

Abstract
Repeat proteins have recently emerged as especially well-suited alternative binding scaffolds due to their modular architecture and biophysical properties. Here we present the design of a scaffold based on the consensus sequence of the leucine rich repeat (LRR) domain of the NOD family of cytoplasmic innate immune system receptors. Consensus sequence design has emerged as a protein design tool to create de novo proteins that capture sequence-structure relationships and interactions present in nature. The multiple sequence alignment of 311 individual LRRs, which are the putative ligand-recognition domain in NOD proteins, resulted in a consensus sequence protein containing two internal and N- and C-capping repeats named CLRR2. CLRR2 protein is a stable, monomeric, and cysteine free scaffold that without any affinity maturation displays micromolar binding to muramyl dipeptide, a bacterial cell wall fragment. To our knowledge, this is the first report of direct interaction of a NOD LRR with a physiologically relevant ligand.
Keywords: leucine rich repeat, NOD, MDP, consensus design, binding scaffold, repeat protein
Introduction
The advancement of protein engineering tools, such as synthetic libraries and evolution and selection technologies, has allowed substantial scientific progress towards the design of protein scaffolds with novel binding specificities.1,2 However, most of the engineered protein scaffolds to date were directed against protein targets.2 Other classes of biomolecules such as carbohydrates and nucleic acids have not been the focus of scaffold development until very recently.3–11 The need for alternative scaffolds with affinity and specificity for carbohydrates is especially pronounced since they are inefficient antigens for the adaptive immune system, and traditional antibodies are difficult, if not impossible, to produce.12 Most readily available carbohydrate binding proteins, such as lectins and antibodies, typically display either broad specificity or low affinity for their antigen. Additionally, carbohydrate-binding proteins are available for only a small fraction of known glycans, in spite of their abundance and importance.13
Recently, repeat proteins emerged as especially well-suited binding scaffolds due to their modular structure that facilitates the binding of a variety of nonrelated protein and peptide ligands. Many repeat proteins, such as ankyrin repeats (ANK), tetratricopeptide repeats (TPR), leucine rich repeats (LRR), and armadillo (ARM) repeats have been successfully used for the development of novel binding scaffolds.5,14–20 The evolutionary advantage of a modular architecture is the possibility to evolve the function through not only point mutations, but also by shuffling, deletion, and/or insertion of repeats. This property makes repeat proteins a particularly attractive system for functional protein engineering.21–28 For example, a specific set of tandem repeats can target a particular cellular compartment, while another set of tandem repeats binds specific endogenous ligands. These types of scaffolds would be beneficial for functional genomics, in vivo imaging, and drug delivery applications.
In contrast to the adaptive immune system, which uses the immunoglobulin scaffolds for ligand binding, the innate immune system relies primarily on LRR protein motifs for target recognition.29 In mammals, two main protein families of such receptors have been identified: extracellular Toll-like receptors (TLRs) and cytoplasmic Nod-like receptors (NLRs). The common feature of both families is the presence of the LRR motif.30 Cocrystal structures of TLR receptors with their ligand indicate that the LRR domain is the ligand binding site.31 In analogy to TLRs, it is proposed that NLRs also bind ligands using their LRR motif.30 Studies of cytoplasmic NLRs showed that these proteins bind a large repertoire of ligands including bacterial cell-wall peptidoglycans, bacterial RNA, uric crystals, and antiviral imidazoquinone.30,32 Thus, we hypothesized that LRR motifs from NLR proteins are especially well poised to function as a framework for development of glycan and nucleotide binding scaffolds since chemically similar types of molecules are within the repertoire of their natural ligands.
Here we describe the design of a peptidoglycan binding protein scaffold based on the LRR domain present in a NOD subgroup of NLR receptors of vertebrates.33 The consensus sequence design resulted in a stable, monomeric, and cysteine free scaffold that without any affinity maturation displays micromolar binding to the muramyl dipeptide, a bacterial cell wall fragment.
Results and Discussion
Repeat protein scaffolds
Repeat proteins are a ubiquitous class of proteins characterized by successive homology motifs that stack in tandem.22,27,34 They are unique in the way that their well-defined three-dimensional structure is dominated by short-range, regularized intra- and inter-repeat hydrophobic interactions. For several classes of repeat proteins, analyses of amino acid variability at different positions within a single repeat have revealed that residues that compose the ligand binding site are significantly more variable than the other positions on the protein surface.35,36 This sequence-function relationship is analogous to the complementarity determining regions (CDR) of antibodies37 and is consistent with the notion that repeat proteins provide a constant framework that displays ligand-binding residues. This spatial separation of framework and ligand-binding function is important for the design of binding scaffolds so that the ligand-binding function does not compromise the overall structure and stability.
Consensus sequence design
Consensus sequence design has emerged as a protein design tool to create de novo proteins that capture sequence-structure relationships and interactions present in nature.34,38 Proteins created in this way are idealized structural motifs optimized for stability.39,40 There are two motivations for using consensus design of repeat proteins as opposed to randomizing the surface of one particular family member. First, consensus design can markedly increase stability of engineered proteins. Second, full-consensus design in which all repeats are the same allows for addition, deletion, and shuffling of repeats.1,41 Additionally, the design of consensus sequences exposes principal features of the protein architecture, which is important for subsequent engineering and chemical coupling.
LRR domains in NLR proteins
In NLRs there is a striking correlation (not observed for analogous TLR proteins) between gene organization and the amino acid sequence of their LRR domain.28,30 Thus based on their gene architecture NLRs are divided into NLRP (α and β) and NOD subgroups.30 Specifically, in a NLRP subgroup, LRR domains are formed by tandem repeats of exons where each exon encodes one central LRR repeat (β) and two halves of the neighboring LRRs (α). In a NOD subgroup, LRR domains are encoded by a single exon per repeat. This modular organization may have important structural and functional consequences and possibly allows extensive alternative splicing of the entire LRR domain. Furthermore, the remarkable gene structure of LRR domains from NLR proteins raises their potential as a modular scaffold for engineering multivalency and multispecificity.
Multiple sequence alignment
We have analyzed the multiple sequence alignment (MSA) of individual repeats in NLR proteins according to the previously published procedure for TPR proteins.42 Briefly, confirmed human NLR proteins were identified in the HUGO Gene Nomenclature Committee (HGNC) database.30 We have chosen this database because it provides the most complete information on both the DNA and protein level together with exon–intron gene structure. Nineteen of the 22 NLR proteins encoded in the human genome that contain LRR domains and follow the typical exon pattern were then blasted, using the blastp algorithm from the National Center for Biotechnology Information (NCBI) to find all homologs mammalian proteins. Only confirmed protein products were selected from this search. At this point the database was curated to exclude alternatively spliced variants of the protein. Through manual searching of selected protein sequences, repeats were extracted and aligned in Microsoft Excel. This procedure resulted in 311 NOD, 255 NLRP-α, and 262 NLRP-β individual LRRs that were then included in the MSA. Using Excel counting functions the greatest percent of occurrence was determined for each position of the repeat. Comparing conserved and variable positions of individual repeats for LRR domains of NLRP and NOD proteins will indicate differences, if any, at the sequence level between the two subgroups.
Consensus sequence of NOD subgroup
MSAs for NOD, NLRP-α, and NLRP-β repeats resulted in three consensus sequences. The sequence conservation for each subgroup is shown by sequence logos.43 The sequence identities for the NOD and NLRP-β consensus sequences are 57%; however, the overall conservation in most positions in the NLRP-β sequence exceeds the NOD. For example, positions 6, 13, 18, 19, 21, 22, 25, 26, and 27 in the NLRP-β MSA all have significantly higher conservation than the NOD sequence [Fig. 1(b)]. The consensus sequence for the NLRP-α subgroup differs from both NOD and NLRP-β, sharing a sequence identity of 25% and 21%, respectively. However, the sequence identity between the tandem α and β consensus sequence and the previously reported consensus protein based on the Ribonuclease Inhibitor (RI) is 61%.28 This sequence similarity between NLRP and RI LRR repeats leads us to conclude that α- and β-repeat types do occur in tandem, that is repeating unit is 57 amino acids long. Hence, we decided to pursue design based on the NOD subgroup that will allow for a shorter (28 amino acid) single repeat, a feature preferred for design of a binding scaffold.
Figure 1.
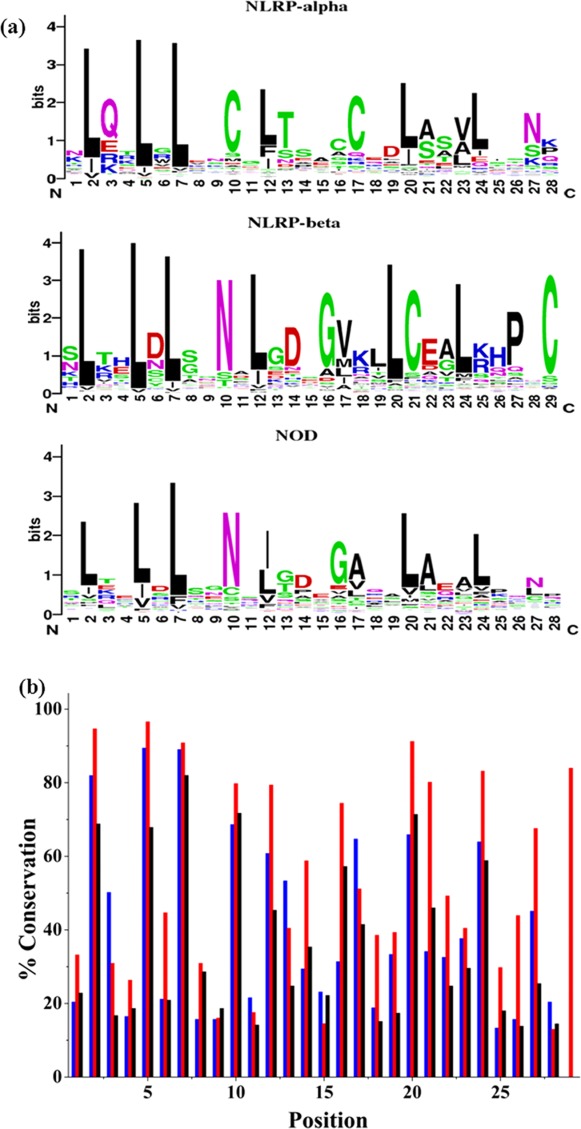
Sequence conservation of NLR subgroups. (a) Sequence logos for each NLR subgroup. The height of individual letters indicates the frequency of occurrence of amino acid at specific position. (b) Percent conservation for each position in the LRR repeat sequence. NLRP-α (blue), NLRP-β (red), and NOD (black).
So-called first generation consensus sequence design proteins were based on the most common amino acid in each of the 28 positions. Proteins consisting of 4, 5, 6, and 10 identical repeats were well expressed in E. coli and soluble, but lacked secondary structure as indicated by the minima at 195 nm in the circular dichroism (CD) spectra (Supporting Information, Fig. 1). This was not surprising because repeat proteins commonly contain N- and C-terminal repeats that act as capping domains to shield the inner hydrophobic core from solvent and aid in solubility and folding and all previous LRR designs contained capping repeats.19,20,44,45 Therefore, we separately constructed MSAs of individual N- and C-terminal repeats. Interestingly, whereas in RI LRRs capping repeats differ from internal repeats in both amino acid composition and length, N- and C-capping repeats for NOD LRRs have exactly the same length as internal repeats, but higher overall conservation (Fig. 2).
Figure 2.
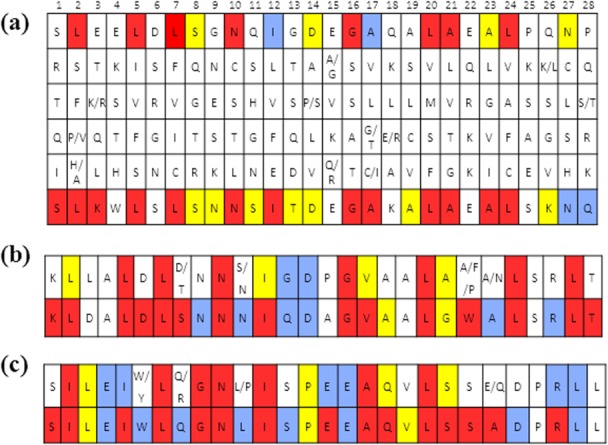
The most frequent amino acid residues found in the statistical analysis of NOD LRRs. Top and bottom row of each table are color coded: conservation of 50% or greater is red, 40% blue, and 30% yellow, and all position less than 30% are white. (a) Internal repeat; rows 2–5 correspond to the second, third, fourth, and fifth most commonly preferred amino acid for each position; (b) N-terminal repeat (c) C-terminal repeat. The top row in each table shows the frequency of occurrence of amino acid identity. The bottom row is the consensus sequence reanalyzed and corrected for preference of amino acid physical properties. Thus, the bottom row of each table is the amino acid sequence of repeats in CLLR2 protein used in this study. CLRR2 protein contains one N-terminal, two internal, and one C-terminal repeat.
Additionally, in the MSAs of internal and terminal repeats for NOD (Fig. 2, top row in each table), only half of the positions are defined with 30% or higher frequency of a specific amino acid. Stumpp et al.28 observed a similar phenomenon for the RI LRR. However, among many positions with less than 40% frequency of one specific amino acid, the position was readily defined by the type, that is, physical properties of the amino acid in that position. Thus we reanalyzed the MSA of the terminal and internal repeats on the basis of conservation of the amino acid's physical properties by considering the hydrophobicity, polarity, size, and charge for the five most common amino acids in each position of the alignment (Fig. 2). The bottom row of each table shows the second generation consensus sequence based on the reanalyzed MSA. The top row of each panel in Figure 2 is color coded for percent conservation of each amino acid while the bottom row is color-coded for conservation of physical properties from the top five consensus sequences. Results indicate that even with amino acid conservation appearing low in many positions, physical properties remain highly conserved. As a result of these findings, the second-generation sequence took into consideration the preferred physical properties in each position of the MSA. The sequence in the top row of each table in Figure 2 was modified in positions where conservation of identity was less than 50%. For example, in position three of the internal repeat [Fig. 2(a)], the frequency of occurrence is less than 30%. However, the Glu, Lys, and Arg residues present are highly favored. Although the frequency of a specific amino acid is low, this position is readily defined with a positively charged residue so Lys was selected for this position. Lys was selected over Glu based on the combined percent of occurrence for Lys and Arg being greater than that of the percent occurrence for Glu. We therefore chose the positively charged Lys over the negatively charged Glu. On the contrary, in position 22 of the internal repeat Lys, Arg, and Glu are also highly favored. However, in this case the percent occurrence of Glu exceeded that of both Lys and Arg together resulting in the selection of Glu at this position.
Additionally, tryptophan residues are in position 22 of the N-terminal repeat, position 4 of the internal repeat, and position 6 of the C-terminal repeat. In the N-terminal repeat, the highest conserved residue of position 22 is a phenylalanine. This preference for an aromatic residue justified the tryptophan substitution. Position 4 in the internal repeat had very low conservation in physical properties, so we selected tryptophan. In position 6 of the C-terminal repeat tryptophan and tyrosine are preferred, each with a conservation of 24%. It is important to note that both position 4 and 6 are within LRR canonical motif that commonly assumes a beta- strand structure. Since, tryptophan has a high propensity for beta-sheet formation, we hypothesized that these substitution will not disrupt the overall fold of the repeat, but will allow us to use fluorescence spectroscopy for further protein analysis.
The second-generation consensus sequence protein consisted of three types of consensus sequence repeats: N-terminal repeat, internal LRR repeat, and C-terminal repeat. We will refer to this consensus protein as CLRRx, where C stands for consensus sequence and x is equal to the number of internal repeats in the protein. For example, CLRR2 protein consists of two internal, and N- and C-terminal consensus LRR repeats. The actual sequence of repeats is shown as the bottom row of each panel in Figure 2.
Biophysical characterization
For further biophysical characterization, we chose the smallest of the designed CLRRs, CLLR2, since smaller proteins are preferred for application as binding scaffolds. CLRR2 was expressed at 37°C in BL-21 cells and purified under denaturing conditions following standard procedure.46 After on-column refolding, CLRR2 elutes from SEC Superdex 75 column in a single symmetrical peak (Supporting Information, Fig. 2). The apparent molecular mass value estimated from molecular mass standards was 17 kDa and is in agreement with the expected molecular mass of 15,977 Da as determined by MALDI, indicating that CLRR2 is monomeric in solution.
Secondary structure
CD spectroscopy was used to measure the secondary structure content, showing two minima at 207 and 220 nm. The relative intensities of the 207 and 220 nm peaks are characteristic of alpha helical and beta sheet secondary structure [Fig. 3(a)]. The CDPro Software's SELCON program predicts a structure comprised of 24.8% alpha helix and 21.5% beta sheet.47,48 The alpha helical content is about 10% lower than that estimated for designed RI LRRs.28 This is not surprising because it has been proposed that while the signature LRR motif contributes to much of the beta structure in LRR proteins, the helical regions may contribute to variability between different LRR families.49 Additionally, we used MUlti-Sources ThreadER (MUSTER) protein threading algorithm to obtain a predicted structural model of CLRR2 [Fig. 4(b)].50 The homology modeling was based on protein NLRX1 that shares 24.1% sequence identity with CLLR2. NLRX1, also known as NOD9, is a mitochondrial protein thought to be involved in an antiviral immune response against viral ribonucleic acid (RNA).51 Hong et al. showed that cNLRX directly binds to single and double stranded RNA.51 The alternating alpha-helical and beta-sheet segments of each repeat in the homology model are consistent with the CD data and CDPro structure prediction confirming that the consensus design retains a structure typical of natural LRRs.
Figure 3.

Biophysical characterization of CLRR2, before (black) and after (blue) incubation at 50°C and irreversible unfolding after heating (red). (a) CD spectrum of CLRR2 normalized to units of mean residue ellipticity indicates a mixed secondary structure of α-helix and β-sheet and irreversible unfolding after denaturation. (b) Thermal denaturation following changes in Trp fluorescence. (c) Thermal denaturation following CD signal at 217 nm. (d) Chemical denaturation of CLRR2 monitoring changes in Trp fluorescence with increasing urea concentration. Inset shows fluorescence spectra at 0M urea (black) and 7M urea (blue).
Figure 4.
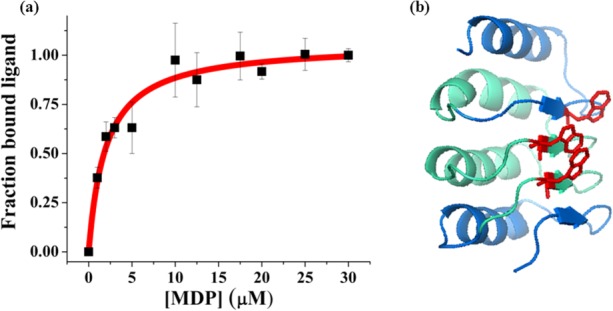
(a) Fluorescence quenching of Trp residues in CLRR2 is shown as a function of MDP concentration. Error bars are the standard deviation from three measurements. The Kd was determined to be 2.0 ± 0.4 µM. (b) The MUSTER model of CLRR2 showing location of Trp residues (red) on the concave face of the protein that are the proposed binding site of MDP. The terminal repeats are shown in blue and the internal repeats are shown in cyan.
Thermal denaturation
The thermal denaturation of the CLRR2 was followed by both tryptophan fluorescence and CD spectroscopy [Fig. 3(b,c)]. Here, it should be taken into consideration that Trp residues are located in both capping repeats and internal repeats and that fluorescence reports on the chemical environment of the indole fluorophore, whereas the CD signal is indicative of the overall amount of the secondary structure. Thermal denaturation of CLRR2 was accompanied by a reduction of fluorescent signal intensity and a shift of the emission maxima to higher wavelengths. The plot of normalized signal change as a function of temperature results in a curve indicative of two transitions [Fig. 3(b)]. To estimate the transition temperatures, we treated each of the transitions as a single two state transition. Although an approximation, this fitting allowed us to estimate transition temperatures of 32 and 57°C. Similarly, thermal unfolding monitored by the change in CD signal at 217 nm shows two transitions at 30 and 68°C [Fig. 3(c)]. After incubating CLLR2 for 10 min at 50°C and then repeating the thermal unfolding experiment, qualitatively different melting curves are observed for fluorescence and CD. The overall shape of the melting curve observed in the fluorescence measurement does not change after incubation, but the melting curve observed in the CD experiment now shows a single sharp transition with a new transition temperature of 60°C [Fig. 3(c)].
Intriguingly, temperature of the first transition in the CD, which disappears after incubation at 50°C, coincides to the first transition observed in fluorescence experiments. Similarly, the temperature of the second transition in the CD experiment is 10° higher than the Tm for the second transition in fluorescence experiment before incubation, but only 3° higher after incubation. The overall CD signal and fluorescence intensity are the same before and after incubation of CLLR2 indicating no change in the overall amount of secondary structure [Fig. 3(a)]. Spectra taken before and after the thermal melt with or without incubation suggest irreversible unfolding [Fig. 3(a)].
Although the thermal stability observed in fluorescence and CD experiments is qualitatively similar, the curves from these two experiments are not superimposable. This leads us to conclude that unfolding does not follow a simple two-state mechanism and that stable intermediates are formed during thermal unfolding. Additionally, the difference in fluorescence and CD curves indicates that the capping repeats and internal repeats may not unfold in a concerted manner. Because of the presence of Trp in each repeat, fluorescence unfolding curves provide a more complete picture of the unfolding process.
Chemical denaturation
The equilibrium denaturation of the designed CLRR2 was followed by observing the change in Trp fluorescence with increasing concentration of urea. The denaturing curve shows a broad transition around 3M urea [Fig. 3(d)]. Fitting this curve to a two state model results in a midpoint of denaturation of 2.6M urea, but cooperativity of the protein is only 2.5 kJ/mol M. This value is less than expected for highly cooperative unfolding proteins of this size, confirming the conclusions from the thermal denaturation. CLRR2 displays similar cooperativity to the designed RI LRR but is much less cooperative than previously studied YopM LRR (22.6 kJ/mol M) and Internalin B LRR (10.25 kJ/mol M).28,52,53
Binding affinity of CLLR2
Proteins of the innate immune system differ from antibodies of the adaptive immune system in that they detect pathogen associated molecular patterns (PAMPS) instead of one specific antigen.54 In other words, NLRs recognize the global features of a family of pathogens. This characteristic of the NLR family led us to hypothesize that the consensus design method may result in a protein that retains similar recognition properties. To test this hypothesis, we measured the affinity of CLLR2 for a muramyl dipeptide (MDP). MDP is a known ligand for the NOD2 protein family of NLRs. Recently, Grimes et al. showed that human NOD2 binds directly to MDP.55 They analyzed the binding of full-length human NOD2 to MDP using surface plasmon resonance and determined an affinity of 51 ± 18 nM.55 However, there is no clear evidence that the LRR domain mediates this interaction. Here we used fluorescence quenching and fluorescence anisotropy to investigate the affinity of dsigned CLRR2 for the biologically relevant L-D isomer of MDP.56
We investigated the ligand binding properties of CLLR2 by observing quenching of the fluorescence signal at 340 nm in the presence of MDP. The binding curve in Figure 4(a) is obtained by plotting the fraction of the bound ligand as a function of MDP concentration. Fitting of this curve to the single-site binding isotherm resulted in a Kd of 2.0 ± 0.4 µM for an average of two trials. Closer inspection of the homology model of CLRR2 structure shows that the Trp residue from the two internal repeats and C-terminal repeat form a cluster on the concave face of the protein [Fig. 4(b)]. Similar carbohydrate recognition through tryptophan residues was seen by Luo et al. in recognition of Thomsen-Friedenreich antigen by a VLR protein.18 Although our observed affinities are an order of magnitude lower than reported for the full-length NOD2 protein55 it is important to note that CLRR2 is representative of the entire NOD family and shares only 70% sequence identity with NOD2.
To confirm that the observed interaction is not an artifact of collisional tryptophan quenching, we performed a fluorescence anisotropy experiment, where we now observed the change in the signal originating on FITC labeled MDP. Fluorescence anisotropy change was measured as a function of increasing concentration of CLRR2 titrated into a solution of FITC-MDP. Fitting of the binding curve to a single-site binding isotherm resulted in a Kd = 20 ± 10 µM (Supporting Information, Fig. 3). This difference in the observed Kd values is not unexpected when comparing two techniques. Additionally, fluorescein is attached to MDP through a flexible linker on the N-acetylmuramic acid and the overall change in the anisotropy signal is low thus affecting the overall signal-to-noise ratio for the anisotropy experiment.
As a control experiment, we investigated the binding of CLRR2 to the D–D isomer of MDP, which is unable to stimulate NOD2 in vivo.56 While results indicate that tryptophans of CLLR2 are quenched in the presence of D–D MDP, the data is representative of random collisional quenching and does not follow the expected trend of a single-site binding isotherm (Supporting Information, Fig. 4). This control experiment leads us to conclude that under the concentration regime tested, CLLR2 is a specific binder for physiologically relevant L–D isomer of MDP2.
Thus, through the consensus sequence design, and without the evolution and selection step, we have developed a scaffold with micromolar affinity for a glycopeptide. This result indicates that consensus sequence LRRs based on the NOD protein family are a good starting point for the design of glycopeptide binding scaffolds. Moreover, this is the first report, to our knowledge, of direct interaction of NOD LRR with a physiologically relevant ligand.
Conclusion
Through consensus-based design, we have developed a novel LRR protein CLRR2. Structural and physical analysis of this protein indicate that it preserves important features of natural LRRs as well as the desired biophysical properties needed for use as a binding scaffold. We have shown that the consensus-based design resulted in a construct that retains binding information of natural NOD proteins.
The mechanism of pathogen sensing by NLRs is still largely unknown. By creating a LRR protein based on structurally and functionally homologous NODs we have developed a protein that can serve as a model structure for this class of NLRs. In the future, analysis of variable residues in the sequence alignment will allow for engineering diverse binding affinities for the development of new binding scaffolds as well as further elucidation of the role of LRR in NLR pathogen recognition.
Material and Methods
Consensus design and multiple sequence alignment
The 22 genes known to encode for NLR proteins in humans were retrieved from the HUGO Gene Nomenclature Committee (HGNC) database. Each confirmed gene sequence was translated into its corresponding protein sequence and input into the National Center for Biotechnology Information (NCBI) basic local alignment search tool (BLAST). Alignment of repeats that followed the canonical LRR motif of XLXXLXLXXNXL(X)nL was accomplished using Microsoft Excel. Repeats were extracted by manually searching the LRR domain of the selected NLR sequences. We aligned repeats following a pattern of XLXXLXLXXNXL(X)nL(X)8. In the canonical motif L is defined as Leu, Ile, Val, or Phe, and X is any other naturally occurring amino acid.30 N can also be defined as Arg, Cys, Ser, or Thr, and n is equal to 7. Repeats were aligned using Microsoft Excel and the consensus sequence was obtained by determining the amino acid with the highest frequency of occurrence in each position of the repeat by using the Microsoft Excel counting function. Consensus amino acid sequences were found for the top 5 most preferred amino acid in each position.
Cloning
For cloning of the LRR protein, a gene was designed consisting of an N-terminal repeat, internal repeat, and C-terminal repeat. Synthetic genes were synthesized by GENEWIZ and cloned into plasmid pProExHtam by ligation of restrictions sites BamHI and HindIII. Gene identity was confirmed by sequencing at the Virginia Tech Bioinformatics Institute.
Protein expression and purification
Overnight cultures of BL21 (DE3) cells were diluted 1:100 in 1 L of Terrific broth media at 37°C, with shaking at 250 rpm, and were grown to an OD600 of 0.5–0.8. Expression was induced with 1 mM IPTG, followed by 4 h of expression at 37°C. The cells were harvested by centrifugation at 5,000 rpm for 15 min and the pellets were frozen at −20°C until purification. To purify proteins, the cell pellet was resuspended in lysis buffer consisting of 50 mM Tris, 300 mM sodium chloride, 0.1% Tween 20, and 8M urea. After 1 min sonication at 30% power using a microtip and Mison sonicator, lysed cells were centrifuged at 16,000 rpm for 30 min and the protein supernatant was collected. Proteins were purified under denaturing conditions using standard Ni-NTA affinity purification protocol and eluted with 300 mM imidazole in lysis buffer with 8M urea. Proteins were then refolded on the size-exclusion column in 50 mM sodium phosphate buffer pH 8 with 150 mM sodium chloride. Protein identity was confirmed with MALDI indicating a molecular weight of 15,977 Da. Proteins were quantified by absorption at 280 nm using an extinction coefficient of 27, 960 M−1 cm−1, calculated from the amino acid sequence using the Expasy Protparam tool.57
Size exclusion chromatography
Akta Prime Plus FPLC was used for size exclusion chromatography. Refolding of denatured proteins after affinity purification was completed on the Superdex 75 16/600 Prep Grade column in 150 mM sodium chloride and 50 mM sodium phosphate buffer pH 8 at a flow rate of 0.5 ml/min. The Superdex 75 10/300 analytical column was used for analysis of molecular weights under the same conditions. A comparison to known standards (Bio-Rad) allowed for determination of the molecular weights and oligomeric states of each LRR protein.
Circular dichroism
CD spectra were acquired using 5–10 µM protein samples in 10 mM phosphate buffer pH 7.4 with 10 mM NaCl using a Jasco J-815 CD spectrometer. Far-UV CD (190–260 nm) spectra were recorded at 25°C to assess the secondary structure of CLRR2. Each sample was recorded three times, from 190 to 260 nm in a 2 mm pathlength cuvette, and averaged. Data collected using a 1 nm bandwidth, 2 nm data pitch, and a data integration time of 1 s, was normalized to units of mean residue ellipticity for all samples. Thermal denaturation curves were recorded by monitoring molar ellipticity at 217 nm while heating from 20 to 90°C in 2°C increments with an equilibration time of 10 min at each temperature.
Urea denaturation
Tryptophan fluorescence was monitored using a Cary Eclipse Fluorometer. Excitation of samples occurred at 295 nm and spectra were recorded from 310 to 380 nm with the excitation and emission slits equal to 5 nm. A 10M urea in 10 µM protein stock solution was titrated into a 10 µM protein sample. After a 10 min equilibration period for each addition, 3 scans were collected and averaged.
Fluorescence anisotropy
To determine the binding affinity, increasing amounts of protein CLLR2, were titrated to a FITC-labeled MDP (Purchased from Invivogen) in 10 mM Na2HPO4 pH 7.4 and 10 mM NaCl, buffer. Binding was performed at 10 nM peptide concentration in a 10 mm path-length cuvette at 25°C, and the fluorescence anisotropy was recorded after a 30 min equilibration period. Fluorescence anisotropy experiments were recorded in a Cary Eclipse Fluorometer equipped with excitation and emission polarizers. Excitation was achieved with a 5 nm slit-width at 495 nm and the emission recorded at 515 nm with a slit-width of 5 nm. For excitation at the vertical orientation (0°) the anisotropy (r) is:
| (1) |
where G is the G-factor, IVV and IVH are the vertical and horizontal emission of the sample, respectively, and IB,VV and IB,VH are the intensity of the emission of the blank with emission polarizer at vertical and horizontal orientation, respectively. The G-factor corrections were calculated using the equation: G = (IHV – IB,HV)/(IHH – IB,HH), where IHV is the vertical emission (0°) of a standard solution with excitation in horizontal orientation (90°), IHH is the horizontal emission (90°) of a standard solution with excitation in vertical orientation (0°), IB,HV is the vertical emission (0°) of a blank solution with excitation in horizontal orientation (90°) and IB,HH is the horizontal emission (90°) of a blank solution with excitation in vertical orientation (0°) using phosphate buffer as a blank solution and a 10 nM FITC labeled MDP as a standard solution. The fraction of ligand bound at each point in the binding curve was calculated by the equation.
| (2) |
where r is the observed anisotropy of the peptide at any protein concentration, rf is the anisotropy of the free peptide, and rb is the anisotropy of the ligand in the plateau region of the binding curve. The data was fit using nonlinear regression analysis with Origin software based on the equation.
| (3) |
where r is equal to the fraction of bound peptide, rb is equal to the maximum signal, Kd is the dissociation constant, and [P] is the concentration of protein in the sample.
Fluorescence quenching
Fluorescence quenching experiments were completed using a Cary Eclipse Fluorometer. Samples of 10 µM protein with MDP (purchased from Invivogen) from 0 to 30 µM were incubated in a 96 well plate for 30 min. Spectra were recorded with an excitation wavelength of 295 nm and excitation and emission slits equal to 10 and 20 nm. Each sample was measured from 310 to 380 nm. Three spectra were recorded and averaged for each trial. The binding curve was obtained by plotting fluorescence signal against MDP concentration where the percent of bound ligand was calculated using the equation.
| (4) |
where Fo is the fluorescent signal without MDP, F is the signal at any ligand concentration, and Fmin is the fluorescent signal at saturation. The data was fit using nonlinear regression analysis with Origin Software using the equation.
| (5) |
where r is equal to the fraction of bound ligand, rb is equal to the maximum signal, Kd is the dissociation constant, and [P] is the concentration of peptide in the sample.
Acknowledgments
The authors are grateful to Dr. Rich Helm and Keith Ray of the VT Biochemistry Department for MALDI analysis. The authors are also thankful to Prof. Webster Santos, Prof. Pablo Sobrado, Dr. Aitziber Cortajarena, and members of the Grove lab for discussion and careful reading of the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.
References
- 1.Boersma YL, Pluckthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Lofblom J, Frejd FY, Stahl S. Non-immunoglobulin based protein scaffolds. Curr Opin Biotechnol. 2011;22:843–848. doi: 10.1016/j.copbio.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Yoshimura H, Inaguma A, Yamada T, Ozawa T. Fluorescent probes for imaging endogenous beta-actin mRNA in living cells using fluorescent protein-tagged pumilio. ACS Chem Biol. 2012;7:999–1005. doi: 10.1021/cb200474a. [DOI] [PubMed] [Google Scholar]
- 4.Filipovska A, Razif MF, Nygard KK, Rackham O. A universal code for RNA recognition by PUF proteins. Nat Chem Biol. 2011;7:425–427. doi: 10.1038/nchembio.577. [DOI] [PubMed] [Google Scholar]
- 5.Hong X, Ma MZ, Gildersleeve JC, Chowdhury S, Barchi JJ, Jr, Mariuzza RA, Murphy MB, Mao L, Pancer Z. Sugar-binding proteins from fish: selection of high affinity “lambodies” that recognize biomedically relevant glycans. ACS Chem Biol. 2013;8:152–160. doi: 10.1021/cb300399s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garg A, Lohmueller JJ, Silver PA, Armel TZ. Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 2012;40:7584–7595. doi: 10.1093/nar/gks404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujimoto YK, Green DF. Carbohydrate recognition by the antiviral lectin cyanovirin-N. J Am Chem Soc. 2012;134:19639–19651. doi: 10.1021/ja305755b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodrum BW, Maxwell JD, Bolia A, Ozkan SB, Ghirlanda G. The antiviral lectin cyanovirin-N: probing multivalency and glycan recognition through experimental and computational approaches. Biochem Soc Trans. 2013;41:1170–1176. doi: 10.1042/BST20130154. [DOI] [PubMed] [Google Scholar]
- 9.Zheng H, Wang F, Wang Q, Gao JM. Cofactor-free detection of phosphatidylserine with cyclic peptides mimicking lactadherin. J Am Chem Soc. 2011;133:15280–15283. doi: 10.1021/ja205911n. [DOI] [PubMed] [Google Scholar]
- 10.Mak ANS, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335:716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mak AN-S, Bradley P, Bogdanove AJ, Stoddard BL. TAL effectors: function, structure, engineering and applications. Curr Opin Struct Biol. 2013;23:93–99. doi: 10.1016/j.sbi.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol. 2009;13:245–255. doi: 10.1016/j.cbpa.2009.04.627. [DOI] [PubMed] [Google Scholar]
- 13.Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. 2009;5:1087–1104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 14.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grutter MG, Pluckthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 15.Cortajarena AL, Kajander T, Pan W, Cocco MJ, Regan L. Protein design to understand peptide ligand recognition by tetratricopeptide repeat proteins. Protein Eng Des Sel. 2004;17:399–409. doi: 10.1093/protein/gzh047. [DOI] [PubMed] [Google Scholar]
- 16.Varadamsetty G, Tremmel D, Hansen S, Parmeggiani F, Pluckthun A. Designed Armadillo repeat proteins: library generation, characterization and selection of peptide binders with high specificity. J Mol Biol. 2012;424:68–87. doi: 10.1016/j.jmb.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 17.Seeger MA, Zbinden R, Flutsch A, Gutte PGM, Engeler S, Roschitzki-Voser H, Grutter MG. Design, construction, and characterization of a second-generation DARPin library with reduced hydrophobicity. Protein Sci. 2013;22:1239–1257. doi: 10.1002/pro.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo M, Velikovsky CA, Yang X, Siddiqui MA, Hong X, Barchi JJ, Jr, Gildersleeve JC, Pancer Z, Mariuzza RA. Recognition of the Thomsen-Friedenreich pancarcinoma carbohydrate antigen by a lamprey variable lymphocyte receptor. J Biol Chem. 2013;288:23597–23606. doi: 10.1074/jbc.M113.480467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wezner-Ptasinska M, Krowarsch D, Otlewski J. Design and characteristics of a stable protein scaffold for specific binding based on variable lymphocyte receptor sequences. Biochim Biophys Acta. 2011;1814:1140–1145. doi: 10.1016/j.bbapap.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Lee SC, Park K, Han J, Lee JJ, Kim HJ, Hong S, Heu W, Kim YJ, Ha JS, Lee SG, Cheong HK, Jeon YH, Kim D, Kim HS. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc Natl Acad Sci U S A. 2012;109:3299–3304. doi: 10.1073/pnas.1113193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortajarena AL, Liu TY, Hochstrasser M, Regan L. Designed proteins to modulate cellular networks. ACS Chem Biol. 2010;5:545–552. doi: 10.1021/cb9002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grove TZ, Cortajarena AL, Regan L. Ligand binding by repeat proteins: natural and designed. Curr Opin Struct Biol. 2008;18:507–515. doi: 10.1016/j.sbi.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grove TZ, Forster J, Pimienta G, Dufresne E, Regan L. A modular approach to the design of protein-based smart gels. Biopolymers. 2012;97:508–517. doi: 10.1002/bip.22033. [DOI] [PubMed] [Google Scholar]
- 24.Grove TZ, Hands M, Regan L. Creating novel proteins by combining design and selection. Protein Eng Des Sel. 2010;23:449–455. doi: 10.1093/protein/gzq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grove TZ, Osuji CO, Forster JD, Dufresne ER, Regan L. Stimuli-responsive smart gels realized via modular protein design. J Am Chem Soc. 2010;132:14024–14026. doi: 10.1021/ja106619w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackrel ME, Valverde R, Regan L. Redesign of a protein-peptide interaction: characterization and applications. Protein Sci. 2009;18:762–774. doi: 10.1002/pro.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13:1435–1448. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stumpp MT, Forrer P, Binz HK, Pluckthun A. Designing repeat proteins: modular leucine-rich repeat protein libraries based on the mammalian ribonuclease inhibitor family. J Mol Biol. 2003;332:471–487. doi: 10.1016/s0022-2836(03)00897-0. [DOI] [PubMed] [Google Scholar]
- 29.Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 30.Istomin AY, Godzik A. Understanding diversity of human innate immunity receptors: analysis of surface features of leucine-rich repeat domains in NLRs and TLRs. BMC Immunol. 2009;10:48. doi: 10.1186/1471-2172-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Liu L, Davies DR, Segal DM. Dimerization of Toll-like receptor 3 (TLR3) is required for ligand binding. J Biol Chem. 2010;285:36836–36841. doi: 10.1074/jbc.M110.167973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 33.Kobe B, Deisenhofer J. The leucine-rich repeat: a versatile binding motif. Trends Biochem Sci. 1994;19:415–421. doi: 10.1016/0968-0004(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 34.Kajander T, Cortajarena AL, Regan L. Consensus design as a tool for engineering repeat proteins. Methods Mol Biol. 2006;340:151–170. doi: 10.1385/1-59745-116-9:151. [DOI] [PubMed] [Google Scholar]
- 35.Magliery TJ, Regan L. Sequence variation in ligand binding sites in proteins. BMC Bioinform. 2005;6:240. doi: 10.1186/1471-2105-6-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magliery TJ, Regan L. Beyond consensus: statistical free energies reveal hidden interactions in the design of a TPR motif. J Mol Biol. 2004;343:731–745. doi: 10.1016/j.jmb.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 37.Johnson G, Wu TT. Kabat Database and its applications: future directions. Nucleic Acids Res. 2001;29:205–206. doi: 10.1093/nar/29.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forrer P, Binz HK, Stumpp MT, Pluckthun A. Consensus design of repeat proteins. Chembiochem. 2004;5:183–189. doi: 10.1002/cbic.200300762. [DOI] [PubMed] [Google Scholar]
- 39.Magliery TJ, Lavinder JJ, Sullivan BJ. Protein stability by number: high-throughput and statistical approaches to one of protein science's most difficult problems. Curr Opin Chem Biol. 2011;15:443–451. doi: 10.1016/j.cbpa.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sullivan BJ, Durani V, Magliery TJ. Triosephosphate isomerase by consensus design: dramatic differences in physical properties and activity of related variants. J Mol Biol. 2011;413:195–208. doi: 10.1016/j.jmb.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Jackrei ME, Valverde R, Regan L. Redesign of a protein-peptide interaction: Characterization and applications. Protein Sci. 2009;18:762–774. doi: 10.1002/pro.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kajander T, Cortajarena AL, Regan L. Consensus design as a tool for engineering repeat proteins. Methods Mol Biol. 2006;340:151–170. doi: 10.1385/1-59745-116-9:151. [DOI] [PubMed] [Google Scholar]
- 43.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kloss E, Courtemanche N, Barrick D. Repeat-protein folding: new insights into origins of cooperativity, stability, and topology. Arch Biochem Biophys. 2008;469:83–99. doi: 10.1016/j.abb.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pancer Z, Amemiya CT, Ehrhardt GR, Ceitlin J, Gartland GL, Cooper MD. Somatic diversification of variable lymphocyte receptors in the agnathan sea lamprey. Nature. 2004;430:174–180. doi: 10.1038/nature02740. [DOI] [PubMed] [Google Scholar]
- 46.Wingfield PT, Palmer I, Liang S-M. Coligan JE, Dunn BM, Ploegh HL, Speicher DW, Wingfield PT. Current Protocols in Protein Science. New York: John Wiley & Sons, Inc; 2001. Folding and Purification of Insoluble (Inclusion Body) Proteins from Escherichia coli; pp. 6.5.1–6.5.27. [Google Scholar]
- 47.Sreerama N, Venyaminov SY, Woody RW. Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 1999;8:370–380. doi: 10.1110/ps.8.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sreerama N, Woody RW. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 2004;383:318–351. doi: 10.1016/S0076-6879(04)83013-1. [DOI] [PubMed] [Google Scholar]
- 49.Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 50.Wu S, Zhang Y. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong M, Yoon SI, Wilson IA. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity. 2012;36:337–347. doi: 10.1016/j.immuni.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Courtemanche N, Barrick D. Folding thermodynamics and kinetics of the leucine-rich repeat domain of the virulence factor Internalin B. Protein Sci. 2008;17:43–53. doi: 10.1110/ps.073166608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kloss E, Barrick D. Thermodynamics, kinetics, and salt dependence of folding of YopM, a large leucine-rich repeat protein. J Mol Biol. 2008;383:1195–1209. doi: 10.1016/j.jmb.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 55.Grimes CL, Ariyananda Lde Z, Melnyk JE, O'Shea EK. The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc. 2012;134:13535–13537. doi: 10.1021/ja303883c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 57.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. The proteomics protocols handbook. Totowa, NJ: Humana Press; 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Figure 3.
Supporting Information Figure 4.