

Abstract
High conservation of glycyl residues in homologous proteins is fairly frequent. It is commonly understood that glycine tends to be highly conserved either because of its unique Ramachandran angles or to avoid steric clash that would arise with a larger side chain. Using a database of aligned 3D structures of homologous proteins we identified conserved Gly in 288 alignment positions from 85 families. Ninety-six of these alignment positions correspond to conserved Gly residue with (φ, ψ) values allowed for non-glycyl residues. Reasons for this observation were investigated by in-silico mutation of these glycyl residues to Ala. We found in 94% of the cases a short contact exists between the Cβ atom of the introduced Ala with the atoms which are often distant in the primary structure. This suggests the lack of space even for a short side chain thereby explaining high conservation of glycyl residues even when they adopt (φ, ψ) values allowed for Ala. In 189 alignment positions, the conserved glycyl residues adopt (φ, ψ) values which are disallowed for Ala. In-silico mutation of these Gly residues to Ala almost always results in steric hindrance involving Cβ atom of Ala as one would expect by comparing Ramachandran maps for Ala and Gly. Rare occurrence of the disallowed glycyl conformations even in ultrahigh resolution protein structures are accompanied by short contacts in the crystal structures and such disallowed conformations are not conserved in the homologues. These observations raise the doubt on the accuracy of such glycyl conformations in proteins.
Keywords: glycyl conformation, homologous proteins, Ramachandran map, steric hindrance
Introduction
It is well known for about 50 years now that allowed conformational space for the glycyl residue is far more than that of the non-glycyl residues.1,2 Much of the (φ, ψ) space with positive φ angle is disallowed for non-glycyl residues due to the steric hindrance involving the Cβ atom in the side chain; that is, there is no space to accommodate a carbon atom in those backbone conformations.3 Therefore, it is not surprising that glycyl residue with (φ, ψ) values disallowed for non-glycyl residues is conserved in homologous proteins. Glycyl residue with such conformations are usually present in loops or β-turns enabling a critical chain reversal or located at the termini of helical or β-strand regions eliciting the start or stop signal for the helical or β-strand region.4–7
Sometimes glycyl residue is conserved in functional sites of proteins. Indeed a comprehensive analysis of enzyme structures suggests that presence of glycyl residues in the active sites provides flexibility to the active sites.8 A well-known example is the Asp-Thr-Gly motif occurring in the catalytic site of aspartic proteinases9 and retroviral proteinases.10 It is also well known that glycine-rich motif plays role in phosphate recognition as seen in protein kinases.11 Given the fact that glycyl residue lacks typical functional groups commonly seen in active sites of proteins it is hardly surprising to note that glycyl residue is conserved more often for the structural reasons than functional reasons.
From comparison of Ramachandran maps for Ala and Gly, it is clear that glycyl residues can adopt conformations which are allowed for alanyl residue. Indeed site-directed mutagenesis of two glycyl residues (Gly 77 and Gly 113) in T4 lysozyme with (φ, ψ) values allowed for Ala resulted in mutants with three dimensional (3D) structures highly similar to that of the wild type.12,13 The (φ, ψ) values at Gly 77 and Gly 113 in the wild type are (−67, −43) and (−71, −18), respectively. In the two Gly → Ala mutants, the (φ, ψ) values at Ala 77 and Ala 113 are (−64, −44) and (−72, −9), respectively. The (φ, ψ) values in the mutant are only slightly different compared to the wild type and the methyl group in the Ala side chain has been accommodated in the 3D scaffold with small adjustments in the structure. Given the fact that all the (φ, ψ) values allowed in the Ala Ramachandran map are also allowed in the Gly Ramachandran map one might expect that a glycyl residue with (φ, ψ) values, for example, in α-helical or β-sheet regions of the Ramachandran map may not be well conserved.
Although it is clear that glycyl residues adopting (φ, ψ) values disallowed for non-glycyl residues is commonly conserved, we addressed the question of conservation of glycyl residues when it adopts (φ, ψ) values allowed for non-Gly residues. Much of the left side of the Ramachandran (φ, ψ) plane (with φ negative) is allowed for non-glycyl residues apart from being allowed for glycyl residue. However, in our current analysis we notice that in many homologous protein families glycyl residue with (φ, ψ) values allowed for non-glycyl residues are conserved. We provide here a comprehensive analysis of such glycyl conformations and explain why a glycyl residue may be conserved though its (φ, ψ) values are allowed for non-glycyl residues. In addition, we provide an analysis of conserved glycyl residues with (φ, ψ) values allowed for Gly and disallowed for non-Gly. Further, we investigated glycyl conformations observed in proteins that are disallowed even for glycyl residue.
Results and Discussion
The database of PALI14 consists of structure-based alignments for 1922 families with each family containing at least two homologous domains of known 3D structure. For the current analysis, we have considered those families containing at least five homologous protein domains each and these yielded 673 families. From these families, we identified 346 families with at least one alignment position in which Gly is completely conserved. From this dataset, we identified 85 families each with at least one constituent member of known 3D structure with a crystallographic resolution 1.2 Å or better. The presence of ultrahigh resolution crystal structure in each family is ensured to have a high confidence especially on the conformations that are disallowed for Gly or non-Gly. It has been shown previously that the extent of occurrence of disallowed conformation in the Ramachandran map is quite low in the ultrahigh resolution structures.15 The number of alignment positions with complete conservation of glycyl residue in 85 families is 288.
In this analysis, we have concentrated on the conserved glycyl residues with (φ, ψ) values allowed for non-glycyl residues. We asked the question “what is the reason for the occurrence of glycyl residue although the (φ, ψ) values are suitable to accommodate a non-glycyl residue?” The number of alignment positions in 85 families with complete conservation of glycyl residues with (φ, ψ) values allowed for non-glycyl residue is 96. Figure 1 shows the distribution of (φ, ψ) values of glycyl residues in this category from protein domain structures with crystallographic resolution of 1.2 Å or better. As can be seen from Figure 1, the (φ, ψ) values are confined to the allowed region of the Ramachandran map of Ala. This category of glycyl residues is referred as “category A.”
Figure 1.
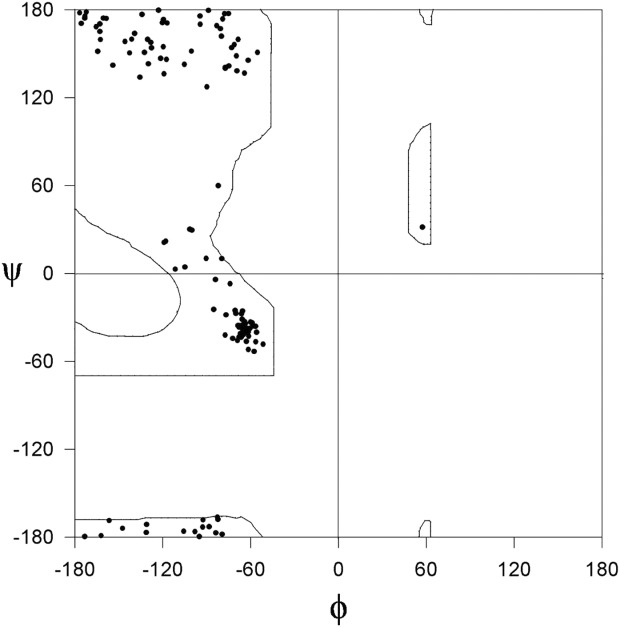
φ, ψ plot of conserved Gly in homologous protein structures determined at ultrahigh resolution with the (φ, ψ) values allowed for non-glycyl residues (Category A). Ala Ramachandran map is also shown.
In 189 alignment positions with complete conservation of Gly, the (φ, ψ) values are allowed in the Gly Ramachandran map and are disallowed in Ala Ramachandran map. These glycyl residues are collectively referred as “category B.” The higher number of alignment positions with Gly conserved with such (φ, ψ) values compared to category A is understandable as (φ, ψ) values of Gly in category B are unsuitable to accommodate non-Gly residues according to Ala Ramachandran map. Figure 2(a,b) show the distribution of glycyl conformations, in ultrahigh resolution protein structures, with (φ, ψ) values allowed for Gly and disallowed for non-Gly. In Figure 2(a), the (φ, ψ) plot is superimposed on the Ala Ramachandran map and in Figure 2(b)the same (φ, ψ) distribution is shown superimposed on Gly Ramachandran map. It can be seen that these glycyl conformations are allowed according to the Gly Ramachandran map and disallowed according to Ala Ramachandran map.
Figure 2.
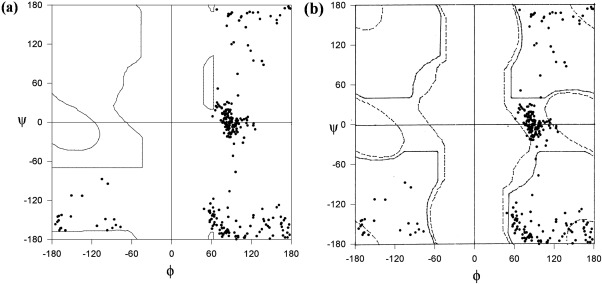
Plot of (φ, ψ) values of conserved Gly in homologous protein structures determined at ultrahigh resolution with the (φ, ψ) values allowed for glycyl residues superimposed on (a) Ala Ramachandran map and (b) Gly Ramachandran map (CategoryB). Solid and dashed lines in (b) show fully and partially allowed regions, respectively.
In our dataset, we found only three examples of protein structures with the ultrahigh resolution in which the (φ, ψ) values at a glycyl residues are disallowed even for glycyl residue (Fig. 3). These glycyl residues are collectively referred as “Category C.”
Figure 3.
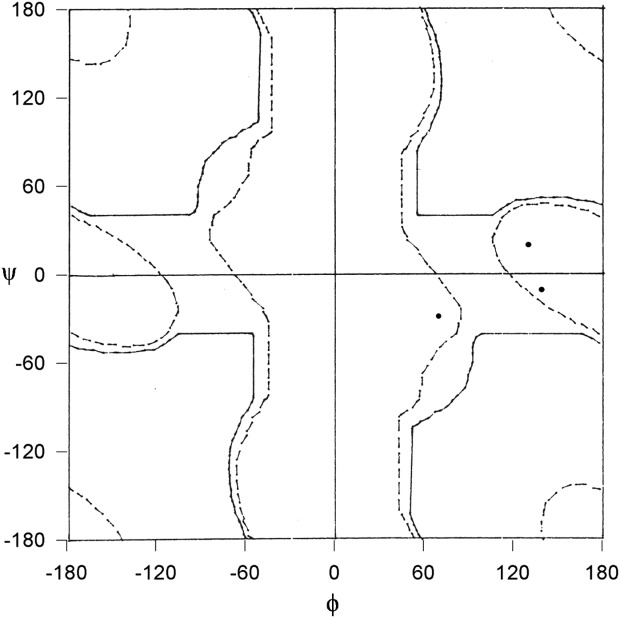
φ, ψ plot of conserved Gly in homologous protein structures determined at ultrahigh resolution with the (φ, ψ) values disallowed in Gly Ramachandran map (Category C). Solid and dashed lines show fully and partially allowed regions, respectively.
Conservation of Gly with (φ, ψ) values allowed for non-Gly—Category A
As mentioned before in 96 alignment positions corresponding to conserved Gly residues, the (φ, ψ) values are allowed for non-Gly residues (Fig. 1). To identify the reason for the conservation of glycyl residues in these alignment positions, we performed in-silico mutations to replace such Gly residues, in ultrahigh resolution structures, to Ala. We investigated if a short contact involving Cβ atom at the sites of mutation is present in the mutant. Interestingly in 85 out of 96 alignment positions, the Cβ atom at the sites of mutation showed short contact. Thus it is clear that the reason for conservation of Gly in these overwhelming majority of cases is the lack of space even to accommodate the second smallest residue, namely, Ala.
Ramachandran et al. did not find any short contact when Gly adopted these (φ, ψ) values allowed for Ala. However, it must be recalled that Ramachandran et al. used a model system comprising of two-linked peptide units with Gly at the linkage. In principle, even when the (φ, ψ) values are allowed for Gly, short contact could exist in specific cases of larger system such as proteins.
One of such example is depicted in Figure 4 is the protein cholesterol oxidase (1N4W)16 the Gly at position 21 occurs in a helix with (φ, ψ) values of (−61.3, −42.9) and if we mutate it to Ala, Cβ is involved in three short contacts all with Thr 473 which is sequentially well separated from Gly 21 but it is proximal to Gly 21 in the 3D structure. The distance of the side chain hydroxyl group and the main chain carbonyl carbon and carbonyl oxygen of Thr 473 from Cβ of A21 in the mutant are 2.6, 2.7, and 2.2 Å, respectively. However, according to Ramachandran et al.1 the shortest approach distance between nonbonded C···O and C···C are 2.7 and 2.9 Å, respectively. Aside from short contacts another unfavorable feature is the proximity of the carbonyl oxygen (polar group) to the methyl (apolar) group in the side chain of Ala 21.
Figure 4.
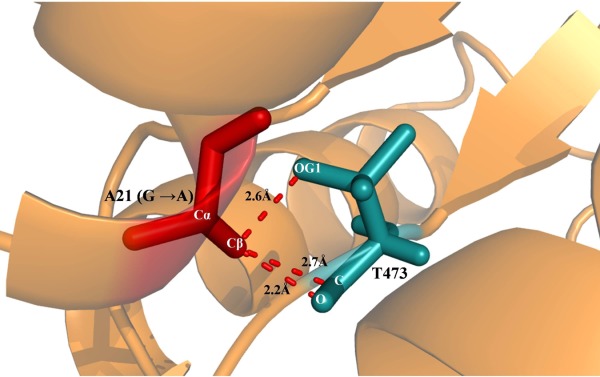
Example showing the conserved Gly mutated to Ala involved in short contacts. Cartoon representation of cholesterol oxidase (1N4W) shown in brown color in which Gly 21 which is mutated to Ala is shown as stick representation in red color. The residue Thr 473 is involved in short contact with Cβ of Ala 21 (G → A mutant) and it is shown in stick representation in blue color. The atoms Cβ of A21 is involved in short contacts with OG1, C and O of Thr 473. The short contact distance is marked and denoted as dotted lines.
However, in small number of 11 out of 96 alignment positions with conserved Gly, the G → A mutant did not show short contact. It should be possible to accommodate at least Ala residue in these positions. As our dataset is confined to homologous proteins of known 3D structure, a protein domain family is not completely represented in our dataset. It is possible that many homologous proteins with yet unknown structure might have accommodated non-Gly residue in these positions. Therefore, we searched the Swiss-prot17 sequence database to identify an elaborate list of homologues of the proteins of known structure and we probed the extent of conservation of Gly by considering all the recognizable homologues. Table I lists the percentage conservation of Gly in 11 alignment positions for which no short contact was found when mutated to Ala. It can be seen from the table in none of the 11 cases the Gly is completely conserved. The occurrence of non-Gly residues in these alignment positions is consistent with our observation that there is no short contact in the 11 cases if Gly is replaced by Ala. Another interesting feature with the list of 11 examples is that in almost all the cases the Gly is observed at the functional site of the protein. It is unknown whether the side chain of a non-Gly residue, if present in the place of Gly, will compromise on the conformational flexibility which may be required in many of the functional sites of proteins.
Table I.
List of Gly Residues, in Ultrahigh Resolution Structures, Corresponding to 11 of the Gly Conserved Alignments Positions in which the φ, ψ Values are Allowed for non-Gly Residues and Gly → Ala In-Silico Mutant has no Short Contact
| S no | PDB code | Residue number | φ | ψ | % conservation of Gly in sequence homologous | Functional significance | References |
|---|---|---|---|---|---|---|---|
| 1 | 1m1n | 87 | −61.5 | −52 | 84 | In functional site | Einsle et al., 200218 |
| 2 | 1m1n | 94 | −56.6 | −36.2 | 83 | In functional site | Einsle et al., 200218 |
| 3 | 1mso | 8 | 56.2 | −136.2 | 76 | In functional site | Smith et al., 200319 |
| 4 | 1n62 | 697 | −62.9 | −40.2 | 87 | In functional site | Dobbek et al., 200220 |
| 5 | 1n62 | 114 | −68.1 | −35.7 | 80 | In functional site | Dobbek et al., 200220 |
| 6 | 1oxd | 127 | −119.2 | 136.2 | 90 | In functional site | Bae et al., 200321 |
| 7 | 1tjx | 374 | −176.7 | 177.8 | 91 | In functional site | Cheng et al., 200422 |
| 8 | 2fn3 | 427 | −68.5 | 159.7 | 86 | In functional site | Magrane, 201117 |
| 9 | 2nmz | 27 | −84 | −4.2 | 94 | In functional site | Tie et al., 200723 |
| 10 | 1kqp | 48 | −94.4 | 170 | 89 | In functional site | Symersky et al., 200224 |
| 11 | 1y93 | 257 | −57.6 | −53.3 | 92 | Not in the functional site | Bertini et al., 200525 |
Conservation of Gly with (φ, ψ) values allowed for Gly but disallowed for non-Gly—Category B
Number of alignment positions in the dataset of homologous protein structures in which glycyl residue is conserved with (φ, ψ) values allowed in Gly Ramachandran map and disallowed in Ala Ramachandran map is 189 [Fig. 2(a,b)]. Given the unique tendency of glycyl residues to adopt such a conformation it is not surprising to find such a large number of Gly conserved alignment positions adopting (φ, ψ) values which are disallowed for non-Gly. On the basis of the work by Ramakrishnan and Ramachandran,2 it is clear that these (φ, ψ) values are disallowed for non-Gly because one or more short contacts are noted involving Cβ atom if we have Ala in the place of Gly. Therefore, one would expect short contact involving Cβ atom in the in-silico Gly → Ala mutants corresponding to 189 Gly conserved alignment positions. Indeed in 184 out of 189 Gly conserved positions we noted that the short contact involving Cβ of Ala at the site of mutation. Therefore, the reason for the conservation of Gly is clearly lack of space to accommodate a side chain.
Interestingly, in 5 out of 189 cases we did not notice short contact in the Gly → Ala mutants. We investigated all the nonbonded interatomic distances in the vicinity of Cβ in the Gly → Ala mutants. Table II provides the list of ultrahigh resolution structures corresponding to this five alignment positions. Table II also lists selected nonbonded distances of Cβ of Ala at the site of mutations along with the shortest approach distance between those nonbonded atoms according to the contact criteria established by Ramachandran et al.1 The last column in Table II shows the difference between the distance of possible shortest approach and the observed distance in the mutant. It can be seen that the difference between the observed distance and threshold distance for the short contact is of the order of 0.01 Å. Therefore, these nonbonded atom pairs managed to have a distance just at the threshold of acceptance of short contact-free distance. In our previous analysis,22 we have shown that genuine disallowed conformations occurring in ultrahigh resolution protein structures and in small peptide structures the short contacts are avoided in the crystal structures by small and acceptable deviations in the bond lengths and bond angles at peptide units from the ideal values proposed by Corey and Pauling.27 These (φ, ψ) values are considered disallowed by Ramachandran et al. using their model system of two-linked peptide units in which they have used ideal bond lengths, bond angles and perfect planarity of the units. Therefore, we believe that these five exceptional cases are actually corresponding to the borderline of allowed and disallowed nature of their (φ, ψ) values.
Table II.
List of Ultrahigh Resolution Structures Corresponding to Conserved Gly Positions with (φ, ψ) Values Disallowed for non-Gly and Shows no Short Contact in Gly → Ala Mutant
| S no | PDB code | Residue number | φ | ψ | One of the nonbonded distance involving Cβ at the site of mutation | Observed distance (Å) | The shortest possible approach distance (normal limit) proposed by Ramachandran et al. (Å) | Difference in the two nonbonded distances (Å) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1od3 | 51 | 72.3 | −165.5 | Cβ51---O50 | 2.81 | 2.8 | 0.01 |
| 2 | 1od3 | 127 | 75.9 | −162.8 | Cβ27---N128 | 2.92 | 2.9 | 0.02 |
| 3 | 1w6s | 2539 | 60.7 | −163.6 | Cβ2539---Cα2540 | 3.02 | 3.0 | 0.02 |
| 4 | 1n4w | 19 | −83.9 | −157.9 | Cβ19---N20 | 2.92 | 2.9 | 0.02 |
| 5 | 1mso | 8 | 56.2 | −136.2 | Cβ8---O7 | 2.82 | 2.8 | 0.02 |
Conservation of Glycyl residue with (φ, ψ) values disallowed in the Gly Ramachandran map—Category C
As mentioned before, there are only three ultrahigh resolution structures in which a Gly residue is present with the (φ, ψ) values disallowed even in Gly Ramachandran map (Fig. 3). Table III lists these three cases. It can be expected from the fact that they occur in disallowed region that these glycyl conformations are associated with short contacts. Indeed in all the three cases we noticed short contacts in the corresponding crystal structures (Table III).
Table III.
List of Conserved Gly Positions with (φ, ψ) Values Disallowed in Gly Ramachandran Map and Shows Short Contact in Crystal Structure
| S no | PDB code | Residue number | φ | ψ | Short contacts | ||
|---|---|---|---|---|---|---|---|
| Short contact distances involving CB at the site of mutation | Observed distance (Å) | The shortest possible approach distance proposed by Ramachandran et al. (Å) | |||||
| 1 | 2g58 | 30 | 70.1 | −28 | Cβ30---Cα23 | 2.5 | 3.0 |
| Cβ30---C23 | 2.1 | 3.0 | |||||
| Cβ30---O23 | 1.8 | 2.8 | |||||
| 2 | 1w6s | 2175 | 130.5 | 20.6 | Cβ2175---O2155 | 2.7 | 2.8, |
| Cβ2175---C2174 | 2.8 | 3.0 | |||||
| Cβ2175---O2174 | 2.4 | 2.8 | |||||
| 3 | 2jhf | 236 | 139.4 | −9.9 | Cβ236---C235 | 2.7 | 3.0 |
| Cβ236---O235 | 2.3 | 2.8 | |||||
In our previous analysis,26 none of the disallowed conformations we noted in ultrahigh resolution protein structures and peptide structures correspond to the short contact in the crystal structure. The expected short contact has been avoided by very small deviations in bond lengths, bond angles, and peptide planarity from ideal values. Therefore, it is surprising to find short contacts in the ultrahigh resolution crystal structures in proteins.
In our earlier work,28 we showed that the disallowed (φ, ψ) values are only moderately conserved in homologous protein structures. Only in very small proportion of cases when the residue is involved in a functional feature the disallowed nature is completely conserved. Figure 5 shows the (φ, ψ) plot of glycyl residues conserved in the three alignment positions. It can be seen in every case many of the glycyl residues in other homologues adopt a (φ, ψ) value that is allowed for glycyl residue. Nonconservation of disallowed nature in the three alignment positions along with the observation of occurrence of short contacts even in ultrahigh resolution protein structures raises doubts on the accuracy of the structure of the local region.
Figure 5.
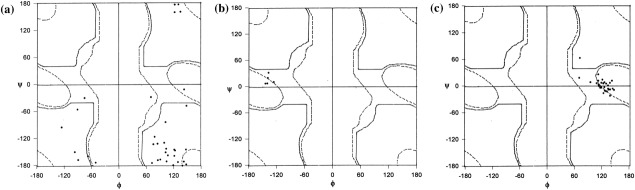
(φ, ψ) plots of conserved glycyl residues in the topologically equivalent positions corresponding to Category C. (a), (b), and (c) correspond to the (φ, ψ) plot of glycyl residues in the topologically equivalent positions in the homologues of 2G58, 1W6S, and 2JHF, respectively.
Conclusions
The Ramachandran map elegantly highlighted the basis of higher backbone conformational freedom of Gly residues over other residues. Impact of the Ramachandran map made us believe the conservation of Gly residues in the homologues proteins is almost always due to the Gly adopting the (φ, ψ) values which are prohibited or unfavorable for non-Gly residues. For the first time here, we have shown that even the glycyl residues adopting (φ, ψ) values which are allowed for non-Gly residues can be highly conserved. We propose on the basis of our analysis that the reasons for this conservation are primarily the lack of space to accommodate the side chain. As shown in our analysis, short contacts involving the Cβ atom of Ala, which is modeled in the place of conserved Gly, are present in overwhelming majority of the cases. Therefore, depending on the tertiary fold, protein structures are constrained for space for accommodating side chain even when the backbone conformation is suitable for accommodating a side chain.
It has been established by Ramachandran et al. that the (φ, ψ) values which are allowed only for the Gly residue would correspond to conformations with short contact(s) involving a Cβ atom, if Ala is present in place of Gly. We have shown from our current analysis on protein structures “this rule” is almost always obeyed. Almost all such Gly residues in protein structure could not be replaced by Ala as there is no sufficient space available to accommodate the methyl group in the Ala side chain.
Our analysis raises a new challenge of prediction of conserved Gly residues with conformations prohibited for non-Gly residues over conserved Gly residues adopting conformations suitable for non-Gly residues.
Despite enormous conformational freedom at glycyl residues and even in the ultrahigh resolution crystal structures of proteins, glycyl conformations with (φ, ψ) values disallowed for Gly have been noticed although the number of such occurrences is very small. In all these cases, steric clashes have been observed in the crystal structures. Occurrence of (φ, ψ) values at topologically equivalent glycyl residues in the homologous proteins at the allowed regions of the Gly Ramachandran map suggest that the disallowed glycyl conformations may correspond to errors in the structures.
Materials and Methods
Dataset generation
The database of Phylogeny and ALIgnment of homologues protein structures (PALI)10 which contains structure-based sequence alignments of protein domain families has been used in the current analysis. In PALI domain family datasets and domain definitions are taken from the database of structural classification of proteins (SCOP version 1.75).29 For the current analysis clearly, orphan (families with only one member of known structure) families are unsuitable and therefore are not considered. We further filtered the PALI dataset using the condition that a family should contain at least five homologues of known structure for inclusion in our analysis. Out of 1922 multimember protein domain families, there are 673 families obtained with at least five homologous structures in each family. It was also ensured in these families that at least one glycyl residue position is conserved among its homologues according to the multiple structural alignment obtained from PALI and it resulted in 346 families. Further, there are 85 families chosen from these 346 families in which each of the family has at least one structure with ultrahigh resolution of better than 1.2 Å.
The (φ, ψ) values at each of the conserved glycyl residue positions are calculated in 85 families. There are 288 alignment positions with conservation of glycyl residue that are considered for further analysis. Many of the glycyl residues from these alignment positions present in ultrahigh resolution structures were mutated in-silico to Ala and the short contacts, if present, involving Cβ atom at Ala were identified using the contact criteria proposed by Ramachandran et al.1 Further these conserved glycyl residues from these 288 alignments positions were classified into following three categories.
Category A: Glycyl residues with (φ, ψ) values that are allowed for non-glycyl residue according to Ala Ramachandran map. There are 96 alignment positions corresponding to this category (Supporting Information Table SI).
Category B: Glycyl residues with (φ, ψ) values that are allowed in Gly Ramachandran map, but, disallowed in Ala Ramachandran map. There are 189 alignments positions in this category (Supporting Information Table SII).
Category C: The glycyl residues having (φ, ψ) values that are even disallowed for glycyl residues according to Gly Ramachandran map. There are only three alignments positions corresponding to this category.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary table I: 96 alignment positions of the glycyl residues having the (φ, ψ) values that are allowed for non-glycyl residue according to Ala Ramachandran map (Category A). For every alignment position PDB code of the entries corresponding to the ultra-high resolution structure, residue number of the glycyl residue of interest and its (φ, ψ) values are provided.
Supplementary table II: 189 alignment positions of the glycyl residues having (φ, ψ) values that are allowed for glycyl residue according to Gly Ramachandran map but disallowed according to Ala Ramachandran map (Category B). For every alignment position PDB code of the entries corresponding to the ultra-high resolution structure, residue number of the glycyl residue of interest and its (φ, ψ) values are provided.
References
- 1.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–99. doi: 10.1016/s0022-2836(63)80023-6. [DOI] [PubMed] [Google Scholar]
- 2.Ramakrishnan C, Ramachandran GN. Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units. Biophys J. 1965;5:909–933. doi: 10.1016/S0006-3495(65)86759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 4.Ramakrishnan C, Srinivasan N, Prashanth D. Conformation of glycyl residues in globular proteins. Intl J Pept Protein Res. 1987;29:629–637. doi: 10.1111/j.1399-3011.1987.tb02293.x. [DOI] [PubMed] [Google Scholar]
- 5.Ramakrishnan C, Srinivasan N. Glycyl residues in proteins and peptides: an analysis. Curr Sci. 1990;59:851–862. [Google Scholar]
- 6.Presta LG, Rose GD. Helix signals in proteins. Science. 1988;240:1632–1641. doi: 10.1126/science.2837824. [DOI] [PubMed] [Google Scholar]
- 7.Richardson JS, Richardson DC. Amino acid preferences for specific locations at the ends of alpha helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- 8.Yan BX, Sun YQ. Glycine residues provide flexibility for enzyme active sites. J Biol Chem. 1997;272:3190–3194. doi: 10.1074/jbc.272.6.3190. [DOI] [PubMed] [Google Scholar]
- 9.Sussman F, Villaverde MC, Dominguez JL, Danielson UH. On the active site protonation state in aspartic proteases: implications for drug design. Curr Pharm Des. 2013;19:4257–4275. doi: 10.2174/1381612811319230009. [DOI] [PubMed] [Google Scholar]
- 10.Ingr M, Uhlikova T, Strisovsky K, Majerova E, Konvalinka J. Kinetics of the dimerization of retroviral proteases: the “fireman's grip” and dimerization. Protein Sci. 2003;12:2173–2182. doi: 10.1110/ps.03171903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- 12.Matthews BW, Nicholson H, Becktel WJ. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholson H, Tronrud DE, Becktel WJ, Matthews BW. Analysis of the effectiveness of proline substitutions and glycine replacements in increasing the stability of phage T4 lysozyme. Biopolymers. 1992;32:1431–1441. doi: 10.1002/bip.360321103. [DOI] [PubMed] [Google Scholar]
- 14.Balaji S, Sujatha S, Kumar SS, Srinivasan N. PALI-a database of Phylogeny and ALIgnment of homologous protein structures. Nucleic Acids Res. 2001;29:61–65. doi: 10.1093/nar/29.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 16.Lyubimov AY, Lario PI, Moustafa I, Vrielink A. Atomic resolution crystallography reveals how changes in pH shape the protein microenvironment. Nat Chem Biol. 2006;2:259–264. doi: 10.1038/nchembio784. [DOI] [PubMed] [Google Scholar]
- 17.Bairoch A, Boeckmann B, Ferro S, Gasteiger E. Swiss-Prot: juggling between evolution and stability. Brief Bioinform. 2004;5:39–55. doi: 10.1093/bib/5.1.39. [DOI] [PubMed] [Google Scholar]
- 18.Einsle O, Tezcan FA, Andrade SL, Schmid B, Yoshida M, Howard JB, Rees DC. Nitrogenase MoFe-protein at 1.16 A resolution: a central ligand in the FeMo-cofactor. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 19.Smith GD, Pangborn WA, Blessing RH. The structure of T6 human insulin at 1.0 A resolution. Acta Cryst D. 2003;59:474–482. doi: 10.1107/s0907444902023685. [DOI] [PubMed] [Google Scholar]
- 20.Dobbek H, Gremer L, Kiefersauer R, Huber R, Meyer O. Catalysis at a dinuclear [CuSMo(==O)OH] cluster in a CO dehydrogenase resolved at 1.1-A resolution. Proc Natl Acad Sci USA. 2002;99:15971–15976. doi: 10.1073/pnas.212640899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harmer NJ, Sivak JM, Amaya E, Blundell TL. 1.15 A crystal structure of the X. tropicalis Spred1 EVH1 domain suggests a fourth distinct peptide-binding mechanism within the EVH1 family. FEBS Lett. 2005;579:1161–1166. doi: 10.1016/j.febslet.2004.11.114. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Y, Sequeira SM, Malinina L, Tereshko V, Sollner TH, Patel DJ. Crystallographic identification of Ca2+ and Sr2+ coordination sites in synaptotagmin I C2B domain. Protein Sci. 2004;13:2665–2672. doi: 10.1110/ps.04832604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tie Y, Kovalevsky AY, Boross P, Wang YF, Ghosh AK, Tozser J, Harrison RW, Weber IT. Atomic resolution crystal structures of HIV-1 protease and mutants V82A and I84V with saquinavir. Proteins. 2007;67:232–242. doi: 10.1002/prot.21304. [DOI] [PubMed] [Google Scholar]
- 24.Symersky J, Devedjiev Y, Moore K, Brouillette C, DeLucas L. NH3-dependent NAD+ synthetase from Bacillus subtilis at 1 A resolution. Acta Cryst D. 2002;58:1138–1146. doi: 10.1107/s0907444902006698. [DOI] [PubMed] [Google Scholar]
- 25.Bertini I, Calderone V, Cosenza M, Fragai M, Lee YM, Luchinat C, Mangani S, Terni B, Turano P. Conformational variability of matrix metalloproteinases: beyond a single 3-D structure. Proc Natl Acad Sci USA. 2005;102:5334–5339. doi: 10.1073/pnas.0407106102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramakrishnan C, Lakshmi B, Kurien A, Devipriya D, Srinivasan N. Structural compromise of disallowed conformations in peptide and protein structures. Protein Pept Lett. 2007;14:672–682. doi: 10.2174/092986607781483921. [DOI] [PubMed] [Google Scholar]
- 27.Corey RB, Pauling L. Fundamental dimensions of polypeptide chains. Proc R Soc Lond B Biol Sci. 1953;141:10–20. doi: 10.1098/rspb.1953.0011. [DOI] [PubMed] [Google Scholar]
- 28.Lakshmi B, Ramakrishnan C, Archunan G, Sowdhamini R, Srinivasan N. Investigations of Ramachandran disallowed conformations in protein domain families. Int J Biol Macromol. 2014;63:119–125. doi: 10.1016/j.ijbiomac.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 29.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table I: 96 alignment positions of the glycyl residues having the (φ, ψ) values that are allowed for non-glycyl residue according to Ala Ramachandran map (Category A). For every alignment position PDB code of the entries corresponding to the ultra-high resolution structure, residue number of the glycyl residue of interest and its (φ, ψ) values are provided.
Supplementary table II: 189 alignment positions of the glycyl residues having (φ, ψ) values that are allowed for glycyl residue according to Gly Ramachandran map but disallowed according to Ala Ramachandran map (Category B). For every alignment position PDB code of the entries corresponding to the ultra-high resolution structure, residue number of the glycyl residue of interest and its (φ, ψ) values are provided.