains connected by a flexible linker (blue), and one RNA molecule (red). (B) Crystallization of the complex may yield a high resolution structure, which, however, may represent a non-native conformation stabilized in the crystal (indicated by the lack of domain motion in the blue protein). NMR together with SAXS and SANS data can provide the conformation and domain arrangements in solution structure by combining high-resolution information available for individual domains with NMR and SAS data. (C) NMR CSPs and cross saturation experiments map binding interface between components, solvent PRE (sPRE) data identify binding interfaces shielded from solvent in the complex, RDCs help to define the relative orientation of subunit and domains, PREs provide long-range distance information >20Å. NMR relaxation data provide information about internal motion, for example, linker flexibility and domain dynamics in the blue protein. (D) SAXS experiments yield a low resolution envelope of the entire complex, while contrast matching with SANS provides information about placement (center-of-mass distances between subunits) and shapes of subunits within the overall complex). The combination of NMR and SAS data with available high-resolution structural data for subunits and domains defines the native solution structure of the complex.
Binding interface mapping based on chemical shift perturbations (CSPs) exploits the dependence of NMR resonance frequencies on the local electronic environment of the nuclear spins. Chemical shifts are obtained from simple and sensitive NMR experiments, such as heteronuclear 1H,15N or 1H,13C correlation experiments. An 1H,15N correlation experiment, for example, correlates the proton and nitrogen Larmor frequencies for all amide groups along a polypeptide chain and, thus, represents an NMR fingerprint of a protein. When a binding interface interacts with a ligand or another protein, its environment changes and the amide signals of residues located in this region will experience changes in their NMR frequencies, that is, CSPs. In case of allosteric conformational changes, residues remote from the binding site can also exhibit CSPs. Such indirect affects have to be considered, when analyzing CSP data, but they are, conversely, an excellent indicator for the presence of allosteric effects and conformational changes associated with ligand binding. Analyzing CSPs is very sensitive and even with large complexes binding interface mapping can be readily performed. For high molecular weight systems, however, proteins need to be deuterated and methyl groups are monitored as they provide greater sensitivity. CSPs can provide structural models that can be further probed by mutational analysis.78 Especially for larger complexes, SAS data can provide information about the overall shape and the relative positioning of subunits within the overall shape. Here, SANS using subunit-selectively deuterated samples in combination with contrast matching yields valuable structural information66 (Fig. 2). Let us assume a ternary protein–protein-RNA complex of rather large size (e.g., protein A 50 kDa, protein B, 100 kDa, and a small 20 nucleotide single-stranded RNA). SAXS data yields the overall shape of the complex. SANS data of a sample, where protein A is perdeuterated, protein B and RNA protonated yields information about the positioning of protein A, and RNA within the complex when recorded in 42% D2O solution, and of proteins within the complex at 70% D2O. This is due to the fact that the (average) scattering density of a protonated protein matches the scattering density of the buffer at 42% D2O, while the scattering density of RNA matches the buffer at 70% D2O. At 70% D2O, protonated protein A has a positive contrast, whereas protein B has a negative contrast. The measurements should be repeated with a sample with perdeuterated protein B and protonated protein A. The more subunits are present in a complex, the more samples need to be prepared to obtain SANS data for all possible combinations. Note, that deuteration is also required for NMR measurements and, thus, protein complexes comprising subunits with NMR isotope labeling (15N,13C) and deuteration can be directly used as well for SANS measurements. As neutron scattering does not damage the protein samples, they can be subsequently reused for additional NMR measurements. From SAS data alone, an ab initio bead model of the complex can be calculated, where the positions of the individual subunits within the overall shape are provided. However, the orientation of the subunits and their binding interfaces remain elusive. Here, NMR derived CSPs can provide such information (Fig. 1). Contrast matching in SAXS is usually difficult due to the small differences in contrast, although a study has been published recently were this possibility was exploited by incorporating heavy-atom labels in 65% aqueous sucrose buffer into the protein and SAXS data was combined with RDCs.79
Figure 2.
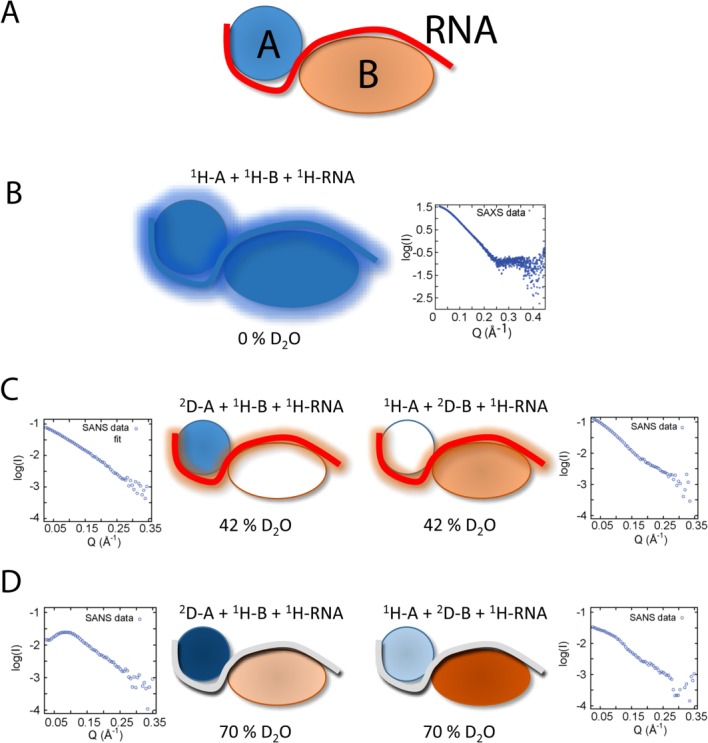
Utility of subunit-selective deuteration of protein complexes with SANS measurements. (A) A ternary complex, consisting of protein A (blue), B (orange), and RNA (red) is assumed. (B) In 0% D2O solution without any deuteration SANS does provide information about the overall shape of the complex, comparable to SAXS (but with lower sensitivity). (C) In 42% D2O solution, deuterated protein components have a positive contrast, while the protonated components match the contrast of the buffer and are “invisible.” Under these conditions the shape and center-of-mass distances between the deuterated component and the RNA can be determined. (D) In 70% D2O solution, the contrast of RNA is matched by the buffer, and the shape and center-of-mass distances between the deuterated components (positive contrast), and protonated components (negative contrast) can be determined.
More precise information can be obtained if CSPs and SAS data are complemented with RDC data, which provide orientational restraints, for example, about relative domain orientations (Fig. 1). RDC data are obtained from weakly aligning a protein in dilute anisotropic phase (e.g. phages, micelles, polyethyleneglycol (PEG) mixtures51,52,80–82), which themselves align in the static magnetic field of the NMR magnet. RDCs are measured as an additional contribution that adds or subtracts to the splitting of NMR signals due to scalar J-couplings in the weakly aligned state. The RDCs yield information about the orientation of a bond vector which reports on the RDC (e.g., HN–N) relative to the principal axes of an alignment tensor, which is obtained and fitted from the experimental RDC data. As all subunits in a rigid complex will have the same alignment tensor, the orientation of the (HN–N) bond vectors in the individual subunits or domains define their relative orientation. Typically at least 20 RDCs are required per domain/subunit to reliably define the relative domain arrangements. Note, however, that this approach assumes the absence of significant domain motion (see later). RDC data from more than one alignment medium are desirable to reduce ambiguities and increase the precision of orientation information. Thus, RDCs are extremely useful and complementary to SAS data.
Long-range distance restraints can be derived from paramagnetic relaxation enhancements (PREs,83–87 Fig. 1).53,57,58,88,89 The side chain thiol group of cysteines can be used to chemically attach a so-called spin label (i.e., paramagnetic compounds with a stabilized electron radical such as a nitroxyl group). The spin label should be switchable between a paramagnetic and a diamagnetic state. In the paramagnetic state, all signals of residues being up to, for example, 20 Å away from the paramagnetic center are attenuated depending on the distance (for a nitroxyl spin label53). Obtaining PRE data from multiple spin labeled samples, where spin labels are attached at different sites are important to obtain reliable structural information. PREs provide a network of long-range distance restraints that allow to define relative domain arrangements [Fig. 3(A)]. For site-specific spin-labeling single-cysteine variants of the protein studied are necessary, where native cysteines are replaced by alanine or serine, while single cysteines are introduced at the surface of the protein to avoid any effects on the structural integrity of the protein. The site of attachment for the spin label is critical, as it should neither be placed too close to an (expected) binding interface, as it can interfere with complex formation, nor should it be too far away, in which case no interdomain distances are obtained [Fig. 3(B)]. Preliminary models obtained, for example, from combining CSPs with HADDOCK modeling may minimize these risks and support a rational design of spin labels possible. Especially, the use of methyl TROSY experiments31,90 allows the measurement of PRE data even for large complexes.
Figure 3.
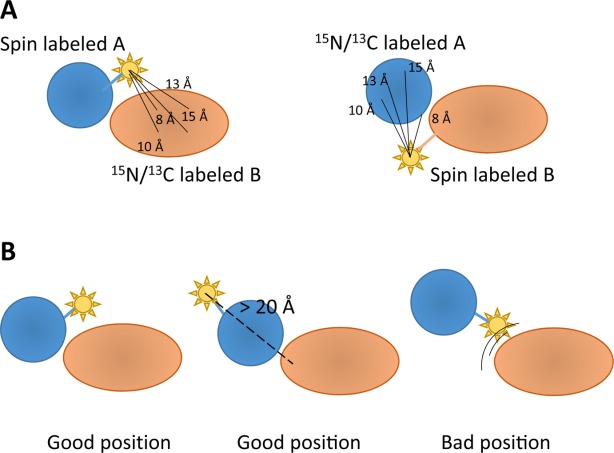
Utility of spin labeling to map interdomain distances from PREs. (A) If protein A is spin labeled (i.e., with a nitroxyl group), PREs lead to an attenuation of NMR signals in the paramagnetic state, which can be detected for the isotope (15N and/or 13C)-labeled protein B for distances up to 20–23 Å. Inverting spin labeling and isotope labeling yields complementary information (right). (B) Placement of the spin label is critical. It is important to place the spin label close to the interface to maximize the number of interdomain PRE effects. However, if the spin label is placed within interface it may interfere with complex formation. Spin labeling at sites remote from the interface will not yield many interdomain PREs, but information from this spin label will still provide useful information as “repelling” restraints, as the lack of any observable PRE indicates that any spin in protein B must be >20 Å away from the paramagnetic center in protein A.
Another strategy to obtain long-range distance restraints is by LBTs, which provide not only PRE effects but also additional orientational restraints, from RDCs and pseudocontact shifts.56,91 The most straightforward approach is for metal-binding proteins, where the native metal ion can be exchanged with the lanthanide.92 It is, however, more challenging for nonmetal binding proteins, where different strategies to introduce an LBT have been proposed and successfully tested (e.g., artificial metal binding motifs in native protein scaffolds,93 coexpressed tags,94 engineering encodable tags into loop regions of proteins,95 or using CLaNP chelators96).
Structural information for protein complex derived from paramagnetic spin labeling or LBTs for one binding partner of the complex are calibrated and validated by analyzing intradomain effects, that is, monitoring PREs induced by the spin label to the NMR signals in the same domain or subunit which is spin labeled. Also, considering that the spin label is often not rigidly attached and the cysteine side chain where it is attached to may exhibit conformational dynamics, the paramagnetic effects and restraints derived thereof are considered as averages of a conformational ensemble. To derive distance restraints from PRE data the internal correlation time of the electron-nuclear spin vectors needs to be known, while the internal flexibility of the spin label should be taken into account in structure calculations by using ensemble-averaged restraints.58 Note, that the use of PREs to define domain arrangements assumes the presence of a rigid and compact structure, that is, the absence of substantial domain motions, which may modulate the PREs. The presence or absence of such motions can be detected from NMR relaxation measurements, using covalent attachment of paramagnetic tags to one of the domains or subunits and/or from SAS data.
Solvent PREs, that is, PREs induced by screening of a protein surface by a chemically inert, soluble spin label provide additional ways to identify binding interfaces in molecular complexes.55,76 The surface of an isolated subunit of a complex will be screened in solution by the paramagnetic cosolute, resulting in PRE effects for residues at or close to the surface. When this subunit is bound to its interaction partner, residues located in the binding interface are no longer accessible to the spin label and thus reduced PREs are observed. The solvent PRE can also be interpreted quantitatively to provide restraints in structure calculations, as has been implemented in an Aria/CNS structure calculation protocol.55
Cross-saturation effects also provide information about binding interfaces.97 Here, the effect of spin diffusion is exploited. Saturation of proton magnetization of an unlabeled component is transferred to protons of the labeled component of the complex by dipolar cross relaxation. As these interactions are strictly mediated through space and fall off with increasing distance between the spins involved only direct effects are observed for residues in the binding interface. Thus, indirect effects due to allosteric conformational changes which, for example, influence CSPs are avoided, rendering cross-saturation experiments as useful complements to CSP analysis.
For structure calculations SAS data can be combined with NMR data in three different ways, (i) models derived from NMR data can be scored against the SAS data, (ii) a grid search of conformational space against a target function that includes NMR and SAS energy terms, and (iii) direct refinement against experimental SAS data implemented in simulated annealing protocols already used for NMR data alone.
On the yet few studies reported on large systems the first method is the most common, perhaps as it is the easiest to implement in practice. For example, SANS data have been successfully used to validate an NMR-based model of K-turn U4 RNA.98 Here, scattering data confirmed that unbound RNA does not form the sharply kinked conformation, which is present in the protein bound form. More recently, an impressive example for the combination of NMR and SAS data has been reported with the structural analysis of the box C/D enzyme involved in ribosomal RNA methylation.99 The authors have combined PRE-derived distance restraints and chemical shift analysis to obtain NMR-derived structural models. These models where then scored and selected against SAXS and extensive SANS data to determine the overall assembly of the 390 kDa multidomain protein-RNA complex. We recently explored whether the structure of a ternary protein–protein-RNA complex can be defined solely based on CSP data.100 Structural model obtained using HADDOCK and CSP data were clustered and scored against SAXS and SANS data.101 Although SANS and SAXS data were able to discriminate between different models, the best model which fitted best all experimental data shows still substantial differences to a crystal structure of the complex, indicating that CSP data alone even when combined with SAS data are not sufficient to obtain reliable structural models. When a more comprehensive set of NMR restraints is combined with SAS data reliable structural models can be obtained, as exemplified with the DH-PH module in complex with nucleotide-free RhoA, where cross-saturation and RDCs have improved structural models.102
Grid-search procedures for combining NMR and SAS data have been developed and used to combine the structures of two single domains to model the structure of the native tandem domain system.103 While the orientation derived from RDCs of both domains toward each other is kept constant, they can be translationally moved within a grid in all three-dimensions. Resulting models are fitted against the experimental restraints. This combination has been extended to model the structure of a 102 nucleotide long RNA.104
The third approach, where NMR and SAS data are used in joint refinement has been introduced by Grishaev et al.,105 where individual domains of γS crystallin have been successfully modeled using RDCs and SAXS data simultaneously. The spherical symmetric shape and similar size of both domains make it difficult if SAXS data are used without supplying RDCs, and significant improvements were obtained on combining RDCs and SAXS data. The utility of this approach has been shown for large single chain proteins, for example, the 82 kDa protein malate synthase G106 or in the structural analysis of the 128 kDa enzyme I dimer.107,108
An alternative approach for direct refinement that is computationally more efficient has been proposed by Gabel et al. Here, individual domains of a complex structure or multidomain protein are treated as rigid-bodies and the complex is refined by using orientational restraints derived from RDC data and shape information from a polynomial fit of the SAXS curve. In the case of a two domain system, a restraint for the distance between the center-of-mass of the two domains can be derived from the radius of gyration.109–111 The domain positions with fixed orientation and distance is then determined from higher angles of the scattering curve. This protocol has also been incorporated into the CNS system.110
When studying high molecular weight complexes and high-resolution crystal structures of the complex or subcomplexes are available, it is important to validate the crystallographic structures by solution experiments, such as SAS, NMR, or fluorescence measurements to confirm that the crystal structure reflects indeed the solution conformation. In case of differences, NMR can help to identify their origin and provide a solution structure by combing the crystallographic structure with solution data.40,112
Thus, Crystallography, NMR, and SAS can be efficiently combined in the study of large complexes. For example, if only parts of the complex can be crystallized due to weaker binding to other components, NMR and SAS can be used to obtain a complete picture of the complex in solution. A beautiful example is a structural investigation of the complement factor H of C3b on self surfaces, where X-ray, SAS, and NMR data has been combined to validate the complex in solution and to confirm protein interfaces by CSPs.113
Combining NMR and SAS to Study Complexes or Multidomain Proteins with Flexible Linkers
X-ray crystallography often fails with systems, which have flexible linkers or other intrinsically unstructured regions. In some cases, these regions may become structured and rigid on complex formation and may still be crystallizable. However, in many cases, the presence of such IDRs prevents crystallization. Nevertheless, the flexible linkers connecting individual structural domains often play essential roles for the kinetics of complex formation and the regulation of molecular interactions of multidomain proteins. This has only been realized in recent years as solution techniques such as NMR and SAS have provided this kind of information that often was not visible from available crystal structures of truncated proteins, where flexible linkers have been removed. Deletion of flexible linkers will alter the available conformational space and, thus, the domain arrangements during crystallization. Also, crystal packing forces may trap a non-native conformation in the crystal lattice, when the domain–domain interactions involved are weak. One recent example is the structure of the splicing factor U2AF65, where it has been shown that the domain orientation in solution is different from the initially proposed crystal structure.40,114
Analysis and interpretation of SAS data is also complicated with increasing number of flexible residues and regions. If structural domains of a protein or complex have no fixed arrangement toward each other, a single structural model will not fit the scattering curve and a structural ensemble has to be used to approximate the scattering curve.88,115,116 Conversely, this information can be very useful as NMR and SAS data can provide information about spatial limitations in movements for two or more domains. To this end, SAS data have been successfully combined with RDCs103 or pseudocontact shifts.117 Computational tools are available to generate or fit an ensemble based on the combination of SAS and NMR.116,118,119 NMR relaxation data, which can be measured to obtain rotational correlation times and residue-wise dynamics have been combined with SAXS to obtain a structural ensemble of a tandem domain protein.115,120–122 Also, RDCs give insight into the dynamic state of regions or residues within a polypeptide chain, where negative RDCs indicate random coil and positive RDCs structured regions.123,124
Understanding the structure and dynamics of multidomain proteins becomes increasingly relevant in biomedical research as they play key roles in physiological processes and malfunction often leads to disease. Structural information is very scarce and are often based on a divide and conquer approach with no information about the domain arrangement. For example, different domains in a multidomain protein may be involved in binding distinct ligands or recruiting a substrate to an enzymatic domain located on the same polypeptide chain. In absence of ligands and/or substrate, the domains might behave like pearls on a string with no fixed relative orientation toward each other. However, on ligand or substrate binding, the individual domains may cooperate and form a specific rigid complex with a specific assembly of all domains.
SANS in combination with segmental labeling and NMR can provide interesting insight into the arrangement of domains in multidomain proteins and their interactions. Segmental deuteration and isotope labeling of multidomain proteins will be useful for such investigations. There are several ways to segmentally label a protein. Native chemical ligation,125 intein-mediated (in vivo and in vitro),126 and sortase-mediated127–129 ligation. The latter has several advantages, in that it needs less optimization, provides higher yields, and has no leakage of labeling. However, due to the nature of the enzymatic reaction, which ligates a recognition sequence in the C-terminus of one domain to an N-terminal glycine residue in the other domain, only one continuous part of the polypeptide chain can be segmentally labeled. A combination of both methods can overcome this problem. Another potential problem is the mutation of linkers from the native sequence into residues which are necessary for the ligation reaction. However, as long as the linker length stays native this should not perturb its structure and function, especially if the linker does not adopt any structure on complex formation. Nevertheless, the functional activity of the ligated protein should be validated experimentally. Now suppose this method is used to determine the overall structure of a multidomain protein. The divide and conquer approach can provide high-resolution structures of the individual, isolated domains by NMR or crystallography. Segmental labeling can be used to obtain NMR data, for example, intermolecular nuclear overhauser effects between domains130 while segmental deuteration of domains and subunits will be useful to reduce spectral complexity for analysis of the NMR spectra. SANS data can be recorded on the same samples, comprising deuterated domains or subunits and provide additional structural information about relative domain arrangements. Assume that, in the absence of any binding partner some domains of a complex are flexible [Fig. 4(A)] and that on binding to another protein the complex adopts a specific and rigid arrangement [Fig. 4(B)]. SAXS data will then provide the overall shape of the complex [Fig. 4(C)]. With segmental and subunit-selective deuteration and contrast variation in SANS measurements, each component of the complex can be located within the overall shape of the complex [Fig. 4(D)]. As mentioned earlier, this is also possible for protein-RNA complexes, where the scattering density of RNA can be matched at 70% D2O. Bead models for the overall shape and the location of subunits and domains can be obtained using available software (MONSA131). Rigid-body modeling can be used to fit available high-resolution structure and define their position and orientation in the complex by combining NMR (PREs, RDC) and SAS data to derive a detailed picture of the entire multidomain protein complex with high-resolution information.
Figure 4.
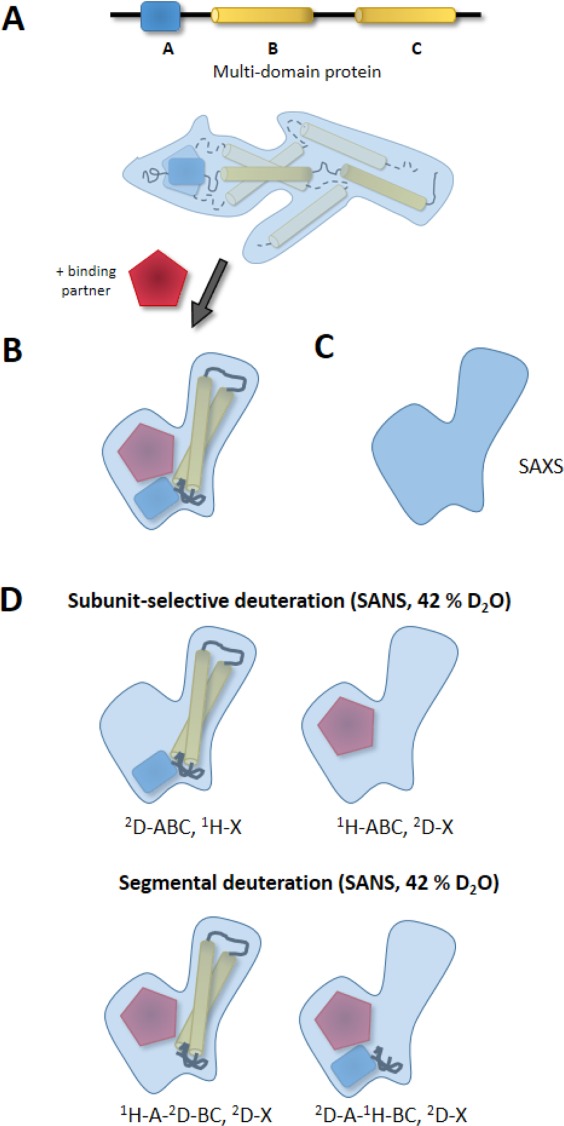
Utility of combining SANS and SAXS data with segmental and subunit-selective deuteration. (A) The approach is exemplified with a multidomain protein comprising domains A, B, and C, which are connected by flexible linkers, assuming that in absence of binding partners the domains have no fixed relative arrangement toward each other. Thus, the SAXS curve will not fit a single structural model but an ensemble of many structures and the radius of gyration and shape of the ensemble would be larger compared to the presence of a single rigid assembly. (B) When a binding partner is added (e.g., substrate X) the domains may rearrange and form a compact, rigid quaternary structure. (C) The envelope of this assembly represents a unique shape which will be reflected by the SAXS data (left). (D) When using subunit-selective and segmental perdeuteration of the proteins involved the individual subunits and domains of the protein complexes can be located within this shape.
Combining NMR and SAS to Study IDPs
One of the current challenges in structural biology is understanding the structure, dynamics, and function of IDPs. Being rich in charged residues and depleted of hydrophobic residues132 IDPs do not form a tangible tertiary structure but nevertheless make up 30–50% of the genome and are more common in eukaryotes.133–135 The problems for structural biology are similar to those posed by multidomain proteins with flexible linkers. Crystallization is not possible and secondary/tertiary structure absent or only transiently formed.136–138 NMR data (PREs and/or relaxation analyses) have been used to identify the transient formation of structural elements. SAS data can contribute information about the overall spatial dimensions occupied by the structural ensemble formed by IDPs.139–142 Although IDPs are found to be unstructured in a laboratory in vitro environment, it is not known whether they are so in the crowded environment of the cell. In this respect, SANS has been recently used to observe a deuterated IDP (N of bacteriophage λ) in a simulated cell environment using high concentrations of bovine pancreatic trypsin inhibitor (BPTI).143 The scattering profiles changed significantly from samples with and without BPTI, where expectantly the conformational space occupied by the IDP was more compact in the presence of BPTI and less prone to aggregation. Similar approaches are under development using in cell NMR144–149 to gain further understanding of IDPs in their native environment. In the study of denatured proteins NMR and SAS were recently combined to characterize the interaction of denatured ubiquitin with the denaturant, revealing that urea binds mainly to the backbone of the protein.150 NMR and SAS were also combined to explore how disulphide bonds influence the unfolded state of proteins.151
IDRs in the context of larger structured regions were also investigated by a combination of NMR and SAS in case of p53 and its IDR, the N-terminal transactivation domain,123 or the domain of the vesicular stomatitis virus N, where a flexible region has been shown to remain flexible in the complex, suggesting a mechanism for viral RNA synthesis152–154 and the IDR at the C-terminal domain of the measles virus nucleocapsid essential for replication of the viral RNA.155
Outlook
In recent years, numerous studies have combined NMR and SAS data together or complementary to crystallographic information and thereby provided unique structural insight into the structure and dynamics of biologically important proteins and complexes, ranging from large protein complexes to IDPs. Given the on-going improvements of both experimental and computational approaches in structural biology it can be expected that multidisciplinary integrated structural biology will greatly advance our understanding of molecular mechanisms involving the structure and dynamics of proteins and protein complexes. Especially, when considering biologically important weak and transient interactions of multidomain proteins and protein complexes with their inherent flexibility, the combination of NMR and SAS has begun to write a new chapter in structural biology. As a “dynamic duo” NMR and SAS will increase our understanding of complex dynamic biological systems.
Acknowledgments
J.H. gratefully acknowledges the Swedish Research Council (Vetenskapsrådet) and the European Molecular Biology Organization (EMBO, ALTF-276-2010) for postdoc fellowships. The authors are grateful for access to high-field NMR at the Bavarian NMR Center, Munich, to synchrotron SAXS measurements at ESRF (Grenoble, France) and DESY/PETRAIII (Hamburg, Germany) and to SANS measurements at ILL (Grenoble, France) and FRMII (Munich, Germany).
Glossary
- BPTI
bovine pancreatic trypsin inhibitor
- CSPs
chemical shift perturbations
- EM
electron microscopy
- IDP
intrinsically disordered proteins
- IDR
intrinsically disordered regions
- LBT
lanthanide binding tag
- NMR
nuclear magnetic resonance spectroscopy
- PRE
paramagnetic relaxation enhancement
- RDC
residual dipolar coupling
- SAS
small angle scattering
- SANS
small angle neutron scattering
- SAXS
small angle X-ray scattering.
References
- 1.Levitt M. Nature of the protein universe. Proc Natl Acad Sci USA. 2009;106:11079–11084. doi: 10.1073/pnas.0905029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selmer M, Dunham CM, Murphy FV, IV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 3.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 2008;9:242–253. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 4.Makino DL, Baumgartner M, Conti E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013;495:70–75. doi: 10.1038/nature11870. [DOI] [PubMed] [Google Scholar]
- 5.Maier T, Jenni S, Ban N. Architecture of mammalian fatty acid synthase at 4.5 A resolution. Science. 2006;311:1258–1262. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 6.Kornberg RD. The molecular basis of eukaryotic transcription. Proc Natl Acad Sci USA. 2007;104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Bushnell DA, Kornberg RD. RNA polymerase II transcription: structure and mechanism. Biochim Biophys Acta. 2013;1829:2–8. doi: 10.1016/j.bbagrm.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sainsbury S, Niesser J, Cramer P. Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature. 2013;493:437–440. doi: 10.1038/nature11715. [DOI] [PubMed] [Google Scholar]
- 9.Lariviere L, Plaschka C, Seizl M, Wenzeck L, Kurth F, Cramer P. Structure of the mediator head module. Nature. 2012;492:448–451. doi: 10.1038/nature11670. [DOI] [PubMed] [Google Scholar]
- 10.Schirle NT, MacRae IJ. The crystal structure of human Argonaute2. Science. 2012;336:1037–1040. doi: 10.1126/science.1221551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakanishi K, Weinberg DE, Bartel DP, Patel DJ. Structure of yeast Argonaute with guide RNA. Nature. 2012;486:368–374. doi: 10.1038/nature11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ, Joshua-Tor L. The structure of human argonaute-2 in complex with miR-20a. Cell. 2012;150:100–110. doi: 10.1016/j.cell.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 15.Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golas MM, Sander B, Bessonov S, Grote M, Wolf E, Kastner B, Stark H, Luhrmann R. 3D cryo-EM structure of an active step I spliceosome and localization of its catalytic core. Mol Cell. 2010;40:927–938. doi: 10.1016/j.molcel.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 17.Diebolder CA, Koster AJ, Koning RI. Pushing the resolution limits in cryo electron tomography of biological structures. J Microsc. 2012;248:1–5. doi: 10.1111/j.1365-2818.2012.03627.x. [DOI] [PubMed] [Google Scholar]
- 18.Grigorieff N. Direct detection pays off for electron cryo-microscopy. Elife. 2013;2:e00573. doi: 10.7554/eLife.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai XC, Fernandez IS, McMullan G, Scheres SH. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Elife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- 21.Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science. 2010;328:98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- 22.Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR, Kay LE. Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science. 2013;339:1080–1083. doi: 10.1126/science.1233066. [DOI] [PubMed] [Google Scholar]
- 23.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 24.Sattler M, Fesik SW. Use of deuterium labeling in NMR: overcoming a sizeable problem. Structure. 1996;4:1245–1249. doi: 10.1016/s0969-2126(96)00133-5. [DOI] [PubMed] [Google Scholar]
- 25.Gardner KH, Rosen MK, Kay LE. Global folds of highly deuterated, methyl-protonated proteins by multidimensional NMR. Biochemistry. 1997;36:1389–1401. doi: 10.1021/bi9624806. [DOI] [PubMed] [Google Scholar]
- 26.Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- 27.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neudecker P, Lundstrom P, Kay LE. Relaxation dispersion NMR spectroscopy as a tool for detailed studies of protein folding. Biophys J. 2009;96:2045–2054. doi: 10.1016/j.bpj.2008.12.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sprangers R, Velyvis A, Kay LE. Solution NMR of supramolecular complexes: providing new insights into function. Nat Methods. 2007;4:697–703. doi: 10.1038/nmeth1080. [DOI] [PubMed] [Google Scholar]
- 30.Lundstrom P, Vallurupalli P, Hansen DF, Kay LE. Isotope labeling methods for studies of excited protein states by relaxation dispersion NMR spectroscopy. Nat Protoc. 2009;4:1641–1648. doi: 10.1038/nprot.2009.118. [DOI] [PubMed] [Google Scholar]
- 31.Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced 1H–13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- 32.Tugarinov V, Kay LE. Methyl groups as probes of structure and dynamics in NMR studies of high-molecular-weight proteins. Chembiochem. 2005;6:1567–1577. doi: 10.1002/cbic.200500110. [DOI] [PubMed] [Google Scholar]
- 33.Riek R, Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K. Solution NMR techniques for large molecular and supramolecular structures. J Am Chem Soc. 2002;124:12144–12153. doi: 10.1021/ja026763z. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Sun X, Watt ED, Al-Hashimi HM. Resolving the motional modes that code for RNA adaptation. Science. 2006;311:653–656. doi: 10.1126/science.1119488. [DOI] [PubMed] [Google Scholar]
- 35.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 36.Bibow S, Mukrasch MD, Chinnathambi S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. The dynamic structure of filamentous tau. Angew Chem Int Ed Engl. 2011;50:11520–11524. doi: 10.1002/anie.201105493. [DOI] [PubMed] [Google Scholar]
- 37.Jensen MR, Ruigrok RW, Blackledge M. Describing intrinsically disordered proteins at atomic resolution by NMR. Curr Opin Struct Biol. 2013;23:426–435. doi: 10.1016/j.sbi.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Ozenne V, Schneider R, Yao M, Huang JR, Salmon L, Zweckstetter M, Jensen MR, Blackledge M. Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. J Am Chem Soc. 2012;134:15138–15148. doi: 10.1021/ja306905s. [DOI] [PubMed] [Google Scholar]
- 39.Bhabha G, Lee J, Ekiert DC, Gam J, Wilson IA, Dyson HJ, Benkovic SJ, Wright PE. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332:234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcarcel J, Sattler M. Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature. 2011;475:408–411. doi: 10.1038/nature10171. [DOI] [PubMed] [Google Scholar]
- 41.Barrett PJ, Chen J, Cho MK, Kim JH, Lu Z, Mathew S, Peng D, Song Y, Van Horn WD, Zhuang T, FD Sönnichsen, CR Sanders. The quiet renaissance of protein nuclear magnetic resonance. Biochemistry. 2013;52:1303–1320. doi: 10.1021/bi4000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacques DA, Trewhella J. Small-angle scattering for structural biology––expanding the frontier while avoiding the pitfalls. Protein Sci. 2010;19:642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mertens HD, Svergun DI. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J Struct Biol. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 45.Madl T, Gabel F, Sattler M. NMR and small-angle scattering-based structural analysis of protein complexes in solution. J Struct Biol. 2011;173:472–482. doi: 10.1016/j.jsb.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Dominguez C, Schubert M, Duss O, Ravindranathan S, Allain FH. Structure determination and dynamics of protein-RNA complexes by NMR spectroscopy. Prog Nucl Magn Reson Spectrosc. 2011;58:1–61. doi: 10.1016/j.pnmrs.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Mackereth CD, Simon B, Sattler M. Extending the size of protein-RNA complexes studied by nuclear magnetic resonance spectroscopy. Chembiochem. 2005;6:1578–1584. doi: 10.1002/cbic.200500106. [DOI] [PubMed] [Google Scholar]
- 48.Tugarinov V, Hwang PM, Kay LE. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu Rev Biochem. 2004;73:107–146. doi: 10.1146/annurev.biochem.73.011303.074004. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez C, Wider G. TROSY in NMR studies of the structure and function of large biological macromolecules. Curr Opin Struct Biol. 2003;13:570–580. doi: 10.1016/j.sbi.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 51.Blackledge M. Recent progress in the study of biomolecular structure and dynamics in solution from residual dipolar couplings. Prog Nucl Magn Reson Spectrosc. 2005;46:23–61. [Google Scholar]
- 52.Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. Chem Rev. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- 53.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 54.Madl T, Felli IC, Bertini I, Sattler M. Structural analysis of protein interfaces from 13C direct-detected paramagnetic relaxation enhancements. J Am Chem Soc. 2010;132:7285–7287. doi: 10.1021/ja1014508. [DOI] [PubMed] [Google Scholar]
- 55.Madl T, Guttler T, Gorlich D, Sattler M. Structural analysis of large protein complexes using solvent paramagnetic relaxation enhancements. Angew Chem Int Ed Engl. 2011;50:3993–3997. doi: 10.1002/anie.201007168. [DOI] [PubMed] [Google Scholar]
- 56.Pintacuda G, John M, Su XC, Otting G. NMR structure determination of protein-ligand complexes by lanthanide labeling. Acc Chem Res. 2007;40:206–212. doi: 10.1021/ar050087z. [DOI] [PubMed] [Google Scholar]
- 57.Simon B, Madl T, Mackereth CD, Nilges M, Sattler M. An efficient protocol for NMR-spectroscopy-based structure determination of protein complexes in solution. Angew Chem Int Ed Engl. 2010;49:1967–1970. doi: 10.1002/anie.200906147. [DOI] [PubMed] [Google Scholar]
- 58.Clore GM, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clore GM, Schwieters CD. Docking of protein-protein complexes on the basis of highly ambiguous intermolecular distance restraints derived from 1H/15N chemical shift mapping and backbone 15N-1H residual dipolar couplings using conjoined rigid body/torsion angle dynamics. J Am Chem Soc. 2003;125:2902–2912. doi: 10.1021/ja028893d. [DOI] [PubMed] [Google Scholar]
- 60.Rieping W, Habeck M, Nilges M. Inferential structure determination. Science. 2005;309:303–306. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 61.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 62.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 63.Lange OF, Rossi P, Sgourakis NG, Song Y, Lee HW, Aramini JM, Ertekin A, Xiao R, Acton TB, Montelione GT, D Baker. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc Natl Acad Sci USA. 2012;109:10873–10878. doi: 10.1073/pnas.1203013109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, A Ignatchenko, CH Arrowsmith, T Szyperski, GT Montelione, D Baker, A Bax. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cusack S, Jacrot B, Leberman R, May R, Timmins P, Zaccai G. Neutron scattering. Nature. 1989;339:330. doi: 10.1038/339330a0. [DOI] [PubMed] [Google Scholar]
- 66.Jacrot B. The study of biological structures by neutron scattering from solution. Rep Prog Phys. 1976;39:911–953. [Google Scholar]
- 67.Capel MS, Engelman DM, Freeborn BR, Kjeldgaard M, Langer JA, Ramakrishnan V, Schindler DG, Schneider DK, Schoenborn BP, Sillers IY, et al. A complete mapping of the proteins in the small ribosomal subunit of Escherichia coli. Science. 1987;238:1403–1406. doi: 10.1126/science.3317832. [DOI] [PubMed] [Google Scholar]
- 68.Ramakrishnan V. Distribution of protein and RNA in the 30S ribosomal subunit. Science. 1986;231:1562–1564. doi: 10.1126/science.3513310. [DOI] [PubMed] [Google Scholar]
- 69.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Round AR, Franke D, Moritz S, Huchler R, Fritsche M, Malthan D, Klaering R, Svergun DI, Roessle M. Automated sample-changing robot for solution scattering experiments at the EMBL Hamburg SAXS station X33. J Appl Crystallogr. 2008;41:913–917. doi: 10.1107/S0021889808021018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacques DA, Guss JM, Svergun DI, Trewhella J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr. 2012;D68:620–626. doi: 10.1107/S0907444912012073. [DOI] [PubMed] [Google Scholar]
- 72.Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trewhella J, Hendrickson WA, Kleywegt GJ, Sali A, Sato M, Schwede T, Svergun DI, Tainer JA, Westbrook J, Berman HM. Report of the wwPDB small-angle scattering task force: data requirements for biomolecular modeling and the PDB. Structure. 2013;21:875–881. doi: 10.1016/j.str.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 74.Spill YG, Kim SJ, Schneidman-Duhovny D, Russel D, Webb B, Sali A, Nilges M. SAXS Merge: an automated statistical method to merge SAXS profiles using Gaussian processes. J Synchrotron Radiat. 2014;21:203–208. doi: 10.1107/S1600577513030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pieper U, Webb BM, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, Khuri N, Spill YG, Weinkam P, Hammel M, JA Tainer, M Nilges, A Sali. ModBase, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2014;42:D336–D346. doi: 10.1093/nar/gkt1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Madl T, Bermel W, Zangger K. Use of relaxation enhancements in a paramagnetic environment for the structure determination of proteins using NMR spectroscopy. Angew Chem Int Ed Engl. 2009;48:8259–8262. doi: 10.1002/anie.200902561. [DOI] [PubMed] [Google Scholar]
- 77.Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- 78.Dominguez C, Bonvin AM, Winkler GS, van Schaik FM, Timmers HT, Boelens R. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure. 2004;12:633–644. doi: 10.1016/j.str.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 79.Grishaev A, Anthis NJ, Clore GM. Contrast-matched small-angle X-ray scattering from a heavy-atom-labeled protein in structure determination: application to a lead-substituted calmodulin-peptide complex. J Am Chem Soc. 2012;134:14686–14689. doi: 10.1021/ja306359z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 81.de Alba E, Tjandra N. NMR dipolar couplings for the structure determination of biopolymers in solution. Prog Nucl Magn Res Spectrosc. 2002;40:175–197. [Google Scholar]
- 82.Rückert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 83.Bloembergen N. Proton relaxation times in paramagnetic solutions. J Chem Phys. 1957;27:572–573. [Google Scholar]
- 84.Bloembergen N, Morgan LO. Proton relaxation times in paramagnetic solutions effects of electron spin relaxation. J Chem Phys. 1961;34:842. [Google Scholar]
- 85.Solomon I. Relaxation processes in a system of 2 spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- 86.Solomon I, Bloembergen N. Nuclear magnetic interactions in the HF molecule. J Chem Phys. 1956;25:261–266. [Google Scholar]
- 87.Bertini I, Luchinat C, Parigi G. Magnetic susceptibility in paramagnetic NMR. Prog Nucl Magn Reson Spectrosc. 2002;40:249–273. [Google Scholar]
- 88.Bernado P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur Biophys J. 2010;39:769–780. doi: 10.1007/s00249-009-0549-3. [DOI] [PubMed] [Google Scholar]
- 89.Gaponenko V, Howarth JW, Columbus L, Gasmi-Seabrook G, Yuan J, Hubbell WL, Rosevear PR. Protein global fold determination using site-directed spin and isotope labeling. Protein Sci. 2000;9:302–309. doi: 10.1110/ps.9.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ollerenshaw JE, Tugarinov V, Kay LE. Methyl TROSY: explanation and experimental verification. Mag Reson Chem. 2003;41:843–852. [Google Scholar]
- 91.Bertini I, Felli IC, Luchinat C. Lanthanide induced residual dipolar couplings for the conformational investigation of peripheral 15NH2 moieties. J Biomol NMR. 2000;18:347–355. doi: 10.1023/a:1026785228634. [DOI] [PubMed] [Google Scholar]
- 92.Bertini I, Luchinat C, Parigi G, Pierattelli R. NMR spectroscopy of paramagnetic metalloproteins. Chembiochem. 2005;6:1536–1549. doi: 10.1002/cbic.200500124. [DOI] [PubMed] [Google Scholar]
- 93.Lu Y, Berry SM, Pfister TD. Engineering novel metalloproteins: design of metal-binding sites into native protein scaffolds. Chem Rev. 2001;101:3047–3080. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 94.Wohnert J, Franz KJ, Nitz M, Imperiali B, Schwalbe H. Protein alignment by a coexpressed lanthanide-binding tag for the measurement of residual dipolar couplings. J Am Chem Soc. 2003;125:13338–13339. doi: 10.1021/ja036022d. [DOI] [PubMed] [Google Scholar]
- 95.Barthelmes K, Reynolds AM, Peisach E, Jonker HR, DeNunzio NJ, Allen KN, Imperiali B, Schwalbe H. Engineering encodable lanthanide-binding tags into loop regions of proteins. J Am Chem Soc. 2011;133:808–819. doi: 10.1021/ja104983t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keizers PH, Saragliadis A, Hiruma Y, Overhand M, Ubbink M. Design, synthesis, and evaluation of a lanthanide chelating protein probe: CLaNP-5 yields predictable paramagnetic effects independent of environment. J Am Chem Soc. 2008;130:14802–14812. doi: 10.1021/ja8054832. [DOI] [PubMed] [Google Scholar]
- 97.Shimada I. NMR techniques for identifying the interface of a larger protein-protein complex: cross-saturation and transferred cross-saturation experiments. Methods Enzymol. 2005;394:483–506. doi: 10.1016/S0076-6879(05)94020-2. [DOI] [PubMed] [Google Scholar]
- 98.Falb M, Amata I, Gabel F, Simon B, Carlomagno T. Structure of the K-turn U4 RNA: a combined NMR and SANS study. Nucleic Acids Res. 2010;38:6274–6285. doi: 10.1093/nar/gkq380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lapinaite A, Simon B, Skjaerven L, Rakwalska-Bange M, Gabel F, Carlomagno T. The structure of the box C/D enzyme reveals regulation of RNA methylation. Nature. 2013;502:519–523. doi: 10.1038/nature12581. [DOI] [PubMed] [Google Scholar]
- 100.Hennig J, Wang I, Sonntag M, Gabel F, Sattler M. Combining NMR and small angle X-ray and neutron scattering in the structural analysis of a ternary protein-RNA complex. J Biomol NMR. 2013;56:17–30. doi: 10.1007/s10858-013-9719-9. [DOI] [PubMed] [Google Scholar]
- 101.Svergun DI, Barberato C, Koch MHJ. CRYSOL–a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- 102.Cierpicki T, Bielnicki J, Zheng M, Gruszczyk J, Kasterka M, Petoukhov M, Zhang A, Fernandez EJ, Svergun DI, Derewenda U, JH Bushweller, ZS Derewenda. The solution structure and dynamics of the DH-PH module of PDZRhoGEF in isolation and in complex with nucleotide-free RhoA. Protein Sci. 2009;18:2067–2079. doi: 10.1002/pro.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang J, Zuo X, Yu P, Byeon IJ, Jung J, Wang X, Dyba M, Seifert S, Schwieters CD, Qin J, AM Gronenborn, YX Wang. Determination of multicomponent protein structures in solution using global orientation and shape restraints. J Am Chem Soc. 2009;131:10507–10515. doi: 10.1021/ja902528f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuo X, Wang J, Yu P, Eyler D, Xu H, Starich MR, Tiede DM, Simon AE, Kasprzak W, Schwieters CD, BA Shapiro, YX Wang. Solution structure of the cap-independent translational enhancer and ribosome-binding element in the 3' UTR of turnip crinkle virus. Proc Natl Acad Sci USA. 2010;107:1385–1390. doi: 10.1073/pnas.0908140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Grishaev A, Wu J, Trewhella J, Bax A. Refinement of multidomain protein structures by combination of solution small-angle X-ray scattering and NMR data. J Am Chem Soc. 2005;127:16621–16628. doi: 10.1021/ja054342m. [DOI] [PubMed] [Google Scholar]
- 106.Grishaev A, Tugarinov V, Kay LE, Trewhella J, Bax A. Refined solution structure of the 82-kDa enzyme malate synthase G from joint NMR and synchrotron SAXS restraints. J Biomol NMR. 2008;40:95–106. doi: 10.1007/s10858-007-9211-5. [DOI] [PubMed] [Google Scholar]
- 107.Schwieters CD, Suh JY, Grishaev A, Ghirlando R, Takayama Y, Clore GM. Solution structure of the 128 kDa enzyme I dimer from Escherichia coli and its 146 kDa complex with HPr using residual dipolar couplings and small- and wide-angle X-ray scattering. J Am Chem Soc. 2010;132:13026–13045. doi: 10.1021/ja105485b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takayama Y, Schwieters CD, Grishaev A, Ghirlando R, Clore GM. Combined use of residual dipolar couplings and solution X-ray scattering to rapidly probe rigid-body conformational transitions in a non-phosphorylatable active-site mutant of the 128 kDa enzyme I dimer. J Am Chem Soc. 2011;133:424–427. doi: 10.1021/ja109866w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gabel F, Simon B, Sattler M. A target function for quaternary structural refinement from small angle scattering and NMR orientational restraints. Eur Biophys J. 2006;35:313–327. doi: 10.1007/s00249-005-0037-3. [DOI] [PubMed] [Google Scholar]
- 110.Gabel F, Simon B, Nilges M, Petoukhov M, Svergun D, Sattler M. A structure refinement protocol combining NMR residual dipolar couplings and small angle scattering restraints. J Biomol NMR. 2008;41:199–208. doi: 10.1007/s10858-008-9258-y. [DOI] [PubMed] [Google Scholar]
- 111.Gabel F. A simple procedure to evaluate the efficiency of bio-macromolecular rigid-body refinement by small-angle scattering. Eur Biophys J. 2012;41:1–11. doi: 10.1007/s00249-011-0751-y. [DOI] [PubMed] [Google Scholar]
- 112.Mackereth CD, Sattler M. Dynamics in multi-domain protein recognition of RNA. Curr Opin Struct Biol. 2012;22:287–296. doi: 10.1016/j.sbi.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 113.Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, Kavanagh D, Mertens HD, Svergun DI, Johansson CM, D Uhrin, PN Barlow, JP Hannan. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. 2011;18:463–470. doi: 10.1038/nsmb.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sickmier EA, Frato KE, Shen H, Paranawithana SR, Green MR, Kielkopf CL. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol Cell. 2006;23:49–59. doi: 10.1016/j.molcel.2006.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bernado P, Modig K, Grela P, Svergun DI, Tchorzewski M, Pons M, Akke M. Structure and dynamics of ribosomal protein L12: an ensemble model based on SAXS and NMR relaxation. Biophys J. 2010;98:2374–2382. doi: 10.1016/j.bpj.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 117.Bertini I, Giachetti A, Luchinat C, Parigi G, Petoukhov MV, Pierattelli R, Ravera E, Svergun DI. Conformational space of flexible biological macromolecules from average data. J Am Chem Soc. 2010;132:13553–13558. doi: 10.1021/ja1063923. [DOI] [PubMed] [Google Scholar]
- 118.Bertini I, Ferella L, Luchinat C, Parigi G, Petoukhov MV, Ravera E, Rosato A, Svergun DI. MaxOcc: a web portal for maximum occurrence analysis. J Biomol NMR. 2012;53:271–280. doi: 10.1007/s10858-012-9638-1. [DOI] [PubMed] [Google Scholar]
- 119.Ozenne V, Bauer F, Salmon L, Huang JR, Jensen MR, Segard S, Bernado P, Charavay C, Blackledge M. Flexible-meccano: a tool for the generation of explicit ensemble descriptions of intrinsically disordered proteins and their associated experimental observables. Bioinformatics. 2012;28:1463–1470. doi: 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- 120.Bertini I, Calderone V, Fragai M, Jaiswal R, Luchinat C, Melikian M, Mylonas E, Svergun DI. Evidence of reciprocal reorientation of the catalytic and hemopexin-like domains of full-length MMP-12. J Am Chem Soc. 2008;130:7011–7021. doi: 10.1021/ja710491y. [DOI] [PubMed] [Google Scholar]
- 121.Bertini I, Fragai M, Luchinat C, Melikian M, Mylonas E, Sarti N, Svergun DI. Interdomain flexibility in full-length matrix metalloproteinase-1 (MMP-1) J Biol Chem. 2009;284:12821–12828. doi: 10.1074/jbc.M809627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grela P, Helgstrand M, Krokowski D, Boguszewska A, Svergun D, Liljas A, Bernado P, Grankowski N, Akke M, Tchorzewski M. Structural characterization of the ribosomal P1A-P2B protein dimer by small-angle X-ray scattering and NMR spectroscopy. Biochemistry. 2007;46:1988–1998. doi: 10.1021/bi0616450. [DOI] [PubMed] [Google Scholar]
- 123.Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bernado P, Blackledge M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys J. 2009;97:2839–2845. doi: 10.1016/j.bpj.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xu R, Ayers B, Cowburn D, Muir TW. Chemical ligation of folded recombinant proteins: segmental isotopic labeling of domains for NMR studies. Proc Natl Acad Sci USA. 1999;96:388–393. doi: 10.1073/pnas.96.2.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Muona M, Aranko AS, Raulinaitis V, Iwai H. Segmental isotopic labeling of multi-domain and fusion proteins by protein trans-splicing in vivo and in vitro. Nat Protoc. 2010;5:574–587. doi: 10.1038/nprot.2009.240. [DOI] [PubMed] [Google Scholar]
- 127.Mao H, Hart SA, Schink A, Pollok BA. Sortase-mediated protein ligation: a new method for protein engineering. J Am Chem Soc. 2004;126:2670–2671. doi: 10.1021/ja039915e. [DOI] [PubMed] [Google Scholar]
- 128.Chen I, Dorr BM, Liu DR. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc Natl Acad Sci USA. 2011;108:11399–11404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Refaei MA, Combs A, Kojetin DJ, Cavanagh J, Caperelli C, Rance M, Sapitro J, Tsang P. Observing selected domains in multi-domain proteins via sortase-mediated ligation and NMR spectroscopy. J Biomol NMR. 2011;49:3–7. doi: 10.1007/s10858-010-9464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Barraud P, Allain FH. Solution structure of the two RNA recognition motifs of hnRNP A1 using segmental isotope labeling: how the relative orientation between RRMs influences the nucleic acid binding topology. J Biomol NMR. 2013;55:119–138. doi: 10.1007/s10858-012-9696-4. [DOI] [PubMed] [Google Scholar]
- 131.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2031;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mukrasch MD, Markwick P, Biernat J, Bergen M, Bernado P, Griesinger C, Mandelkow E, Zweckstetter M, Blackledge M. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J Am Chem Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 137.Jensen MR, Markwick PR, Meier S, Griesinger C, Zweckstetter M, Grzesiek S, Bernado P, Blackledge M. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure. 2009;17:1169–1185. doi: 10.1016/j.str.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 138.Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, Blackledge M. NMR characterization of long-range order in intrinsically disordered proteins. J Am Chem Soc. 2010;132:8407–8418. doi: 10.1021/ja101645g. [DOI] [PubMed] [Google Scholar]
- 139.Guttman M, Weinkam P, Sali A, Lee KK. All-atom ensemble modeling to analyze small-angle x-ray scattering of glycosylated proteins. Structure. 2013;21:321–331. doi: 10.1016/j.str.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johansen D, Trewhella J, Goldenberg DP. Fractal dimension of an intrinsically disordered protein: small-angle X-ray scattering and computational study of the bacteriophage lambda N protein. Protein Sci. 2011;20:1955–1970. doi: 10.1002/pro.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gabel F. Small angle neutron scattering for the structural study of intrinsically disordered proteins in solution: a practical guide. Methods Mol Biol. 2012;896:123–135. doi: 10.1007/978-1-4614-3704-8_8. [DOI] [PubMed] [Google Scholar]
- 142.Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RW, Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc Natl Acad Sci USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Johansen D, Jeffries CM, Hammouda B, Trewhella J, Goldenberg DP. Effects of macromolecular crowding on an intrinsically disordered protein characterized by small-angle neutron scattering with contrast matching. Biophys J. 2011;100:1120–1128. doi: 10.1016/j.bpj.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ito Y, Selenko P. Cellular structural biology. Curr Opin Struct Biol. 2010;20:640–648. doi: 10.1016/j.sbi.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 145.Selenko P, Wagner G. NMR mapping of protein interactions in living cells. Nat Methods. 2006;3:80–81. doi: 10.1038/nmeth0206-80. [DOI] [PubMed] [Google Scholar]
- 146.Selenko P, Serber Z, Gadea B, Ruderman J, Wagner G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc Natl Acad Sci USA. 2006;103:11904–11909. doi: 10.1073/pnas.0604667103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Serber Z, Selenko P, Hansel R, Reckel S, Lohr F, Ferrell JE, Jr, Wagner G, Dotsch V. Investigating macromolecules inside cultured and injected cells by in-cell NMR spectroscopy. Nat Protoc. 2006;1:2701–2709. doi: 10.1038/nprot.2006.181. [DOI] [PubMed] [Google Scholar]
- 148.Selenko P, Frueh DP, Elsaesser SJ, Haas W, Gygi SP, Wagner G. In situ observation of protein phosphorylation by high-resolution NMR spectroscopy. Nat Struct Mol Biol. 2008;15:321–329. doi: 10.1038/nsmb.1395. [DOI] [PubMed] [Google Scholar]
- 149.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, M Shirakawa. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458:106–109. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 150.Huang JR, Gabel F, Jensen MR, Grzesiek S, Blackledge M. Sequence-specific mapping of the interaction between urea and unfolded ubiquitin from ensemble analysis of NMR and small angle scattering data. J Am Chem Soc. 2012;134:4429–4436. doi: 10.1021/ja2118688. [DOI] [PubMed] [Google Scholar]
- 151.Silvers R, Sziegat F, Tachibana H, Segawa S, Whittaker S, Gunther UL, Gabel F, Huang JR, Blackledge M, Wirmer-Bartoschek J, H Schwalbe. Modulation of structure and dynamics by disulfide bond formation in unfolded states. J Am Chem Soc. 2012;134:6846–6854. doi: 10.1021/ja3009506. [DOI] [PubMed] [Google Scholar]
- 152.Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA, Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. Structure of the vesicular stomatitis virus N(0)-P complex. PLoS Pathog. 2011;7:e1002248. doi: 10.1371/journal.ppat.1002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leyrat C, Jensen MR, Ribeiro EA, Jr, Gerard FC, Ruigrok RW, Blackledge M, Jamin M. The N(0)-binding region of the vesicular stomatitis virus phosphoprotein is globally disordered but contains transient alpha-helices. Protein Sci. 2011;20:542–556. doi: 10.1002/pro.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Leyrat C, Schneider R, Ribeiro EA, Jr, Yabukarski F, Yao M, Gerard FC, Jensen MR, Ruigrok RW, Blackledge M, Jamin M. Ensemble structure of the modular and flexible full-length vesicular stomatitis virus phosphoprotein. J Mol Biol. 2012;423:182–197. doi: 10.1016/j.jmb.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 155.Jensen MR, Communie G, Ribeiro EA, Jr, Martinez N, Desfosses A, Salmon L, Mollica L, Gabel F, Jamin M, Longhi S, RW Ruigrok, M Blackledge. Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci USA. 2011;108:9839–9844. doi: 10.1073/pnas.1103270108. [DOI] [PMC free article] [PubMed] [Google Scholar]