

Abstract
Integrative structural biology attempts to model the structures of protein complexes that are challenging or intractable by classical structural methods (due to size, dynamics, or heterogeneity) by combining computational structural modeling with data from experimental methods. One such experimental method is chemical crosslinking mass spectrometry (XL-MS), in which protein complexes are crosslinked and characterized using liquid chromatography-mass spectrometry to pinpoint specific amino acid residues in close structural proximity. The commonly used lysine-reactive N-hydroxysuccinimide ester reagents disuccinimidylsuberate (DSS) and bis(sulfosuccinimidyl)suberate (BS3) have a linker arm that is 11.4 Å long when fully extended, allowing Cα (alpha carbon of protein backbone) atoms of crosslinked lysine residues to be up to ∼24 Å apart. However, XL-MS studies on proteins of known structure frequently report crosslinks that exceed this distance. Typically, a tolerance of ∼3 Å is added to the theoretical maximum to account for this observation, with limited justification for the chosen value. We used the Dynameomics database, a repository of high-quality molecular dynamics simulations of 807 proteins representative of diverse protein folds, to investigate the relationship between lysine–lysine distances in experimental starting structures and in simulation ensembles. We conclude that for DSS/BS3, a distance constraint of 26–30 Å between Cα atoms is appropriate. This analysis provides a theoretical basis for the widespread practice of adding a tolerance to the crosslinker length when comparing XL-MS results to structures or in modeling. We also discuss the comparison of XL-MS results to MD simulations and known structures as a means to test and validate experimental XL-MS methods.
Keywords: chemical crosslinking/mass spectrometry, molecular dynamics simulations, distance restraints, hybrid modeling, integrative structural biology
Introduction
The integration of experimentally derived restraints with the computational prediction of protein structure (integrative or hybrid modeling) is a promising avenue for investigating the structures of proteins and multiprotein complexes.1,2 This is especially true for proteins and complexes that have proved to be refractory to the more direct determination of structure using X-ray crystallography and/or nuclear magnetic resonance spectroscopy (NMR) analysis. Integrative structural biology approaches can make use of a number of experimental methods, such as small-angle X-ray scattering, cryoelectron microscopy, and chemical crosslinking/mass spectrometry (XL-MS). XL-MS approaches (reviewed in Refs. 3–6) consists of covalently crosslinking residues adjacent in three-dimensional (3D) space within a protein or complex by reaction with a bifunctional crosslinking reagent, followed by proteolytic digestion of the sample, and detection and identification of crosslinked peptides by mass spectrometry. Tandem mass spectrometry can ideally reveal both the sequence of the crosslinked peptides and the amino acid residues involved in crosslinking. The most commonly used crosslinkers are N-hydroxysuccinimide esters such as bis(sulfosuccinimidyl)suberate (BS3) [Fig. 1(A)], which primarily target amine groups (lysine residues and protein N-termini), although they can also react with hydroxyl-containing residues.7,8 The sites of crosslinking combined with the known length of the crosslinker reagent (11.4 Å for DSS/BS3) provide site-specific structural restraints that can then be used for fold identification,9 identification of protein–protein interactions,10,11 characterization of the subunit architecture of protein complexes12 or atomic-resolution hybrid structural modeling.13–15 Structural restraints from XL-MS can also be used in combination with restraints derived from other methods such as single-particle cryoelectron microscopy, small-angle X-ray scattering, and others,1 for further improvements in structural modeling.
Figure 1.
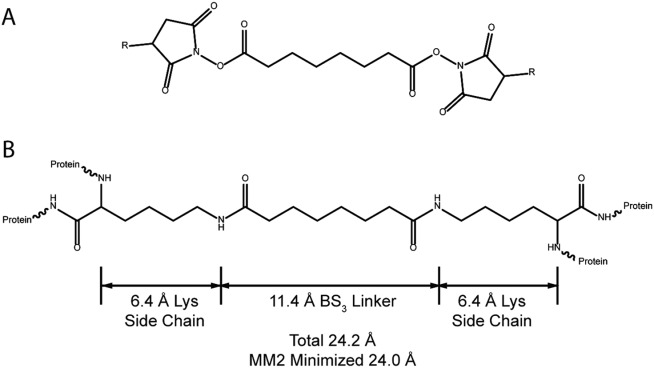
Chemical structure of NHS ester crosslinker reagents and crosslinked lysine residues. (A) R = H, structure of disuccinimidylsuberate (DSS); R = SO3—, bis(sulfosuccinimidyl)suberate (BS3). These two compounds differ only in the leaving group; the crosslinked product is the same. (B) two lysine residues crosslinked by BS3 or DSS through their ζ-amino groups. Models were constructed and subjected to molecular mechanics minimization, and distances measured with ChemDrawBioUltra and ChemDrawBioUltra 3D (CambridgeSoft, Cambridge, MA, USA).
Many studies demonstrating the application of XL-MS to proteins of known structure have been reported, with a large majority of them using BS3 or its nonsulfonated analog disuccinimidyl suberate [DSS; Fig. 1(A)]. In the fully extended conformation, the linker chain of BS3 has a length of 11.4 Å. This is the maximum distance between crosslinked lysine side-chain amino nitrogen [zeta-nitrogen atom (Nζ) of lysine side-chain (also known as the “epsilon amino group” because it is attached to the epsilon carbon)] atoms after the crosslinking reaction has occurred. In other words, the Nζ atoms that become crosslinked must approach to within this distance for a sufficient amount of time for the crosslinking reaction to occur. However, in many XL-MS studies examining proteins of known structure, a small proportion of confidently identified crosslinked peptides indicate crosslinking between pairs of lysine residues with Nζ atoms further apart than the expected fully extended length of 11.4 Å as determined in the solved protein structure. These observed crosslinks may sometimes arise from unintended intermolecular crosslinking or from perturbed structures, but the observation is sufficiently common to suggest that an alternative explanation is needed. Protein dynamics or conformational flexibility is often cited as such an explanation. To account for dynamics of the lysine side chains, an alternative maximum distance of ∼24 Å between Cα (alpha carbon of protein backbone) atoms is often used, based on the distance between peptide backbone Cα atoms when the lysine side chains and the linker are all fully extended [Fig. 1(B)]. This threshold distance implicitly accounts for side-chain dynamics, assuming that the backbone motions are negligible. However, because the crystal structure Cα–Cα distances of some observed crosslinks still exceed this threshold, many authors add an additional tolerance of ∼3–6 Å. The assumption of a dynamic backbone is evidently required to interpret these results. The magnitude of this tolerance is somewhat arbitrary, although there is some empirical justification based on distributions of crosslink distances in individual studies (e.g., refs. 12 and 16). Thus, there is a need to realistically estimate the influence of protein flexibility on Nζ–Nζ and Cα–Cα distances.
A small number of studies have used a variety of computational methods17–21 to characterize the influence of protein dynamics on XL-MS results for individual proteins, but a general analysis of how protein motions influence XL-MS results has not yet been presented. In this study, we use an extensive repository of molecular dynamics (MD) simulations, the Dynameomics database,22–25 to calculate the time-dependent distances between intramolecular pairs of lysine Nζ or Cα atoms. MD is a classical mechanics simulation method that has been extensively used to characterize the motions of proteins and to generate ensembles of protein conformations. The output of an MD simulation is a trajectory of the coordinates of every atom contained in the simulation through time. Thus, lysine–lysine distances from simulation can be compared with both static experimental structures and the results of XL-MS experiments.
We have used the MD simulations of the Dynameomics database in two ways. First, we compared simulated and experimental distances between reported sites of crosslinking for a single well-studied protein, equine cytochrome c, which is included in the Dynameomics database. We find that in evaluating XL-MS results for a protein of known structure, MD simulations can be of great value in estimating the conformational distribution of crosslinkable side-chains. If multiple experimental structures are available, these can be used to validate the potential for crosslinking as well. The second use of the Dynameomics database was to analyze Nζ–Nζ and Cα–Cα distances across the entire simulation set. Specifically we analyzed 766 protein simulations (those containing more than one lysine residue) from the 807 protein simulations of the Dynameomics Consensus Domain Dictionary (CDD) release set. The CDD contains a simulation of at least one representative protein for essentially all known protein fold families having a structure suitable for simulation.26 Our approach is distinct from previous studies: rather than directly comparing theory and experiment for the same protein, we make use of the unprecedented breadth of sampling of protein dynamic behavior in the Dynameomics database to characterize general trends. Our results suggest that pairs of lysine Nζ atoms much further apart than the 11.4-Å length of the crosslinker (up to ∼40 Å) can approach to within crosslinking distance due to native-state protein dynamics. Dynameomics simulation data also show that the typical Cα–Cα distance cutoff of 27–30 Å, applied to the simulation starting structures, accounts for the vast majority of the Nζ–Nζ pairs that approach to within the crosslinker length (11.4 Å) during a simulation. Since the values estimated from the Dynameomics simulations are consistent with the distance tolerances commonly found in current XL-MS studies, our work provides an improved theoretical justification for a common “expert-driven” practice.
At present, users of mass-spectrometry crosslinking data can be roughly divided into two groups. Both groups want to know if reported crosslinks are correct, but each group approaches the problem from a different direction. The first group (modelers) consists of computational structural biologists who use crosslinking derived distance constraints in structural modeling. These researchers need accurate distance constraints (both in terms of atoms/residues constrained and the constraint distance) to ensure that computational conformational searches reach the correct solution. The second group (experimentalists) consists of those researchers—mass spectrometrists, bioinformaticians, biochemists, and chemists—contributing to the development of new techniques for crosslinking and utilizing the data to design confirmatory experiments such as site-directed mutagenesis. Many new crosslinkers, mass spectrometric detection schemes, and software platforms for XL-MS have been introduced in recent years, and the results of these newly introduced procedures require validation. That validation usually takes the form an XL-MS analysis of a protein of known structure, followed by mapping the observed crosslinks onto that structure to determine whether the observed crosslinks are consistent with the structure. Such validation is usually viewed as a prerequisite to modeling of unknown structures. It is our hope that by providing a solid basis for the magnitude of XL-MS distance constraints, this work will assist both groups of researchers. In particular, we anticipate that the results presented here will be helpful to experimentalists seeking to compare XL-MS results to a known structure without access to MD simulations or other means of generating a structural ensemble.
Results
Distances between crosslinked residues from the XL-MS literature
Because XL-MS is a relatively new technique, many studies have focused on evaluating methods. A large number of XL-MS experiments on model proteins with known 3D structures have been published (e.g., see Refs. 12, 14, 16, and 27–29). These studies often find a subset of crosslinks between lysine Nζ atoms that are notably further apart than the 11.4 Å length of the crosslinker [Fig. 2(A)]. Even after accounting for the well-known flexibility of the long lysine side chain, by adding the length of two lysine side chains to the linker length (giving a maximum distance of∼24 Å between Cα atoms, as in Fig. 1(B)), a number of reported crosslinks still have Cα–Cα distances exceeding this new threshold [Fig. 2(B)].
Figure 2.
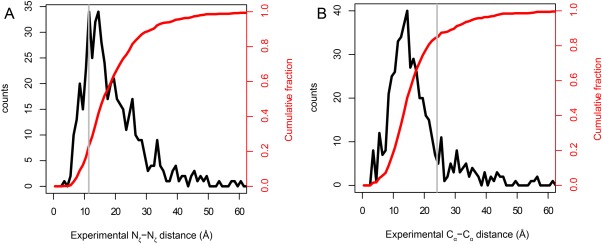
Distances between crossed-linked atoms in XL-MS experiments using BS3 or DSS and proteins with known experimental structures. The data come from the compilation of Kahraman et al.30 Distributions (black) and cumulative distributions (red) are shown with 1-Å bins. The gray lines show the conventional cutoff distances of 11.4 Å for Nζ and 24.4 Å for Cα. The total number of crosslinks included in the data is 502. (A) Experimental Nζ–Nζ distances. Note that the central tendency of the Nζ–Nζ distances is considerably larger than the 11.4-Å span of the fully extended BS3 linker, and that the distribution has a heavy tail at larger distances. (B) Experimental Cα–Cα distances. The Cα–Cα distance distribution peaks at distances considerably lower than the maximum theoretical Cα–Cα distance (24 Å, see Fig. 1).
The recently published curated database of chemical crosslinks from the literature, called XLdb, by Kahraman et al.30 has enabled a quantitative assessment of this phenomenon across a large number of protein structures. Figure 2 shows the Nζ–Nζ and Cα–Cα distance distributions from this compilation of literature data. For both inter- and intramolecular cases, only ∼19% of observed crosslinks had a Nζ–Nζ distance shorter than the crosslinker arm of BS3 or DSS (11.4 Å). Given this large proportion of the results, and the number of independent studies considered, it is unlikely that the primary reason for this finding is an artifact such as aggregation.4 A more likely explanation is that because of dynamics, the Nζ–Nζ distances probed by the XL-MS experiment are different than the distances seen in the experimental structures [Fig. 3(A)]. These data, taken at face value, suggest that an upper bound crosslinking distance between Nζ atoms of 35–40 Å may be more appropriate than 11.4 Å.
Figure 3.
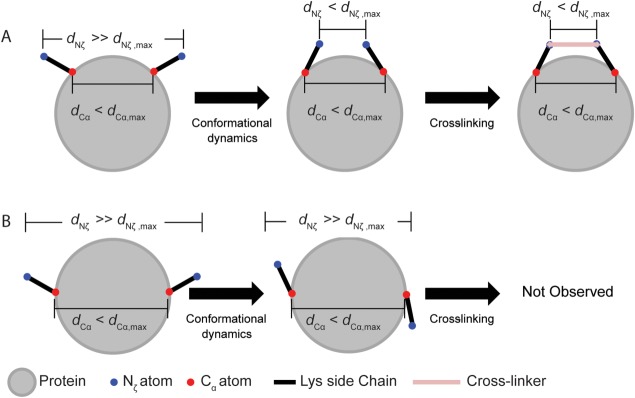
Schematic of dynamics-dependent crosslinking. A, lysine residues with Nζ atoms further apart than the linker length dNζ,max can be brought into apposition by side chain motions. This is only possible if the Cα atoms are within dCα,max as defined in Figure 1(B). (B) Cα–Cα distances shorter than dCα,max do not necessarily imply that crosslinking is possible. Even though the Cα atoms are nominally in range, the Nζ atoms cannot be brought close enough for crosslinking to occur. This situation highlights how a Cα distance criterion may be insufficient for evaluating whether a crosslink is consistent with a known structure.
In contrast to the Nζ data, only, ∼84% of the observed intramolecular crosslinks and ∼86% of the observed intermolecular crosslinks have Cα distances smaller than the theoretical (i.e., both linker and side chains fully extended) Cα–Cα distance of 24 Å [Fig. 2(B)], suggesting that the common 24-Å criterion is in fairly close agreement with the data [Fig. 2(B), gray line]. The contrast between Nζ and Cα highlights the strength of the Cα-based criterion. The summed lengths of the linker and the two lysine side chains [24 Å; Fig. 1(B)] represent a hard upper limit to the Cα–Cα distance in the crosslinked conformation. Figure 2(B) and current practice both suggest that an additional tolerance should be added to account for backbone dynamics that cause the Cα–Cα distance in the crosslinked structure to differ from that seen in the experimental structure. The tail of the distribution in Figure 2(B) implies that this difference can be quite large in some cases.
A case study in validating XL-MS results by comparison to experimental and MD structural ensembles: horse heart cytochrome c
To illustrate how the combination of MD simulations and multiple experimental structures can be used to explain XL-MS results, we present an analysis of previously reported MD and XL-MS results for equine cytochrome c. We selected cytochrome c both because it is part of the Dynameomics database, and because it has been well studied by XL-MS.28,31 Figure 4 shows a comparison of simulated and experimental Cα–Cα and Nζ–Nζ distances for four of the five most confidently identified crosslinks in the protein.28 (Confidence of crosslink assignments was based on the number and quality of MS/MS spectra of peptides containing the specified crosslink.) The experimental measurements are the distances taken from the 40 structures of the NMR solution structure ensemble (PDB code 2giw). This view of the data emphasizes that the interatomic distances are time dependent and can fluctuate due to any type of motion in the protein, whether those motions are global or local, involving side-chains or backbone. The range of distances represented in the simulations is comparable to the range of distances seen in the NMR ensemble, supporting the fidelity of the simulations to the starting structures. (However, lysine residues in NMR structures often have very few experimental NOE restraints, and their conformation in reported structures may be largely determined by a molecular mechanics potential function similar to that used in our simulations.)
Figure 4.
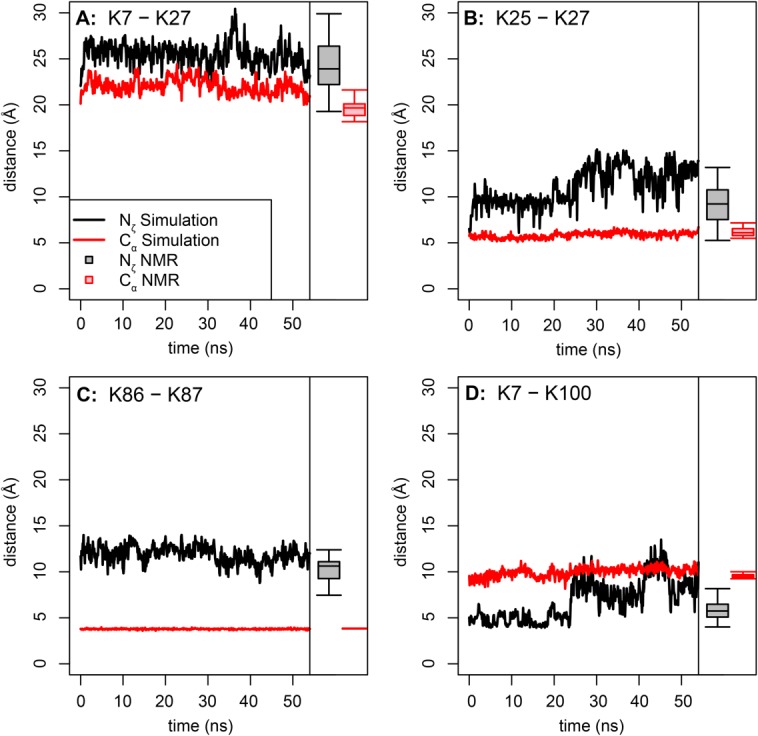
Comparison of simulation and experimental distances for four XL-MS identified crosslinks from equine cytochrome c. (A) K7–K27 distances; (B) K25–K27 distances; (C) K86–K87; (D) K7–K100 distances. The experimental distances, taken from the 40 structures of the 2giw NMR ensemble, are shown as box plots at the right of each plot. The first structure of the NMR ensemble was used, after minimization, as the starting structure for this simulation. The maximum extent of the BS3 crosslinker is 11.4 Å from Nζ to Nζ or 24 Å from Cα to Cα, as shown in Figure 1. Confident crosslinked sites were identified in Ref. 28. Note the good agreement between simulation and experiment. See text for further discussion.
The crosslinkable residue pairs K25–K27, K86–K87, and K7–K100 all spend a substantial fraction of the simulation with their respective pairs of Nζ atoms closer than the 11.4-Å fully extended linker threshold distance. K86 and K87 are adjacent, but point in opposite directions, in a “tail-to-tail” arrangement. Since the residues are adjacent, the Cα atoms are separated by only three covalent bonds, which constrains the Cα–Cα distance. Therefore, the fluctuations in the Nζ–Nζ distance are due to side-chain motions and rotations of the backbone. Side chain motions are also likely of importance for K7–K100. The large jump in the Nζ distance around 23.7 ns is due to rotameric shifts of both lysine residues, with little involvement of the backbone, as these residues are both in stable α-helices. Residues K7 and K27 are always further apart than the threshold, even though crosslinked peptides containing this link were detected at the same level of confidence as the others.28 The K7–K27 Nζ–Nζ distance is always far greater than the conventional cutoff distance, but the Cα–Cα distance is not, suggesting that it is possible for a crosslink to form by additional side-chain motions. Examination of the simulation, however, shows that this is not the case. The two residues are simply too far apart throughout the simulation for crosslinking to occur. The K7–K27 Nζ–Nζ distance in the simulation agrees well with the distances in the 2giw NMR ensemble [Fig. 4(A)], which is expected since the simulation used the first structure in this ensemble (after minimization) as its starting structure. Consequently, we also investigated the lysine–lysine distances in other cytochrome c structures.
Figure 5 compares the distributions of the MD-derived distances between residues reported to be crosslinked28,31 with the distances from eight experimentally derived structures (or structural ensembles), including both NMR and crystal structures. These structures were identified in a search of the PDB for structures of equine cytochrome c that did not contain another protein chain. The agreement between simulation and experiment is excellent: every pairwise distance displays at least some overlap between the experimental and simulated distances. The largest discrepancies are found for lysine 87, which is in a flexible loop that undergoes a conformational change early in the simulation. As expected, since the backbone is more rigid than the side-chains, the Cα–Cα distances distributions are narrower than the Nζ–Nζ distances. More surprising is the heterogeneity of the experimental distances. There are many reasons why interatomic distances in experimental protein structures can vary, including crystal packing contacts, experimental conditions such as pH and temperature (for instance, many modern crystal structures are determined at cryogenic temperatures, whereas NMR experiments are carried out at a variety of temperatures, some in excess of physiological), and even differences in data processing and refinement protocols. In the case of homomultimers or structures with multiple copies of a molecule in the same crystallographic asymmetric unit, multiple conformations can be present in a single structure. Thus, if the various experimental structures are considered as samples from the same underlying native-state distribution of conformations, it follows that this native-state distribution is relatively broad. The simulations also aim to sample the same distribution and, therefore, they also reflect that conformational breadth. Therefore, when evaluating the agreement between XL-MS and a known protein structure, it can be advantageous to consider both MD simulations and all of the available experimental structures. To return to the example of cytochrome c, if the K7- K27 crosslink (labeled in red in the Fig. 5) is compared only with the 2giw ensemble of structures (experimental and simulated), the Nζ–Nζ distance will seem to exceed the 11.4 Å cutoff by ∼10 Å. However, the Nζ–Nζ distances for K7–K27 are much closer to the cutoff in the 2crc and 3o20 structures. Hence, the degree to which the results of an XL-MS experiment seem to “agree” with experimental structures can be dependent on the choice of experimental structure. Using several crystal structures reveals that the K7 and K27 Nζ atoms can approach to within the reach of the BS3 linker arm.
Figure 5.
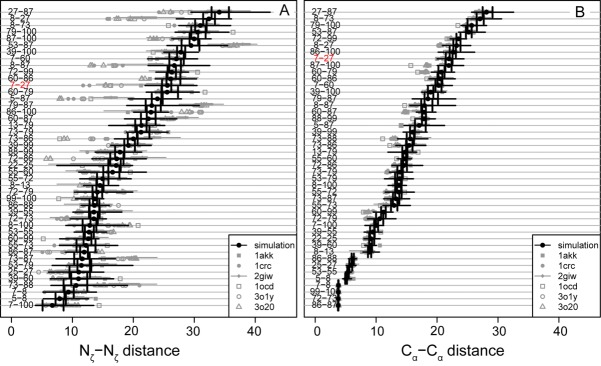
Experimental and simulation lysine–lysine distances for residues reported to be crosslinked with BS3. (A) Nζ–Nζ data for cytochrome c from horse. (B) Cα–Cα data for cytochrome c from horse. Black circles, simulation median values; vertical black lines, first and third quartile distances; heavy gray line, maximum and minimum distances; gray symbols, experimental distances derived from x-ray crystallography or NMR spectroscopy from the structures indicated by PDB code in the legend. 1crc, 1hrc, 3o1y and 3o20, are crystal structures containing one, three, and three molecules in the asymmetric unit, respectively. 1giw, 2giw, 1akk, 2frc, and 1ocd are NMR structures. 2giw is an ensemble with 40 structures; the others are all single minimized average structures. Crosslinking data from28,31. Note that both the experimental and simulation ensembles are quite broad, and for most residues, there is substantial overlap between experiment and simulation. Many of the exceptions involve lysine 87, which is located in a flexible loop that undergoes a conformational change in conformation early in the simulation. In both A and B, the K7-K27 link, discussed in the text, is highlighted in red.
Deriving general tolerances for crosslinking distances with the Dynameomics database
Although MD simulations have previously been used to evaluate XL-MS data from individual proteins,17,18,21 our approach is fundamentally different. Rather than comparing simulations and experiments of the same protein, we are using the Dynameomics simulations to estimate general protein conformational properties. (The 807 protein targets in the Dynameomics Database represent 95% of all known protein folds.) Therefore, taking the Dynameomics simulations as representative of all proteins, we attempt to estimate the maximum distance in an experimental structure that is likely to allow crosslinking, as defined by pairs of Nζ atoms approaching within 11.4 Å (the length of the fully extended DSS linker).
We analyzed 766 relevant (i.e., containing more than one lysine residue) simulations from the 807 simulations in the Dynameomics CCD release set, for a total of 43,511 lysine–lysine pairs. The starting distances, d0, between these pairs range from ∼4 Å to more than ∼100 Å. Most starting distances (which are approximately equal to the experimental distances) are much greater than the 11.4 Å threshold, as are the corresponding simulation median distances. As expected, the median and starting distances are highly correlated (Pearson correlation coefficient R = 0.94; Fig. 6). However, many pairs with starting distances much greater than the conventional cutoff still spend at least some time within this cutoff [Fig. 6(B)]. Using a “distance of closest approach” criterion as in Ref. 18, two residues are judged to be in range if the minimum distance during the course of the simulation is less than the crosslinker length (11.4 Å). By this criterion, the data in Figure 6 suggest that some lysine–lysine pairs with starting distances as great as ∼35–40 Å could still become crosslinked, in keeping with the experimental results in Figure 2(A). It is not known what proportion of time lysine Nζ atoms needs to remain within 11.4 Å of each other in order for the crosslinking reaction to proceed. Crosslinking reactions rates are influenced not only by intrinsic chemical kinetics, but also by the Nζ–Nζ distance, solvent accessibility, and the local electrostatic environment of the lysine amino group.32–34 Therefore, a maximum allowable starting distance based on the distance of closest approach may be an overestimate.
Figure 6.
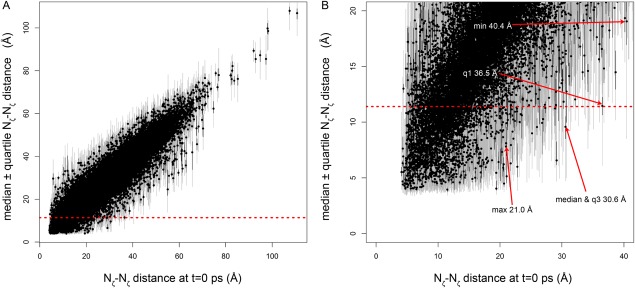
Nζ–Nζ distances for 43,511 lysine–lysine pairs from the Dynameomics database as a function of the simulation starting distance (approximately equal to the crystal structure). Black circles indicate median values, black lines stretch from first to third quartile distances, and gray lines indicate the extrema. Red dotted line indicates the 11.4-Å span of the BS3 crosslinker. (A) All data; (B) close-up of the data in A. Red arrows indicate the pairs with the greatest starting distance for which the indicated distance measure is less than the 11.4-Å cutoff. “Min,” “q1,” “median,” “q3,” and “max” indicate the minimum, first quartile, median, third quartile, and maximum of the Nζ–Nζ distance distribution for the indicated pair. These data indicate that because of dynamics, it may be possible for pairs of lysine Nζ atoms as far apart as ∼40 Å in the starting structure to approach to within 11.4 Å and thus potentially become crosslinked.
There are clear advantages to converting XL-MS results to Cα-based distance restraints for purposes of structural modeling. However, since the Nζ atoms actually participate in the crosslinking reaction, the potential for the reaction to occur is directly determined by the Nζ–Nζ distance, and only secondarily by the Cα–Cα distance. Since the simulations directly track the Nζ atoms, we can estimate the Cα–Cα distance criterion that would account for the actual Nζ–Nζ distances observed in the simulations. To estimate this upper-bound Cα–Cα distance, we divide the observed Cα d0 values into 1-Å bins and calculate the fraction of lysine–lysine pairs having Nζ–Nζ distance less than 11.4 Å in each bin. The distance at which this fraction approaches zero is taken as the maximum distance at which crosslinking is possible (Fig. 7). Figure 7(A) shows that the fraction in range approaches zero in the range of 24–30 Å. The values at which the fraction in range reaches exactly zero are 22, 26, 26, 28, or 38 Å when the distance metric used is the maximum, third quartile, median, first quartile, or minimum of the distance distribution for the given lysine–lysine pair, respectively. These values correspond to the Cα–Cα simulation starting distances that would allow crosslinking to occur, assuming that the crosslinking reaction requires residues to be in range during 100, ≤75, ≤50, ≤25, or >0% (equivalent to using the distance of closest approach) of the duration of the simulation, respectively. In other words, according to the simulations, if a pair of Cα atoms is within 38 Å in the starting structure, the instantaneous distance between the corresponding Nζ atoms is <11.4 Å at some point during of the simulation. This corresponds to the distance of closest approach criterion [black line in Fig. 7(A)]. If a pair of Cα atoms is within 26 Å of each other, there is a small but nonzero probability that the corresponding Nζ atoms are within 11.4 Å for 50% of the simulation. The time for a crosslinking reaction to occur is not well understood, but Figure 7(A) shows that the likely upper distance limit of 24–30 Å is robust to time fractions between 0 and 50% of an approximately 50-ns trajectory. For Cα starting distances greater than 30 Å, the time lysine pairs spend time with their Nζ atoms within the cutoff distance becomes vanishingly rare [Fig. 7(B)].
Figure 7.
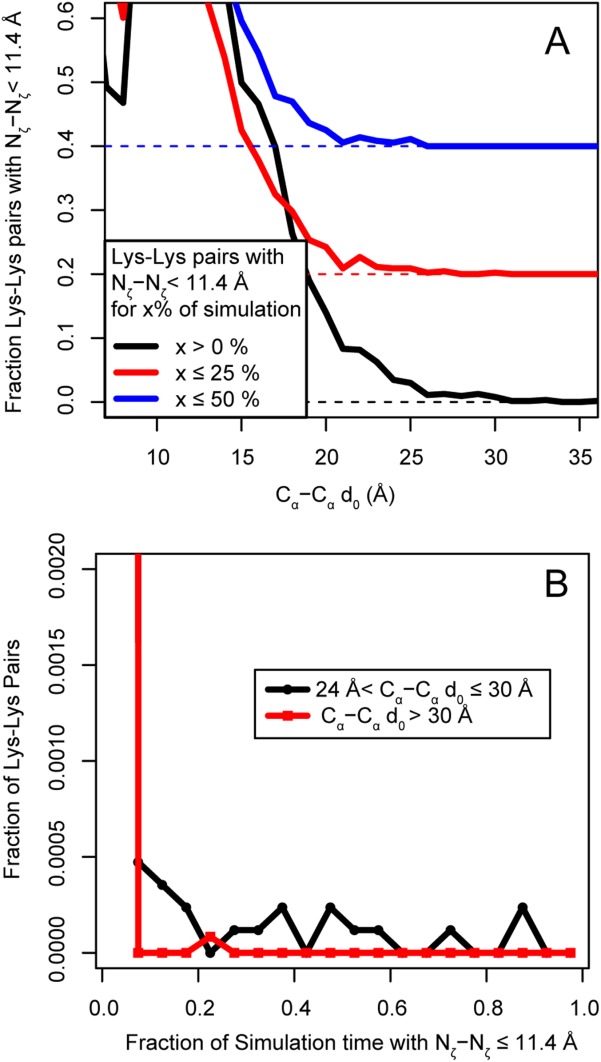
(A) Histograms showing the relationship between Cα–Cα distances in the simulation starting structures d0 and the fraction of simulation time in which the corresponding Nζ atoms are within then 11.4 Å length of the BS3 crosslinker. Black line, fraction of pairs with Nζ atoms that come within range at any point in the simulation; red line, fraction of pairs in range for up to 25% of the simulation; blue line, fraction of pairs in range for up to 50% of the simulation. For clarity, the red trace has been shifted vertically by 0.2 units and the blue trace by 0.4 units. The red and blue dotted horizontal lines show the respective zero points. The value of the Cα–Cα simulation starting distance where the fraction of pairs in range approaches zero is between 25 and 30 Å for the black and red traces and between 21 and 26 Å for the blue trace. These distances represent the Cα–Cα simulation starting distances that would allow crosslinking to occur, assuming that the crosslinking reaction requires residues to be in range greater than 0, 25, or 50% of the simulation time. (B) Histogram of the fraction of Lys–Lys pairs having Nζ atoms closer than 11.4 Å for the indicated fraction of simulation time. Black trace, pairs with Cα–Cα d0 between 24 and 30 Å; red trace, pairs with Cα–Cα greater than 30 Å. Pairs with distances between Cα–Cα d0 between 24 and 30 Å can still come into range due to dynamics, although rarely. Beyond 30 Å, this is vanishingly rare, supporting the choice of 24–30 Å as the upper bound for Cα–Cα d0 of a crosslinkable pair. Note that this range overlaps well with the 27–30 Å Cα–Cα cutoff used in the literature.
An analysis similar to that in Figure 7(A) comparing fraction of the time Nζ–Nζ are within 11.4 Å with the Nζ–Nζ distances in the starting structures (not shown) gives a range of 24–31 Å. This range, representing the maximum experimental Nζ–Nζ distance that allows Nζ atoms to become closer than 11.4 Å during the simulations, is somewhat shorter than the range suggested by the extreme values in Figure 6, possibly because the extreme values represent outliers. This range is also shorter than that suggested by the distribution in Figure 2(A). The reason for the lower relative occurrence of very long Nζ–Nζ starting distances in the simulation data compared to the experimental data is not clear, but may relate to the size of the proteins in each dataset (the domain-centric approach of the Dynameomics project favors smaller proteins), or to the influence of protein motions that occur on longer time-scales (microseconds to milliseconds), outside the time-scale probed by our ∼50 ns simulations.
Discussion
Using experimental and simulated structures for validating XL-MS results
Numerous XL-MS studies report methodological innovations in linker reagents, mass spectrometric techniques, and data processing. Frequently, the authors of these studies seek to demonstrate the validity of their techniques by applying them to a protein of known structure. We have discussed the advantages of using all available experimental structures as well as MD simulations where available. Each type of comparison has advantages and disadvantages.
For experimental structures, the positions of lysine Nζ atoms may not be highly reliable. The dynamic nature of lysine side chains is well known. The long aliphatic chain, with its four rotatable bonds, can assume up to 81 different low-energy conformations [Ref. 35 and available at: http://www.dynameomics.org/rotamer/LYS.aspx]. Lysine residues also occur on the surface of proteins, where their conformation can be relatively unconstrained by contacts with other residues. Further, the conformations of surface lysine side chains may not be experimentally well determined. In crystal structures, electron density may not be adequate to precisely define side chain orientation, and, as surface residues, the atoms of a lysine residue will tend to have higher B-factors than buried atoms. Perhaps more importantly, lysine side-chains may be influenced by crystal packing contacts. Similarly, the conformations of lysine residues in NMR structures may be determined by only a few NOE restraints, or maybe even none at all for the Cε atoms. Still, the conformations in experimental structures are not meaningless, and usually at least some experimental data supports the reported conformations. These conformations, while not necessarily representing the equilibrium or average solution conformations, can still be considered as samples from the ensemble of possible conformations.
MD, Monte Carlo simulation, or other computational techniques can also be used to generate more extensive sampling of the conformational ensemble. The best evaluation of XL-MS results is probably achieved by comparison with a high-quality MD ensemble. However, not all researchers have easy access to the necessary computational resources or expertise. This work aims to make some of the conclusions of MD simulations available to all XL-MS researchers. Furthermore, MD simulations are not perfect, being limited by the computationally accessible time scale and influenced by the quality and conformations of the starting structures. Therefore, XL-MS results should also be compared to simulations and to multiple structures of the protein under study, if available, including, for example, all of the members of an NMR ensemble, or all the copies of a protein in the asymmetric unit of a crystal structure, or even ligand-bound and ligand-free structures. Crystallographic B-factors, which encode information on the fluctuation and uncertainty of atomic coordinates, can also be considered.36,37
When comparing XL-MS derived crosslinks to a known structure, the criterion of Cα–Cα distance, should be combined with visual inspection of the experimental structure, and with consideration of the possible Nζ–Nζ distances due to likely dynamics. Lysine side chains often point outward into solution, away from the center of the protein. Thus, for some small proteins such as those used in many XL-MS validation studies, the Cα atoms could be much closer together than the Nζ atoms. Some Cα pairs could even lie well within the 26–30 Å cutoff, but with the Cα–Cα vector passing through the tightly packed center of the protein [Fig. 3(B)], which is a physically unrealistic path for a crosslinker molecule. Another excellent method is the solvent accessible surface distance (SASD) calculated by the program Xwalk.38 SASD measures the path a crosslinker would take along the solvent-excluded surface of the protein, thus accounting for both changes in side-chain conformation and solvent accessibility. Xwalk uses a SASD cutoff of 30 Å between Cβ atoms in order to account for protein flexibility. Since the goal of this study was to explicitly model protein flexibility, we have not pursued SASD calculations. Furthermore, while Xwalk is well suited to analyzing individual proteins, SASD would be computationally expensive to calculate for an entire simulation, and prohibitively so for the entire Dynameomics database.
Comparison of MD-derived threshold values with literature
Our estimated maximum crosslinking distance values (24–30 Å between Cα atoms) agree well with the experimental distance distribution (Fig. 2). For Cα–Cα distances, 89.3% of the observed crosslinks fall below our recommended maximum threshold of ∼30 Å. Our recommended Cα threshold value is similar to the values applied by Aebersold and coworkers (30 Å)14,16,39 and only slightly longer than the value applied by Sinz and coworkers (27.4 Å).29 Rappsilber and coworkers have used 27.4 Å,12 but more recently they suggest a range (25–29 Å).40 Thus, the Dynameomics-based maximum Cα recommendation is in line with the empirically determined threshold values already in common use.
The Nζ atoms of a crosslinked pair must approach and remain within the crosslinker length for a sufficient amount of time in order for the crosslinking reaction to take place. Exactly what fraction of time is sufficient remains unclear. Crosslinking may occur between lysine pairs that rarely sample such a conformation. This situation is known as a “kinetic trap” as described by Fabris and Yu:4 the crosslinking reaction captures a transient or rare conformation of the protein that does not reflect the equilibrium conformation. These authors suggest experimental approaches to identify and avoid kinetic traps, such as maintaining a low crosslinker concentration, and repeating experiments under slightly varying conditions to ensure that the results are robust. However, comparing the data in Figure 2 with the data in Figure 7 suggests that many crosslinks may reflect kinetic trapping to some extent. (Many crosslinks are detected at Cα–Cα d0 values for which the fraction of time the Nζ atoms are in range is very low.) However, since one goal of XL-MS is to gain structural restraints that are useful for modeling, the key question may not be to what extent an observed crosslink reflects the equilibrium structure, but rather, what distance restraint should be used in modeling protocols in order to adequately account for dynamics? The simulation results imply that a distance restraint between 24 and 30 Å is appropriate.
This finding is important for two reasons. First, we have obtained theoretical support for the current and largely unexamined practice of adding a tolerance to the linker length, and confirmed that the commonly employed values of that tolerance are appropriate. Second, since the tolerance was derived from protein dynamics, the values provide a way to estimate the importance of dynamics in determining which residues become crosslinked. Only a minority of crosslinked residues in the experimental dataset appear to be within range of the conventional Nζ–Nζ 11.4-Å cutoff, but 89.3% of the pairs in Figure 1(B) have Cα–Cα distances less than our MD-based estimate of the maximum. Thus, the majority of the observed crosslinks are in a distance regime that is well explained by the type of dynamics observed in the Dynameomics simulations. These dynamics include relaxation from a crystalline to a solution conformation, local motions, such as side-chain rotations, small fluctuations of the backbone, some flexing of secondary structures, and movements of flexible loops and tails. These types of motions occur on time scales of up to 10 s of nanoseconds.41 Large backbone deviations are not usually observed in native-state simulations, and simulations containing large backbone deviations did not pass quality control metrics and were removed from the Dynameomics database.25 Therefore, there is no need to invoke dramatic structural rearrangements or distortions to explain the observed distances between crosslinked residues for the majority of cases, providing confidence that the experimental protocols are successfully probing the native state without substantially perturbing it. There are several possible reasons why, in the remaining ∼10% of cases, the observed distances are greater than the MD-estimated maximum. First, excessive crosslinking or other suboptimal experimental conditions may have induced structural distortions. Second, large amplitude, low-frequency motions that happen on a timescale slower than the 10 s of nanoseconds of the MD simulations could bring the crosslinked residues into apposition. (Crosslinking reactions are typically allowed to proceed for tens of minutes to 2 h.) These slower motions may be functionally relevant, as was the case for a recent XL-MS and normal mode analysis study of calmodulin.20 In that study, the authors showed that motion along a prominent normal mode of the protein brought previously distant crosslinked sites into range. Furthermore, the structural changes inferred from motion along this normal mode resembled the well-known calcium-induced conformational change of calmodulin. Third, the experimental structures may not accurately or completely sample the native state ensemble, as was described above for equine cytochrome c. In this context, it is important to note that the starting conformation for an MD simulation can bias the simulation toward similar conformations, particularly in short simulations.
Implications for structural modeling
A major goal of XL-MS is to provide distance constraints that can be used for structural modeling of protein complexes, in particular docking problems where individual structures may be known but the quaternary structure of the complex is not. A shorter distance restraint results in a smaller number of models that satisfy it. The upper range of our MD-derived distance restraint estimates is greater than some commonly used values and, therefore, may not be as effective at reducing the search space during a modeling or docking calculation.42 However, it has been shown that even loose distance constraints can significantly improve modeling if there are enough of them.42
Figure 3(B) suggests that even when appropriate restraint values are used, simple Euclidean distances may not be sufficient to prevent calculation of spurious models, since a straight-line distance between two atoms can take a path through the center of a protein that could never be traversed by an actual crosslinker molecule. The SASD calculated by Xwalk (see above) effectively prevents this problem. However, SASD is too computationally expensive to use in model-generating calculations in which the distance must be recalculated for each iteration. The more appropriate application of SASD is to use the SASD between crosslinked residues in the final models and used as a postmodeling filter, as described by Kahraman et al.30
In principle, it is possible to use either the aggregate XL-MS data from the literature (Fig. 2)30 or the simulation data to construct statistical restraining potentials for use in modeling. In such an approach, models could be scored by, for example, the sum of their distance-dependent crosslinking probabilities. However, this approach (using the experimental data) resulted in no improvement in the RMSD between models and the targets (A.K., unpublished data). Simply using the Xwalk SASD as a postmodeling filter proved more effective.30 Attempting such an approach with the simulation data is outside the scope of this study.
Conclusions
We have used 766 MD simulations from the Dynameomics Database to investigate the motions of lysine side chains on the time scale of 10s of nanoseconds. This investigation was motivated by a common observation in many XL-MS studies: a subset of confidently identified crosslinked peptides involve linked residues that are too far apart in the known structures for the span of the linker. Our results suggest that the comparison of XL-MS results with an ensemble of structures, whether from simulation or experiment, is important for validation. If a simulation ensemble is available, then the crosslinker length can be used directly as the upper distance limit between crosslinked atoms. If no computational analysis is available, then an upper bound of 26–30 Å for Cα atoms should be used. Distances between Nζ atoms that are much longer than the linker length, even up to 35–40 Å, are not necessarily cause for concern if the Cα atoms fall within the recommended range and analysis of the structure suggests a path between the linked Nζ atoms that does not need to pass through the center of the protein. Our estimated Cα distance constraints are in agreement with those typically used in modeling studies.
Methods
Compilation of XL-MS data from the literature
XL-MS data from equine cytochrome c was taken from the studies of Xu et al.28 and Lackner et al.31 Selected Nζ and Cα atom coordinates from six structures of equine cytochrome c (PDB codes: 1akk, 1crc, 2giw, 1ocd, 3o1y, and 3o2o) were retrieved from the Protein Data Bank and interatomic distances were calculated using a custom Perl script (available on request). Distributions of experimental crosslink distances were taken from the extensive compilation of XL-MS data of Kahraman et al.30 This database was then filtered to contain only XL-MS results that used either BS3 or disuccinimidyl suberate (DSS), a reagent of identical length, as the crosslinker. Crosslinks were flagged as either intramolecular or intermolecular. Only the intramolecular set was used for comparison to the Dynameomics simulations, since the Dynameomics database contains only simulations of monomers. However, the difference between inter- and intramolecular Nζ–Nζ distance distributions was not significant (p=0.32, Welch's two-sample t-test), suggesting that the results are relevant to protein complexes as well. For Cα–Cα distances, the inter- and intramolecular distance distributions were significantly different (p=0.035.), and only the intramolecular set was used. Crosslinks involving residues with missing Cα or Nζ atoms were also removed, and in a few cases, the identity of the crosslinked subunit was changed to either make the crosslinked distance shorter or account for a missing atom. The final set included 486 intramolecular crosslinks with distances between crosslinked residues calculated from 40 different protein structures.
Dynameomics MD simulations
Simulations were conducted using the in lucem Molecular Mechanics (ilmm43) software package using the Levitt et al. potential function,44 and the F3C water model.45 Detailed protocols for selection of starting structures, preparation and simulation, and quality assurance of the Dynameomics targets have been described elsewhere.25,46 Using SQL queries (available upon request), we extracted the distances between all lysine Nζ atom pairs and all lysine Cα atom pairs from every simulation at 100-ps intervals from the Dynameomics database.24 This sampling frequency was chosen because it was the least-frequent sampling that maintained the distribution of distances for a representative simulation. Of the 807 CCD simulations, 766 simulations contained more than one lysine residue and were subsequently analyzed, comprising a total of 43,364 lysine–lysine pairs. All aggregate measures of simulation distance (median, etc.) were calculated after omitting the first 2 ns of the simulation, to allow for relaxation from the starting conformation. The simulation length varied from 50.999 to 75.522 ns, (average 52.529 ns), for a combined total of 40 µs of simulation time. The simulation starting distances d0 are the distance in the simulated structure at simulation time zero. These starting distances closely approximate the experimental distances of the simulated protein structures, having been only slightly altered (on the order of 0.1 Å Cα RMSD) by the minimization and other protocols used to prepare the structure for simulation. Data were imported into the R statistical computing environment47 for analysis and plotting. Custom R scripts used for the analysis are available upon request.
Acknowledgments
Dynameomics simulations were performed using computer time through the DOE Office of Biological and Environmental Research as provided by the National Energy Research Scientific Computing Center. The authors would like to thank Michael Daily for helpful comments on the manuscript and Kevin Crowell for preliminary work on database queries.
Glossary
- MD
molecular dynamics simulation
- XL-MS
chemical crosslinking/mass spectrometry.
References
- 1.Schneidman-Duhovny D, Rossi A, Avila-Sakar A, Kim SJ, Velázquez-Muriel J, Strop P, Liang H, Krukenberg KA, Liao M, Kim HM, Sobhanifar S, Dötsch V, Rajpal A, Pons J, Agard DA, Cheng Y, Sali A. A method for integrative structure determination of protein-protein complexes. Bioinformatics. 2012;28:3282–3289. doi: 10.1093/bioinformatics/bts628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward AB, Sali A, Wilson IA. Integrative structural biology. Science. 2013;339:913–915. doi: 10.1126/science.1228565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinz A. Chemical crosslinking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 4.Fabris D, Yu ET. Elucidating the higher-order structure of biopolymers by structural probing and mass spectrometry: MS3D. J Mass Spectrom. 2010;45:841–860. doi: 10.1002/jms.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rappsilber J. The beginning of a beautiful friendship: crosslinking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merkley ED, Cort JR, Adkins JN. Crosslinking and mass spectrometry methodologies to facilitate structural biology: finding a path through the maze. J Struct Funct Genomics. 2013;14:77–90. doi: 10.1007/s10969-013-9160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalkhof S, Sinz A. Chances and pitfalls of chemical crosslinking with amine-reactive N-hydroxysuccinimide esters. Analyt Bioanalyt Chem. 2008;392:305–312. doi: 10.1007/s00216-008-2231-5. [DOI] [PubMed] [Google Scholar]
- 8.Mädler S, Bich C, Touboul D, Zenobi R. Chemical crosslinking with NHS esters: a systematic study on amino acid reactivities. J Mass Spectrom. 2009;44:694–706. doi: 10.1002/jms.1544. [DOI] [PubMed] [Google Scholar]
- 9.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular crosslinks and mass spectrometry. Proc Natl Acad Sci USA. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng CX, Yang L, Hoopmann MR, Eng JK, Tang XT, Weisbrod CR, Bruce JE. Crosslinking measurements of in vivo protein complex topologies. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.006841. M110.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisbrod CR, Chavez JD, Eng JK, Yang L, Zheng C, Bruce JE. In vivo protein interaction network identified with a novel teal-time crosslinked peptide identification strategy. J Proteome Res. 2013;12:1569–1579. doi: 10.1021/pr3011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills J-C, Nilges M, Cramer P, Rappsilber J. Architecture of the RNA polymerase II-TFIIF complex revealed by crosslinking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanowar S, Singh P, Pfuetzner RA, Andre I, Zheng HJ, Spreter T, Strynadka NCJ, Gonen T, Baker D, Goodlett DR, Miller SI. Interactions of the transmembrane polymeric rings of the Salmonella enterica serovar typhimurium type III secretion system. MBio. 2010;1 doi: 10.1128/mBio.00158-10. pii: e00158–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl F-U, Ban N, Malmström L, Aebersold R. Structural probing of a protein phosphatase 2A network by chemical crosslinking and mass spectrometry. Science. 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- 15.Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walzthoeni T, Claassen M, Leitner A, Herzog F, Bohn S, Forster F, Beck M, Aebersold R. False discovery rate estimation for crosslinked peptides identified by mass spectrometry. Nat Meth. 2012;9:901–903. doi: 10.1038/nmeth.2103. [DOI] [PubMed] [Google Scholar]
- 17.Green NS, Reisler E, Houk KN. Quantitative evaluation of the lengths of homobifunctional protein crosslinking reagents used as molecular rulers. Protein Sci. 2001;10:1293–1304. doi: 10.1110/ps.51201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobsen RB, Sale KL, Ayson MJ, Novak P, Hong JH, Lane P, Wood NL, Kruppa GH, Young MM, Schoeniger JS. Structure and dynamics of dark-state bovine rhodopsin revealed by chemical crosslinking and high-resolution mass spectrometry. Protein Sci. 2006;15:1303–1317. doi: 10.1110/ps.052040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelter A, Hoopmann MR, Vernon R, Baker D, MacCoss MJ, Davis TN. Isotope signatures allow identification of chemically crosslinked peptides by mass spectrometry: a novel method to determine inter-residue distances in protein structures through crosslinking. J Proteome Res. 2010;9:3583–3589. doi: 10.1021/pr1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Wells SA, Jimenez-Roldan JE, Römer RA, Zhao Y, Sadler PJ, O'Connor PB. Protein flexibility is key to cisplatin crosslinking in calmodulin. Protein Sci. 2012;21:1269–1279. doi: 10.1002/pro.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pettelkau J, Schroder T, Ihling CH, Olausson BES, Kolbel K, Lange C, Sinz A. Structural insights into retinal guanylylcyclase-GCAP-2 interaction determined by crosslinking and mass spectrometry. Biochemistry. 2012;51:4932–4949. doi: 10.1021/bi300064v. [DOI] [PubMed] [Google Scholar]
- 22.Beck DA, Jonsson AL, Schaeffer RD, Scott KA, Day R, Toofanny RD, Alonso DO, Daggett V. Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations. Protein Eng Des Sel. 2008;21:353–368. doi: 10.1093/protein/gzn011. [DOI] [PubMed] [Google Scholar]
- 23.Kehl C, Simms AM, Toofanny RD, Daggett V. Dynameomics: a multi-dimensional analysis-optimized database for dynamic protein data. Protein Eng Des Sel. 2008;21:379–386. doi: 10.1093/protein/gzn015. [DOI] [PubMed] [Google Scholar]
- 24.Simms AM, Toofanny RD, Kehl C, Benson NC, Daggett V. Dynameomics: design of a computational lab workflow and scientific data repository for protein simulations. Protein Eng Des Sel. 2008;21:369–377. doi: 10.1093/protein/gzn012. [DOI] [PubMed] [Google Scholar]
- 25.van der Kamp MW, Schaeffer RD, Jonsson AL, Scouras AD, Simms AM, Toofanny RD, Benson NC, Anderson PC, Merkley ED, Rysavy S, Bromley D, Beck DAC, Daggett V. Dynameomics: a comprehensive database of protein dynamics. Structure. 2010;18:423–435. doi: 10.1016/j.str.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaeffer RD, Jonsson AL, Simms AM, Daggett V. Generation of a consensus protein domain dictionary. Bioinformatics. 2011;27:46–54. doi: 10.1093/bioinformatics/btq625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rinner O, Seebacher J, Walzthoeni T, Mueller L, Beck M, Schmidt A, Mueller M, Aebersold R. Identification of crosslinked peptides from large sequence databases. Nat Meth. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H, Hsu P-H, Zhang L, Tsai M-D, Freitas MA. Database search algorithm for identification of intact crosslinks in proteins and peptides using tandem mass spectrometry. J Proteome Res. 2010;9:3384–3393. doi: 10.1021/pr100369y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritzsche R, Ihling CH, Gotze M, Sinz A. Optimizing the enrichment of crosslinked products for mass spectrometric protein analysis. Rapid Commun Mass Spectrom. 2012;26:653–658. doi: 10.1002/rcm.6150. [DOI] [PubMed] [Google Scholar]
- 30.Kahraman A, Herzog F, Leitner A, Rosenberger G, Aebersold R, Malmström L. CrossLink guided molecular modeling with ROSETTA. Plos One. 2013;8:e73411. doi: 10.1371/journal.pone.0073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee YJ, Lackner LL, Nunnari JM, Phinney BS. Shotgun crosslinking analysis for studying quaternary and tertiary protein structures. J Proteome Res. 2007;6:3908–3917. doi: 10.1021/pr070234i. [DOI] [PubMed] [Google Scholar]
- 32.Guo X, Bandyopadhyay P, Schilling B, Young MM, Fujii N, Aynechi T, Guy RK, Kuntz ID, Gibson BW. Partial acetylation of lysine residues improves intraprotein crosslinking. Anal Chem. 2008;80:951–960. doi: 10.1021/ac701636w. [DOI] [PubMed] [Google Scholar]
- 33.Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Goshe MB. Combinatorial electrostatic collision-induced dissociative chemical crosslinking reagents for probing protein surface topology. Anal Chem. 2010;82:6215–6223. doi: 10.1021/ac101030w. [DOI] [PubMed] [Google Scholar]
- 35.Scouras AD, Daggett V. The dynameomics rotamer library: amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci. 2011;20:341–352. doi: 10.1002/pro.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clifford-Nunn B, Showalter HDH, Andrews PC. Quaternary diamines as mass spectrometry cleavable crosslinkers for protein interactions. J Am Soc Mass Spectrom. 2012;23:201–212. doi: 10.1007/s13361-011-0288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merkley ED, Baker ES, Crowell KL, Orton DJ, Taverner T, Ansong C, Ibrahim YM, Burnet MC, Cort JR, Anderson GA, Smith RD, Adkins JN. Mixed-isotope labeling with LC-IMS-MS for characterization of protein-protein interactions by chemical crosslinking. J Am Soc Mass Spectrom. 2013;24:444–449. doi: 10.1007/s13361-012-0565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kahraman A, Malmström L, Aebersold R. Xwalk: computing and visualizing distances in crosslinking experiments. Bioinformatics. 2011;27:2163–2164. doi: 10.1093/bioinformatics/btr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Förster F, Aebersold R. Expanding the chemical crosslinking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014126. M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer L, Chen ZA, Rappsilber J. Quantitative crosslinking/mass spectrometry using isotope-labelled crosslinkers. J Proteomics. 2013;88:120–128. doi: 10.1016/j.jprot.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 42.Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Probing native protein structures by chemical crosslinking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. il, mm, in lucem molecular mechanics, Seattle: Beck DA, Alonso DOV, Daggett V; 2000–2008.
- 44.Levitt M, Hirshberg M, Sharon R, Daggett V. Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput Phys Commun. 1995;91:215–231. [Google Scholar]
- 45.Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J Phys Chem B. 1997;101:5051–5061. [Google Scholar]
- 46.Beck DAC, Daggett V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods. 2004;34:112–120. doi: 10.1016/j.ymeth.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 47.R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]