

Abstract
Unusual di- and trideoxysugars are often found on the O-antigens of Gram-negative bacteria, on the S-layers of Gram-positive bacteria, and on various natural products. One such sugar is 3-acetamido-3,6-dideoxy-d-glucose. A key step in its biosynthesis, catalyzed by a 3,4-ketoisomerase, is the conversion of thymidine diphosphate (dTDP)−4-keto-6-deoxyglucose to dTDP-3-keto-6-deoxyglucose. Here we report an X-ray analysis of a 3,4-ketoisomerase from Thermoanaerobacterium thermosaccharolyticum. For this investigation, the wild-type enzyme, referred to as QdtA, was crystallized in the presence of dTDP and its structure solved to 2.0-Å resolution. The dimeric enzyme adopts a three-dimensional architecture that is characteristic for proteins belonging to the cupin superfamily. In order to trap the dTDP-4-keto-6-deoxyglucose substrate into the active site, a mutant protein, H51N, was subsequently constructed, and the structure of this protein in complex with the dTDP–sugar ligand was solved to 1.9-Å resolution. Taken together, the structures suggest that His 51 serves as a catalytic base, that Tyr 37 likely functions as a catalytic acid, and that His 53 provides a proton shuttle between the C-3′ hydroxyl and the C-4′ keto group of the hexose. This study reports the first three-dimensional structure of a 3,4-ketoisomerase in complex with its dTDP–sugar substrate and thus sheds new molecular insight into this fascinating class of enzymes.
Keywords: dideoxysugar; 3,4-ketoisomerase; cupin superfamily; enzyme mechanism; O-antigen; S-layer; 3-acetamido-3,6-dideoxy-d-glucose; protein structure
Introduction
The bacterial 3,4-ketoisomerases play key roles in the production of unusual 3,6-dideoxysugars such as 3-acetamido-3,6-dideoxy-d-galactose (Fuc3NAc), 3-acetamido-3,6-dideoxy-d-glucose (Qui3NAc), and d-mycaminose (Scheme 1). The first two sugars, 3-acetamido-3,6-dideoxy-d-galactose and 3-acetamideo-3,6-dideoxy-d-glucose, have been observed in the S-layer of Aneurinibacillus thermoaerophilus and Thermoanaerobacterium thermosaccharolyticum, respectively, and have also been identified on the lipopolysaccharides of several Gram-negative bacteria.1–6 The third sugar, d-mycaminose, is found attached to a variety of macrolide rings including tylosin, an antibiotic produced by Streptomyces fradiae.7
Scheme 1.

Examples of 3,6-dideoxysugars.
The complete biosynthetic pathways for the production of 3-acetamido-3,6-dideoxy-d-galactose and 3-acetamido-3,6-dideoxy-d-glucose were first reported by the Messner laboratory in 2003 and 2008, respectively, and are shown in Scheme 2.1,2 As can be seen, the 3,4-ketoisomerases (QdtA or FdtA) catalyze the third steps in the pathways. Whereas both QdtA and FdtA function on the same substrate [thymidine diphosphate (dTDP)−4-keto-6-deoxyglucose], the products of their reactions differ with respect to the orientations of the hydroxyl groups about the hexose C-4′ carbons. Specifically, QdtA functions as a retaining enzyme whereas FdtA inverts the configuration about the C-4′ carbon.
Scheme 2.
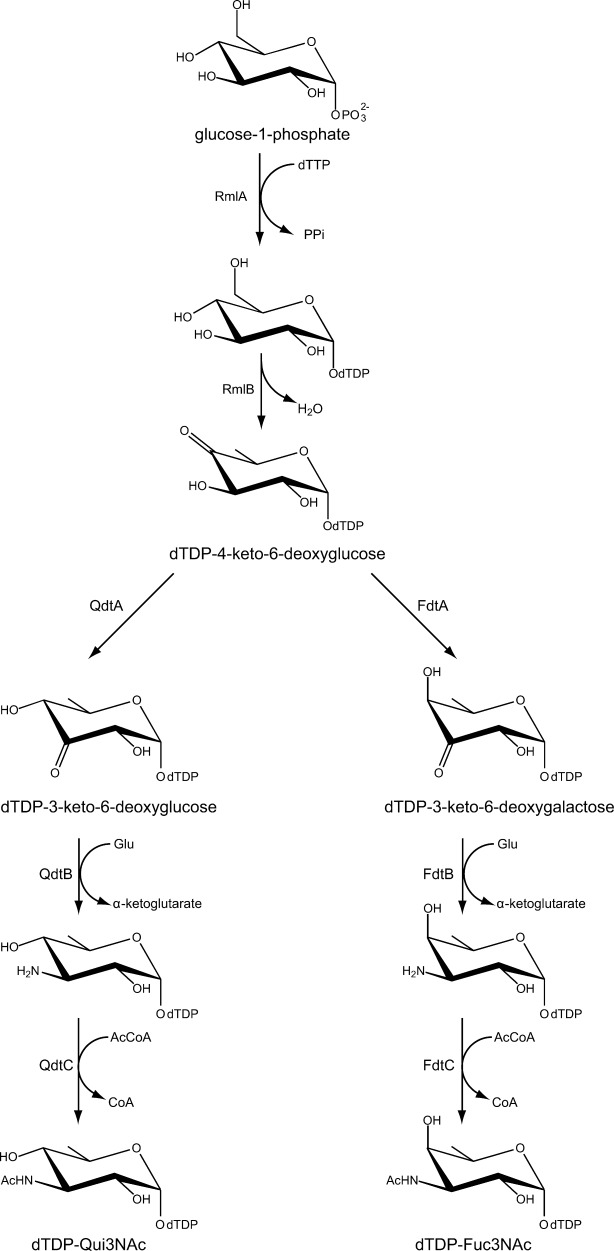
Biosynthetic pathway for the production of dTDP-Qui3NAc and dTDP-Fuc3NAc.
In 2007, our laboratory determined the first structure of a sugar 3,4-ketoisomerase, namely that of FdtA from A. thermoaerophilus.8 Each subunit of the dimeric enzyme is characterized by a β-barrel motif with one α-helix. A cluster of three histidine residues lines the active site region, two of which appear to be strictly conserved amongst the 3,4-ketoisomerases. Subsequent site-directed mutagenesis experiments suggested that one of these histidines, His 49, functions as an active site base. Whereas this X-ray analysis of a 3,4-ketoisomerase revealed the overall fold of the enzyme, the study was somewhat limited because it was never possible to determine the structure of FdtA with a bound dTDP–sugar substrate or analog.
Here we report the molecular architecture of QdtA from T. thermosaccharolyticum. For this investigation, crystals were grown of both the wild-type enzyme in complex with dTDP and of a site-directed mutant protein (H51N) in complex with dTDP-4-keto-6-deoxyglucose. The structures reveal that His 51 is located in an ideal position to serve as the catalytic base, that Tyr 37 likely functions as the active site acid, and that His 53 is correctly situated to function as a proton shuttle between the C-3′ hydroxyl group and the C′-4 keto functionality. The investigation described herein provides additional insight into the sugar 3,4-ketoisomerases.
Results and Discussion
Biochemical analysis of T. thermosaccharolyticum QdtA
The amino acid sequence of the T. thermosaccharolyticum QdtA utilized in this investigation differs from that deposited in GenBank (A52S, F94L, L95V, and C116Y). Amino acid sequence alignments with other QdtA (or putative QdtA) enzymes demonstrate that Position 52 is either an alanine or serine, Positions 94 and 95 show variability, and Position 116 is always a tyrosine rather than a cysteine. In the model described here, Ser 52 is ∼7 Å and Leu 94 and Val 95 are over 10 Å from the active site. Importantly, Tyr 116, as described below, forms a stacking interaction with the thymine ring of the dTDP–sugar substrate.
The version of T. thermosaccharolyticum QdtA described here is fully functional. Indeed, a kinetic analysis showed that the Km for dTDP-4-keto-6-deoxyglucose is 0.32 ± 0.04 mM and the kcat is 2.8 ± 0.3 s−1. The overall catalytic efficiency of QdtA is 8.8 x 103 M−1 s−1.
Structure of the wild-type T. thermosaccharolyticum QdtA
The QdtA/dTDP complex crystals grew from pentaerythritol propoxylate (pH 8.0) and contained one dimer in the asymmetric unit. The structure was solved and refined to 2.0-Å resolution. Shown in Figure 1(a) is the electron density corresponding to the bound dTDP ligand in Subunit 1. Overall the quality of the electron density for both subunits in the asymmetric unit was excellent with no breaks in the polypeptide chain backbones from Met 1 to Phe 136. Interestingly, there was electron density positioned on a crystallographic dyad that corresponded to one molecule of tetraerythritol propoxylate, a contaminant of pentaerythritol propoxylate.
Figure 1.

The three-dimensional architecture of QdtA. The structure of QdtA was solved in the presence of dTDP. Shown in (a) is the electron density corresponding to the bound ligand in Subunit 1 of the dimer. The map was calculated with coefficients of the form Fo − Fc, where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. The map was contoured at 4σ. The ligand was not included in the map calculation or in the model refinement. A ribbon representation of the QdtA dimer is displayed in (b) with Subunits 1 and 2 depicted in purple and teal, respectively. The arrow indicates the position of the twofold rotational axis that relates the two subunits. The N- and C-termini are located at ∼12 Å from each other. A close-up stereo view of the region surrounding the dTDP ligand in Subunit 1 is presented in (c). The dashed lines indicate potential hydrogen bonds. Ordered water molecules are depicted as red spheres. All figures were created using PyMOL.19
A ribbon representation of the QdtA dimer is displayed in Figure 1(b). The molecular architecture of QdtA places it into the well-characterized cupin superfamily.9–11 Members of this superfamily include both metal-dependent and metal-independent enzymes as well as seed storage and sugar-binding proteins. Each subunit of the dimer consists of 11 β-strands and a C-terminal α-helix. The N- and C-termini are separated by ∼12 Å. The β-strands form an antiparallel flattened β-barrel that can be envisioned as two layers of four and seven β-strands, respectively. Classical domain swapping occurs as the second and third β-strands from one subunit form part of the seven-stranded β-sheet in the second subunit. In light of the fact that the α-carbons for the two subunits superimpose with a root-mean-square deviation of 0.19 Å, the following discussion refers only to Subunit 1 in the X-ray coordinate file.
A close-up view of the dTDP-binding site is presented in Figure 1(c). The thymine ring lies within hydrogen bonding distance to the guanidinium group of Arg 35 and two water molecules. In addition, Tyr 31 and Tyr 116 form T-shaped and parallel stacking interactions, respectively, with the thymine ring. The pyrophosphoryl group of the ligand hydrogen bonds with the backbone amide group of Gly 49, the side chains of Arg 48 His 51, Tyr 121, and Arg 123, and three water molecules.
As noted in the Introduction, the first structural model of a sugar 3,4-ketoisomerase to be reported was that of FdtA from A. thermoaerophilus.8 FdtA and QdtA demonstrate an amino acid sequence identity of 55% and a sequence similarity of 68%. Not surprisingly, the two enzymes superimpose with a root-mean-square deviation of 0.90 Å for 125 α-carbons. A superposition of the active site regions for QdtA and FdtA is presented in Figure 2. The dTDP ligands adopt slightly different conformations. In QdtA the pyrophosphoryl group is directed toward the interior of the protein whereas in FdtA it projects outward. Overall the active sites are similar with the only major difference being the replacement of His 95 in FdtA with Arg 97 in QdtA. This difference may play a role in determining the overall stereochemical outcome of the dTDP-product as discussed below.
Figure 2.

Comparison of the QdtA and FdtA active sites. Those amino acid residues corresponding to QdtA and FdtA are displayed in teal and white, respectively. The top labels refer to QdtA residues whereas the bottom labels correspond to FdtA residues.
Structure of the H51N mutant protein of T. thermosaccharolyticum QdtA
Previous site-directed mutagenesis experiments implicated His 49 in FdtA as the catalytic base that initiates catalysis by abstracting the C-3′ proton from the sugar.8 This residue is structurally conserved in QdtA as His 51 (Fig. 2). Thus, in an attempt to prepare a QdtA/dTDP–sugar complex, the H51N site-directed mutant protein was subsequently prepared and shown to be catalytically inactive. Its three-dimensional structure was analyzed in the presence of the substrate, dTDP-4-keto-6-deoxyglucose (Scheme 2). The model was refined to a nominal resolution of 1.9 Å. The electron densities corresponding to the bound dTDP–sugars in Subunits 1 and 2 of the dimer are presented in Figure 3(a,b), respectively. In both subunits, the electron density corresponding to the C-4′ position of the pyranosyl moiety demonstrates that the keto-form of the sugar, with sp2 hybridization, has been trapped within the active site cleft.
Figure 3.

Electron density for the dTDP–sugars. The electron density maps shown in (a) and (b) correspond to the dTDP–sugar ligands found in the active sites of Subunits 1 and 2, respectively. The maps, contoured at 4σ, were calculated with coefficients of the form Fo − Fc, where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. The ligands were not included in the map calculations or in the initial model refinements.
Overall the polypeptide chains for the wild-type QdtA and the H51N mutant protein are similar such that their α-carbons superimpose with a root-mean-square deviation of 0.18 Å. Only two amino acid residues change position significantly upon dTDP–sugar binding [Fig. 4(a)]. Specifically, the side chain of Arg 97 shifts in the active site to accommodate the C-4′ keto group of the dTDP–sugar. Likewise, the side chain of Gln 58 reorients such that Nε2 lies within 2.5 Å to Oη of Tyr 37. In the wild-type enzyme/dTDP complex model, this distance is 3.7 Å. A close-up view of the active site in Subunit 1 is provided in Figure 4(b). The interactions between the protein and the thymine ring, the deoxyribose, and the pyrophosphoryl group of the dTDP–sugar are virtually identical to those observed for the wild-type enzyme with bound dTDP. Key residues that anchor the pyranosyl group of the dTDP–sugar to the protein include His 53 and Arg 97.
Figure 4.

Active site of the H51N mutant protein. Shown in (a) is a superposition of the active site of the H51N mutant protein (gray bonds) onto that of the wild-type enzyme (purple bonds). Only two residues, Gln 58 and Arg 97, shift significantly upon binding the dTDP–sugar ligand. The hydrogen-bonding pattern surrounding the dTDP–sugar substrate in the H51N mutant protein is displayed in (b). The dashed lines indicate distances of 3.2 Å or shorter. An ethylene glycol molecule, labeled EG, was observed binding near the pyrophosphoryl group of the ligand.
Implications for the catalytic mechanism
As can be seen in Figure 4(a), substituting an asparagine for a histidine residue at Position 51 resulted in little three-dimensional perturbation of the surrounding region. Thus, by combining the two structures described in this investigation, it is possible to obtain a reasonable model for the Michaelis complex as depicted in Figure 5. In this model, His 51 lies within ∼4 Å of the C-3′ carbon. Tyr 37, situated on the opposite side of the pyranosyl ring, is positioned within 3.2 Å from the C-4′ carbon. The imidazole side chain of His 53 sits between the C-3′ and C-4′ oxygens, and the guanidinium group of Arg 97 lies within hydrogen bonding distance of the C-4′ keto oxygen.
Figure 5.

Model of the Michaelis Complex. In the complex, His 51 and Tyr 37 are located on opposite sides of the hexose moiety and are ideally positioned to serve as a catalytic base and acid, respectively (red dashed lines). There is a hydrogen-bonding network, indicated by the dashed black lines, between Arg 97, Gln 58, and Tyr 37. This may help to lower the pKa of Tyr 37.
A catalytic mechanism can thus be envisioned whereby His 51 abstracts the C-3′ proton leading to the formation of an enolate intermediate as indicated in Scheme 3. The side chain of Tyr 37 functions as an active site acid by protonating the C-4′ carbon, thereby leading to retention of the “glucose” configuration of the product. The role of His 53 is most likely to shuttle protons from the C-3′ hydroxyl to the C-4′ keto group (Fig. 5 and Scheme 3). Possible side chains that might stabilize the enolate intermediate include Arg 97 and/or Gln 58. Whereas Gln 58 is absolutely conserved amongst the 3,4-ketoisomerases, the equivalent position for Arg 97 is sometimes occupied by a histidine or a glycine residue.12 The mechanism outlined in Scheme 3 is proposed as step wise, but it is also possible that proton transfer occurs in a concerted manner.
Scheme 3.

Possible mechanism for QdtA.
The 3,4-ketoisomerases are, indeed, an intriguing class of enzymes in that they utilize the same substrate, dTDP-4-keto-6-deoxyglucose, but some function as retaining enzymes such as QdtA or inverting enzymes such as FdtA (Scheme 2). An elegant biochemical investigation of another 3,4-ketoisomerase from S. fradiae suggested similar catalytic roles for His 63 (His 51 in QdtA) and Tyr 49 (Tyr 37 in QdtA), although there are no published crystal structures of this enzyme thus far. Like QdtA, the S. fradiae protein is a retaining enzyme.13
Very recently we reported a combined biochemical and structural investigation on the 3,4-ketoisomerase from Shewanella denitrificans. As observed for FdtA, this enzyme inverts the stereochemistry about the C-4′ carbon.12 Importantly, whereas His 51 and His 53 are conserved in the S. denitrificans enzyme, Tyr 37 is replaced with a phenylalanine.12 Based on amino acid sequence alignments of the 3,4-ketoisomerases, there is obviously a subset of these enzymes that contain a phenylalanine rather than a tyrosine residue in the active site. Thus a different mechanism must be operative in these inverting enzymes, most likely involving the two conserved histidines. Deprotonation of the C-3′ carbon and protonation of the C-4′ carbon on the same side of the pyranosyl ring would result in inversion of configuration.
Clearly, the S. denitrificans enzyme functions as an inverting enzyme because it lacks the required catalytic acid oriented on the opposite side of the pyranosyl ring. The reason why FdtA is an inverting enzyme is less clear, however. As in QdtA, it contains a tyrosine residue at Position 37 (Fig. 2). Indeed, the only significant difference between FdtA and QdtA is the replacement of Arg 97 in QdtA for a histidine residue in FdtA. Such a replacement could result in a charge change in the active site, thus dramatically affecting the pKa of catalytically important residues and/or the manner in which the substrate binds. It could also affect a proton shuttling network. For example, in QdtA, Arg 97 is bridged to Tyr 37, the putative catalytic acid, via the side chain of Gln 58 (Fig. 5). In FdtA, this hydrogen-bonding network is broken, which may perturb the pKa of the corresponding tyrosine enough that it can no longer function as a catalytic acid. Experiments designed to more fully explore the reaction mechanisms of the inverting and retaining 3,4-ketoisomerases are in progress.
Materials and Methods
Cloning of QdtA
The gene encoding QdtA was polymerase chain reaction (PCR)-amplified from T. thermosaccharolyticum E207-71 genomic DNA via standard procedures using primers such that the forward (5′-AAACATATGATTTTGTATAACGTT GCGTTAATAAAATTTAAGGATATAGCAGATAAATATGGTCATTTGACACC) and the reverse (5′-AAACTCGAGAAATCTTTTTTTAGCTTCATCAATATAAAA ATCATAATTTCTTATGTAGTCCGTTTCATCAC) added NdeI and XhoI cloning sites, respectively. The purified PCR product was subsequently A-tailed and ligated into the pGEM-T vector (Promega), which was used to transform DH5-α Escherichia coli cells for subsequent screening and sequencing. A pGEM-T-qdtA vector construct of the correct sequence was digested and ligated into the pET31 vector for protein expression. DH5-α E. coli cells were transformed with the resulting plasmid and streaked onto lysogeny broth agar plates supplemented with ampicillin. Multiple colonies were tested for the presence of the qdtA gene.
Protein expression and purification
The purified pET31-qdtA plasmid was used to transform Rosetta (DE3) E. coli cells (Novagen). Cultures were grown in terrific broth medium supplemented with ampicillin and chloramphenicol at 37°C with shaking until an optical density of 0.9 was reached at 600 nm. The flasks were cooled to room temperature, and the cells were induced by the addition of 0.5 mM isopropyl-β-d-1-thiogalactopyranoside. They were allowed to express protein at 23°C for 24 h following induction.
The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and QdtA was purified utilizing Ni nitrilotriacetic acid resin (Qiagen) according to the manufacturer's instructions. The protein was dialyzed against 10 mM Tris–HCl (pH 8.0) and 200 mM NaCl and concentrated to 20 mg mL−1 based on an extinction coefficient of 0.77 mg−1 mL cm−1.
Preparation of dTDP-4-keto-6-deoxyglucose
dTDP-4-keto-6-deoxyglucose was prepared by dissolving dTDP–glucose in 1 mL of 50 mM N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid (HEPPS; pH 8.5) containing 10 mg of E. coli RmlB (4,6-dehydratase) to a final concentration of 50 mM. This reaction was incubated at 37°C for 6 h. The resulting dTDP-4-keto-6-deoxyglucose was used without further purification. Note that high-performance liquid chromatography (HPLC) analysis of the mixture at this time point showed that the reaction was essentially complete and negligible amounts of dTDP–glucose were present. RmlB was purified in the laboratory as a C-terminal His6 construct (unpublished procedures).
Crystallization of QdtA
Prior to crystallization trials, the protein was incubated with 10 mM dTDP. Crystallization conditions were then surveyed by the hanging drop method of vapor diffusion using a sparse matrix screen developed in the laboratory. Single crystals of QdtA were subsequently grown against a precipitant solution containing 100 mM HEPPS (pH 8.0) and 22–28% pentaerythritol propoxylate. The crystals belonged to the space group P4212 with unit cell dimensions of a = b = 95.8 Å and c = 94.7 Å. The asymmetric unit contained one dimer.
Site-directed mutagenesis and crystallization
The site-directed mutant protein, H51N, was generated using the QuikChange method of Stratagene. The protein variant was expressed and purified as described above for the wild-type enzyme.
Crystals were grown under conditions similar to that of the wild-type enzyme, except that no ligands were included in the experiments. The ligand-free crystals were transferred to a synthetic mother liquor containing 30% pentaerythritol propoxylate, 200 mM NaCl, 100 mM HEPPS (pH 8.0), and 10 mM dTDP-4-keto-6-deoxyglucose. They were allowed to soak for 18 h.
Structural analysis of QdtA
Prior to X-ray data collection, crystals of the wild-type enzyme were transferred to a cryoprotectant solution containing 32% pentaerythritol propoxylate, 200 mM NaCl, 10 mM dTDP, and 100 mM HEPPS (pH 8.0). X-ray data were collected at 100 K with a Bruker AXS Platinum 135 CCD detector controlled by the Proteum software suite (Bruker AXS). The X-ray source was Cu Kα radiation from a Rigaku RU200 X-ray generator equipped with Montel optics and operated at 50 kV and 90 mA. These X-ray data were processed with SAINT version 7.06A (Bruker AXS) and internally scaled with SADABS version 2005/1 (Bruker AXS). Relevant X-ray data collection statistics are listed in Table I. The structure was solved with PHASER using the coordinates for FdtA (PDB code 2PA7) as a search model.8,14 Iterative rounds of model building with COOT and refinement with REFMAC reduced the Rwork and Rfree to 19.7 and 23.9%, respectively, from 30 to 2.0 Å resolution.15,16
Table I.
X-Ray Data Collection Statistics
| QdtA with bound dTDP | QdtA H51N with bound dTDP-4-keto-6-deoxyglucose | |
|---|---|---|
| Resolution limits | 30–2.0 (2.05–2.0)a | 50–1.90 (2.0–1.9) |
| Number of independent reflections | 28,352 (2040) | 33,577 (4259) |
| Completeness (%) | 98.2 (97.9) | 95.7 (87.1) |
| Redundancy | 6.5 (5.1) | 7.7 (2.1) |
| Avg I/avg σ(I) | 18.5 (3.7) | 14.9 (1.9) |
| Rsym (%)b | 6.9 (28.2) | 7.9 (37.4) |
Statistics for the highest resolution bin.
Rsym = (Σ|I −  |/ΣI) × 100.
|/ΣI) × 100.
Crystals of the H51N variant were transferred to a cryoprotectant solution containing 36% pentaerythritol propoxylate, 200 mM NaCl, 6% ethylene glycol, 10 mM dTDP-4-keto-6-deoxyglucose, and 100 mM HEPPS (pH 8.0). X-ray data were collected and processed in a similar manner as that described above for the wild-type enzyme. Relevant statistics are presented in Table I. Iterative cycles of model-building with COOT and refinement with REFMAC reduced the Rwork and Rfree to 20.1 and 24.1%, respectively, from 30 to 1.90 Å resolution.15,16 Model refinement statistics are listed in Table II for both the wild-type enzyme and the H51N mutant protein.
Table II.
Refinement Statistics
| QdtA with bound dTDP | QdtA H51N with bound dTDP-4-keto-6-deoxyglucose | |
|---|---|---|
| Resolution limits (Å) | 30.0–2.0 | 30–1.9 |
| aR-Factor (overall)%/no. reflections | 19.9/28352 | 20.3/33577 |
| R-Factor (working)%/no. reflections | 19.7/26835 | 20.1/31868 |
| R-Factor (free)%/no. reflections | 23.9/1517 | 24.1/1709 |
| Number of protein atoms | 2283 | 2316 |
| Number of heteroatoms | 195 | 274 |
| Average B values | ||
| Protein atoms (Å2) | 36.1 | 27.8 |
| Ligand (Å2) | 47.4 | 21.7 |
| Solvent (Å2) | 42.1 | 57.8 |
| Weighted RMS deviations from ideality | ||
| Bond lengths (Å) | 0.006 | 0.009 |
| Bond angles (°) | 2.4 | 1.4 |
| Planar groups (Å) | 0.012 | 0.006 |
| Ramachandran regions (%)b | ||
| Most favored | 89.4 | 89.9 |
| Additionally allowed | 9.3 | 9.7 |
| Generously allowed | 1.2 | 0.4 |
| Disallowed | 0.0 | 0.0 |
R-Factor = (Σ|Fo − Fc|/Σ|Fo|) × 100 where Fo is the observed structure factor amplitude and Fc. is the calculated structure factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.20
QdtA activity
The 3,4-ketoisomerase activity of QdtA was verified via a coupled assay. Specifically, a 1.0 mL reaction mixture was set up that contained 1 mM dTDP–glucose, 2 mg mL−1 of E. coli RmlB, 1 mg mL−1 QdtA, 4 mg mL−1 T. thermosaccharolyticum QdtB, 40 mM sodium glutamate, and 100 mM HEPPS (pH 8). The functions of RmlB and QdtB are shown in Scheme 2. The reaction was incubated at 37°C for 2 h. Enzymes were removed, and the reaction products were evaluated via HPLC. The amino sugar product had the same retention time as that of a standard dTDP-3-amino-6-deoxyglucose sample. QdtB was purified as previously described.17
Kinetic analysis
The kinetic constants for the 3,4-ketoisomerase reaction were determined via a coupled assay using a Beckman Coulter DU-640 spectrophotometer according to Scheme 4. The activity of QdtA was measured by monitoring the decrease in absorbance at 340 nm as NADPH is oxidized to NADP+ due to the action of KijD10. Previous studies from the laboratory have verified that KijD10 from Actinomadura kijaniata functions as an NADPH-dependent C-3′ ketoreductase.18 The reaction mixtures contained 50 mM HEPES (pH 7.5), 0.2 mM NADPH, 1 mg mL−1 KijD10, and dTDP-4-keto-6-deoxyglucose varying from 0.025 to 5.0 mM. The reactions were performed at 23°C and initiated by the addition of 0.005 mg mL−1 QdtA. No measurable activity was observed for the H51N mutant protein.
Scheme 4.

Coupled assay used to measure QdtA activity.
Sequencing data
The amino acid sequence of QdtA from T. thermosaccharolyticum was deposited into GenBank (AAR85518.1) in 2008. Surprisingly, our multiple and independent PCRs using Platinum Pfx polymerase always showed the following four amino acid substitutions in the PCR products: A52S, F94L, L95V, and C116Y. To verify that these variations from the published sequence were correct and not a function of the polymerase, PCRs were subsequently performed using PfuTurbo DNA Polymerase (Stratagene). The resulting products also contained the same changes from the deposited sequence.
Acknowledgments
The authors thank Professor Grover L. Waldrop for helpful discussions.
Glossary
- dTDP
thymidine diphosphate
- HEPPS
N-2-hydroxyethylpiperazine-N′-3-propanesulfonic acid
- HPLC
high-performance liquid chromatography
- PCR
polymerase chain reaction.
References
- 1.Pfoestl A, Hofinger A, Kosma P, Messner P. Biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-D-galactose in Aneurinibacillus thermoaerophilus L420-91T. J Biol Chem. 2003;278:26410–26417. doi: 10.1074/jbc.M300858200. [DOI] [PubMed] [Google Scholar]
- 2.Pfostl A, Zayni S, Hofinger A, Kosma P, Schaffer C, Messner P. Biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-D-glucose. Biochem J. 2008;410:187–194. doi: 10.1042/BJ20071044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aspinall GO, McDonald AG, Pang H, Kurjanczyk LA, Penner JL. Lipopolysaccharide of Campylobacter coli serotype O:30. Fractionation and structure of liberated core oligosaccharide. J Biol Chem. 1993;268:6263–6268. [PubMed] [Google Scholar]
- 4.Perry MB, MacLean LL. Structural characterization of the antigenic O-chain of the lipopolysaccharide of Escherichia coli serotype O65. Carbohydr Res. 1999;322:57–66. doi: 10.1016/s0008-6215(99)00212-8. [DOI] [PubMed] [Google Scholar]
- 5.Kocharova NA, Blaszczyk A, Zatonsky GV, Torzewska A, Bystrova OV, Shashkov AS, Knirel YA, Rozalski A. Structure and cross-reactivity of the O-antigen of Providencia stuartii O18 containing 3-acetamido-3,6-dideoxy-D-glucose. Carbohydr Res. 2004;339:409–413. doi: 10.1016/j.carres.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 6.Zych K, Perepelov AV, Siwinska M, Knirel YA, Sidorczyk Z. Structures of the O-polysaccharides and classification of Proteus genomospecies 4, 5 and 6 into respective Proteus serogroups. FEBS J. 2005;272:5536–5543. doi: 10.1111/j.1742-4658.2005.04958.x. [DOI] [PubMed] [Google Scholar]
- 7.Bryskier AJ. Macrolides: chemistry, pharmacology, and clinical uses. Paris: Arnette Blackwell. 1993 [Google Scholar]
- 8.Davis ML, Thoden JB, Holden HM. The X-ray structure of dTDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase. J Biol Chem. 2007;282:19227–19236. doi: 10.1074/jbc.M702529200. [DOI] [PubMed] [Google Scholar]
- 9.Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Evolution of functional diversity in the cupin superfamily. Trends Biochem Sci. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- 10.Dunwell JM, Purvis A, Khuri S. Cupins: the most functionally diverse protein superfamily? Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Galperin MY, Koonin EV. Divergence and convergence in enzyme evolution. J Biol Chem. 2012;287:21–28. doi: 10.1074/jbc.R111.241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chantigian DP, Thoden JB, Holden HM. Structural and biochemical characterization of a bifunctional ketoisomerase/N-acetyltransferase from Shewanella denitrificans. Biochemistry. 2013;52:8374–8385. doi: 10.1021/bi401170t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tello M, Rejzek M, Wilkinson B, Lawson DM, Field RA. Tyl1a, a TDP-6-deoxy-D-xylo-4-hexulose 3,4-isomerase from Streptomyces fradiae: structure prediction, mutagenesis and solvent isotope incorporation experiments to investigate reaction mechanism. Chembiochem. 2008;9:1295–1302. doi: 10.1002/cbic.200800021. [DOI] [PubMed] [Google Scholar]
- 14.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 17.Thoden JB, Schaffer C, Messner P, Holden HM. Structural analysis of QdtB, an aminotransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-D-glucose. Biochemistry. 2009;48:1553–1561. doi: 10.1021/bi8022015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubiak RL, Holden HM. Combined structural and functional investigation of a C-3″-ketoreductase involved in the biosynthesis of dTDP-L-digitoxose. Biochemistry. 2011;50:5905–5917. doi: 10.1021/bi200514b. [DOI] [PubMed] [Google Scholar]
- 19.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 20.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]