

ABSTRACT
Obese pregnant women have increased levels of proinflammatory cytokines in maternal circulation and placental tissues. However, the pathways contributing to placental inflammation in obesity are largely unknown. We tested the hypothesis that maternal body mass index (BMI) was associated with elevated proinflammatory cytokines in maternal and fetal circulations and increased activation of placental inflammatory pathways. A total of 60 women of varying pre-/early pregnancy BMI, undergoing delivery by Cesarean section at term, were studied. Maternal and fetal (cord) plasma were collected for analysis of insulin, leptin, IL-1beta, IL-6, IL-8, monocyte chemoattractant protein (MCP) 1, and TNFalpha by multiplex ELISA. Activation of the inflammatory pathways in the placenta was investigated by measuring the phosphorylated and total protein expression of p38-mitogen-activated protein kinase (MAPK), c-Jun-N-terminal kinase (JNK)-MAPK, signal transducer-activated transcription factor (STAT) 3, caspase-1, IL-1beta, IkappaB-alpha protein, and p65 DNA-binding activity. To determine the link between activated placental inflammatory pathways and elevated maternal cytokines, cultured primary human trophoblast (PHT) cells were treated with physiological concentrations of insulin, MCP-1, and TNFalpha, and inflammatory signaling analyzed by Western blot. Maternal BMI was positively correlated with maternal insulin, leptin, MCP-1, and TNFalpha, whereas only fetal leptin was increased with BMI. Placental phosphorylation of p38-MAPK and STAT3, and the expression of IL-1beta protein, were increased with maternal BMI; phosphorylation of p38-MAPK was also correlated with birth weight. In contrast, placental NFkappaB, JNK and caspase-1 signaling, and fetal cytokine levels were unaffected by maternal BMI. In PHT cells, p38-MAPK was activated by MCP-1 and TNFalpha, whereas STAT3 phosphorylation was increased following TNFalpha treatment. Maternal BMI is associated with elevated maternal cytokines and activation of placental p38-MAPK and STAT3 inflammatory pathways, without changes in fetal systemic inflammatory profile. Activation of p38-MAPK by MCP-1 and TNFalpha, and STAT3 by TNFalpha, suggests a link between elevated proinflammatory cytokines in maternal plasma and activation of placental inflammatory pathways. We suggest that inflammatory processes associated with elevated maternal BMI may influence fetal growth by altering placental function.
Keywords: cytokines, innate immune response, obesity
INTRODUCTION
In the United States, over two-thirds of women of reproductive age have a high body mass index (BMI; >25 kg/m2), and more than one-third are obese (BMI > 30 kg/m2) [1]. Pregravid obesity represents significant health risks to both the mother and the baby, with an increased risk of miscarriage, pre-eclampsia, gestational diabetes, stillbirth, and Cesarean delivery [2]. Infants born to overweight and obese mothers are more likely to be large for gestational age, macrosomic, and insulin resistant [3, 4]. In addition, epidemiological and experimental studies have demonstrated associations between maternal obesity and increased risk of developing cardiovascular and metabolic diseases in the offspring later in life [5]. As increasing numbers of women enter pregnancy with high BMI, this represents a major public health issue [1, 2].
Chronic inflammation has emerged as a common pathophysiology in obesity and obesity-related disorders, such as insulin resistance in nonpregnant individuals [6]. Recent findings indicate that the systemic and placental inflammatory responses associated with normal pregnancy are exaggerated by obesity [7–9]. This is reflected in elevated maternal circulating levels of proinflammatory cytokines [3, 8–11] and increased mRNA expression of IL-1β, IL-8, and monocyte chemoattractant protein (MCP) 1 in the placenta [10]. The increase in placental inflammatory mediators may be secondary to infiltration of maternal macrophages, which are elevated in the maternal circulation in obesity, and known to release the proinflammatory cytokines IL-1, IL-6, and TNFα [9, 12]. Alternatively, inflammatory stimuli, such as endotoxin [8], lipids [13, 14], reactive oxygen species [15], and oxidized lipids and sterols [16], may directly activate placental inflammatory pathways leading to local production of proinflammatory cytokines. Placental syncytiotrophoblast cells are in direct contact with maternal blood, and express many isoforms of Toll-like receptors and Nod-like receptors, which detect extracellular and intracellular inflammatory stimuli [17, 18]. These inflammatory receptors, along with receptors for proinflammatory cytokines, activate downstream signaling pathways, including nuclear factor (NF) κB, signal transducer-activated transcription factor (STAT) 3, caspase-1, and the stress/mitogen-activated protein kinases (MAPK) p38 and p46/p54 (c-Jun-N-terminal kinase [JNK]), which are functionally expressed in the placenta [19]. However, it is currently unknown which of these pathways are involved in the placental inflammatory response to maternal obesity.
Many studies have examined the effect of maternal obesity on maternal systemic inflammatory profile [3, 8–11], but few reports have investigated the impact of obesity on the fetal [3, 20] or placental [9, 10] inflammatory milieu. Furthermore, the impact of maternal BMI on maternal and fetal inflammatory status, as well as specific inflammatory signaling pathways in the placenta, is largely unknown. Moreover, the impact of BMI on maternal, placental, and fetal inflammation has not been simultaneously studied in a single cohort. Therefore, in this study, we determined the influence of maternal BMI on maternal and fetal inflammatory mediators, and placental inflammatory pathways. We hypothesized that increased maternal BMI is associated with elevated proinflammatory cytokines in the maternal and fetal circulation, as well as activation of placental p38-MAPK, JNK-MAPK, STAT3, caspase-1, IL-1β, and NFκB signaling pathways.
MATERIALS AND METHODS
Study Subjects
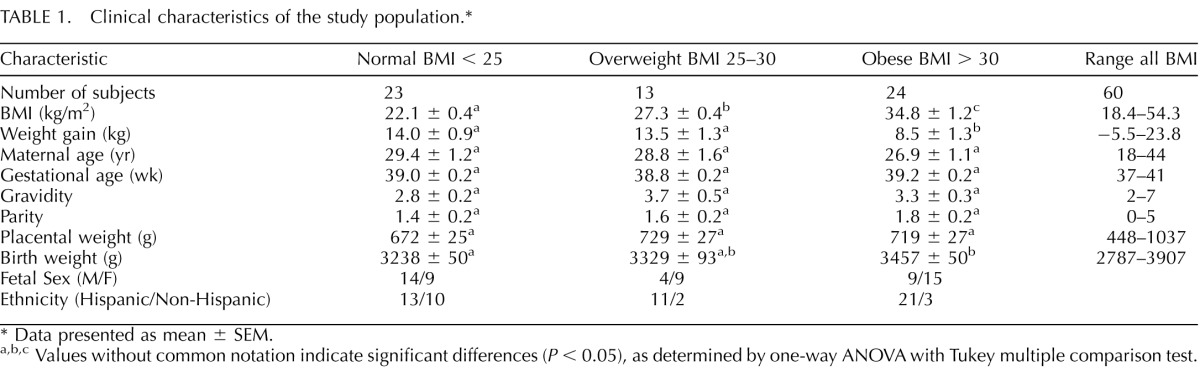
Maternal and umbilical blood samples and placental tissue were collected after informed and written consent; the protocol was approved by the Institutional Review Board at University of Texas Health Science Center, San Antonio. A total of 60 women with varying BMI (range, 18.4–54.3 kg/m2) based on pregravid or early gestation weights and uncomplicated, singleton term pregnancies (>37 wk of gestation) were studied. BMI was calculated in the clinic based on height and weight measurements from prepregnancy medical records where available, or at the first visit to maternity clinic (<14 wk). All deliveries were elective Cesarean section and performed before the onset of labor. The clinical characteristics of the study participants are presented in Table 1. While the maternal and fetal blood samples were matched, we were only able to obtain a match of 21 subjects for blood and placental samples. The demographics of the subjects for each sample set (i.e., blood plasma and placental analysis) were very similar (Supplemental Table S1; available online at www.biolreprod.org). Exclusion criteria were smoking, concurrent disease, such as diabetes or hypertension, development of pregnancy complications, including gestational diabetes, pregnancy-induced hypertension, and pre-eclampsia, and the delivery of small for gestational age (<10th centile) or large for gestational age (>90th centile) infants according to published growth curves [21].
TABLE 1.
Clinical characteristics of the study population.*
Data presented as mean ± SEM.
Values without common notation indicate significant differences (P < 0.05), as determined by one-way ANOVA with Tukey multiple comparison test.
Cytokine Analysis
Maternal fasting blood samples were collected from a subset of the recruited women (n = 49) prior to Cesarean section and the corresponding venous cord blood obtained within 15 min of the delivery of the placenta. For the collection of venous cord blood, the umbilical cord was doubly clamped immediately after the delivery of the neonate. With the placenta attached, the umbilical vein was identified (thin walled, single vessel overlying the two umbilical arteries) and punctured approximately 10 cm from the site of placental attachment. Blood samples were collected in BD Vacutainer tubes containing 5.4 mg K2EDTA (BD Bioscience, San Jose, CA). After 30-min incubation at room temperature, tubes were centrifuged for 15 min at 800 × g at 4°C, and plasma collected, aliquoted, and frozen at −80°C. Plasma samples were analyzed for insulin, leptin, IL-1β, IL-6, IL-8, MCP-1, and TNFα by multiplex ELISA with a Milliplex MAP kit (EMD Millipore). Coefficient of variation for interassay comparisons ranged from 3.6% to 10% and intra-assay comparisons ranged from 5.5% to 11.5%. The lowest detection limit for all multiplex analytes was 0.5 pg/ml.
Placental Collection and Tissue Processing
Placentas were collected from a subset of the recruited women (n = 32). Within 15 min of delivery, the decidua basalis and chorionic plate were removed and villous tissue was dissected and rinsed in cold physiological saline. The villous tissue was transferred to cold buffer D (250 mM sucrose, 10 mM hepes, pH 7.4) containing 1:100 dilution of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO) and homogenized on ice with a Polytron (Kinematica, Luzern, Switzerland). The placental homogenates were frozen in liquid nitrogen and stored at −80°C until further processing.
Western Blotting
Placental homogenates were thawed on ice and centrifuged at 16 000 × g, 4°C for 15 min. Supernatants were collected and used for protein expression analysis. Protein concentrations were determined with Pierce BCA Protein Assay kit (Thermo Scientific, Rockford, IL). Equal amounts of protein (10 μg) were loaded into each well and separated on Any kD Mini-PRO-TEAN TGX precast polyacrylamide gels from Bio-Rad (Hercules, CA). Separated proteins were transferred onto PVDF membranes (Thermo Scientific) and blocked with 5% nonfat milk for 1 h at room temperature. After washing in Tris-buffered saline containing 0.1% Tween (TBS-T), membranes were incubated in primary antibodies overnight at 4°C in TBS-T containing 2.5% BSA. Primary antibodies were purchased from Cell Signaling Technology (Danvers, MA): (pro)caspase-1 (#2225), inhibitor of kappa Bα (IκBα; #4812), JNK (#9252), phospho-JNK (#4668; T183/Y185), p38-MAPK (#9212), phospho-p38 MAPK (#4511; T180/Y182), STAT3 (#9139), and phospho-STAT3 (#9145; Y705); from Abcam (Cambridge, MA): IL-1β (ab2105); and from Sigma: β-actin (A2228). The membranes were then washed and incubated with the appropriate peroxidase-labeled secondary antibody diluted in 2% nonfat milk powder and visualized by enhanced chemiluminescence using Super Signal Dura West (Thermo Scientific). Resultant images were captured on a G:Box ChemiXL1.4 (Syngene, Cambridge, U.K.) and bands quantified using GeneTools (version 4.03; Syngene, Cambridge, U.K.). Target protein expression was normalized to β-actin expression.
NFĸB Activation Assay
Nuclei were extracted from homogenates of placental villous tissue using a nuclear isolation kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. Total nuclear protein concentrations were not different between the BMI groups. NFκB p65 DNA-binding activity was measured in 20 μg of nuclear extract protein with a transcription factor assay kit (Active Motif). Each sample was measured in duplicate.
Primary Human Trophoblast Culture and Treatments
Placentas from healthy pregnant women at term were collected for isolation of primary human trophoblast (PHT) cells. PHT cells were isolated by trypsin digestion and Percoll purification, as previously described [22, 23]. Cells were cultured in Dulbecco modified Eagle medium (Sigma-Aldrich) and Ham F-12 nutrient mixture (Life Technologies) containing 10% fetal calf serum (Atlanta Biological), 50 μg/ml gentamicin, 60 μg/ml benzyl penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich). Cells were plated in 35-mm dishes at a density of 2 × 106 for subsequent protein analyses, and incubated in a 5% CO2 humidified atmosphere at 37°C. After 18 h, attached PHT cells were washed twice in warm Dulbecco PBS, and subsequently culture media were changed daily. PHT cell purity was confirmed by high protein expression of Cytokeratin-7 (epithelial cell marker) and absence of Vimentin (fibroblast cell marker) expression as previously described [22].
After 66 h in culture, PHT cells were treated with MCP-1 (100 pg/ml) and TNFα (10 pg/ml), and insulin (5.8 ng/ml) at 87 h of culture, and protein lysates collected at 90 h for protein analysis. The concentrations of cytokines used for in vitro experiments were based on high physiological concentrations observed in maternal plasma. The concentration of insulin used corresponds to postprandial insulin levels in pregnant women at term [24]. The timing of insulin and cytokine stimulations were based on time points previously determined to produce the most robust effects [22]. Cell culture experiments were repeated on trophoblast cells isolated from four different placentas.
Data Presentation and Statistical Analysis
Demographic and clinical characteristics of the study population were categorized according to maternal BMI as normal (BMI < 25), overweight (BMI 25–30), or obese (BMI > 30), and one-way ANOVA, followed by Tukey post hoc multiple comparison test, performed. Summary data are presented as mean ± SEM or interquartile range. D'Agostino and Pearson normality test was initially performed and relationships between maternal BMI or birth weight and maternal/fetal cytokines or placental inflammatory signaling were determined using Pearson or Spearman correlation coefficients, as appropriate. Influence of insulin and cytokines on inflammatory activation in PHTs was investigated by one-way ANOVA followed by Dunnet post hoc test. P values below 0.05 were considered statistically significant.
RESULTS
Clinical Characteristics
Maternal demographics and placental and fetal weights are presented in Table 1. Data presented in this table represent the combined information of all recruited subjects. Separate analysis of the maternal/fetal plasma and placental samples (Supplemental Table S1) demonstrates similar demographics between the two sample sets. Obese women gained less weight than overweight and normal BMI women, and obese women delivered heavier babies than normal BMI women (Table 1). Maternal age, gestational age at delivery, gravidity and parity, and placental weight were not statistically different between groups.
Maternal BMI and Circulating Proinflammatory Cytokine Concentrations
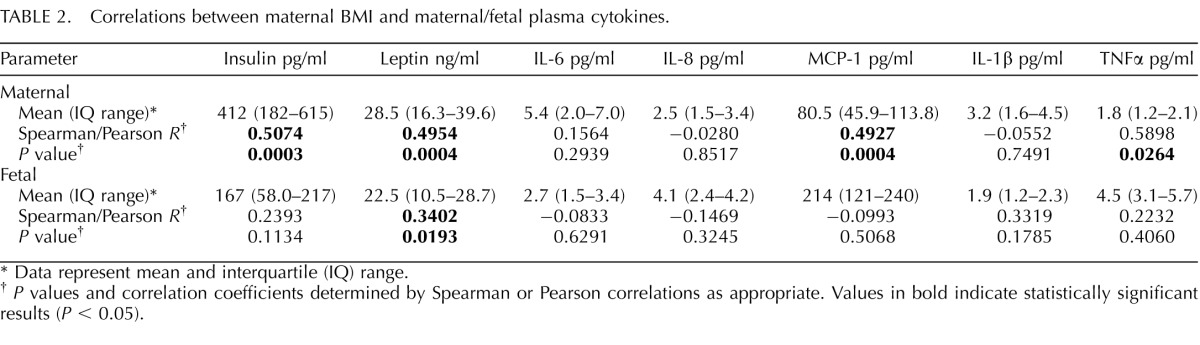
Circulating levels of proinflammatory cytokines as well as insulin and leptin are presented in Table 2. As expected, maternal plasma levels of insulin and leptin were increased with maternal BMI. The proinflammatory cytokines MCP-1 and TNFα in the maternal plasma were also positively correlated with maternal BMI. The other cytokines measured in the maternal circulation (IL-1β, IL-6, and IL-8) did not vary with maternal BMI. Importantly, fetal plasma concentrations of cytokines were not related to maternal BMI, but fetal leptin levels were positively correlated with maternal BMI. None of the maternal or fetal plasma cytokines correlated significantly with birth weight (data not shown).
TABLE 2.
Correlations between maternal BMI and maternal/fetal plasma cytokines.
Data represent mean and interquartile (IQ) range.
P values and correlation coefficients determined by Spearman or Pearson correlations as appropriate. Values in bold indicate statistically significant results (P < 0.05).
Maternal BMI and Placental Inflammatory Pathways
The inflammatory pathways caspase-1, STAT3, NFκB, and MAPKs (p38 and JNK) were analyzed in placental protein homogenates and regression analyses were performed with maternal BMI. Phosphorylation of p38-MAPK (T182/Y180) was positively correlated with maternal BMI (Fig. 1B); however JNK-MAPK (T183/Y185) phosphorylation was not associated with maternal BMI (Fig. 1D). The total expression of p38-MAPK or JNK-MAPK did not change with maternal BMI (Fig. 1, C and E). Analysis of phosphorylated and total JNK p46 and p54 proteins separately also did not result in significant correlation with maternal BMI (data not shown).
FIG. 1.

Placental p38-MAPK and JNK-MAPK signaling in relation to maternal BMI. A) Representative Western blots of phosphorylated p38-MAPK (T182/Y180) and JNK-MAPK (T183/Y185) and total p38-MAPK and JNK-MAPK protein expression in homogenates of placentas from pregnancies with varying maternal BMI (n = 32). Scatter plots demonstrate relationship between maternal BMI and placental p38-MAPK (T182/Y180) phosphorylation (B), p38-MAPK expression (C), p46/54 JNK-MAPK (T183/Y185) phosphorylation (D), and p46/54 JNK-MAPK expression (E). Line of best fit indicates significant correlation with maternal BMI.
Phosphorylation of STAT3 (Y705) was significantly increased with maternal BMI (Fig. 2B), while total STAT3 expression did not change with maternal BMI (Fig. 2C). NFκB activation, as measured by IκBα degradation (Fig. 2E) and p65 DNA-binding activity (Fig. 2F), was not correlated with maternal BMI.
FIG. 2.

Placental STAT3 and NFκB activity in relation to maternal BMI. Representative Western blots of phosphorylated STAT3 (Y705) and STAT3 (A), and IκBα (D) protein expression in homogenates of placentas from pregnancies with varying maternal BMI (n = 32). Scatter plots demonstrate relationship between maternal BMI and placental STAT3 (Y705) phosphorylation (B), STAT3 expression (C), IκBα expression (E), and nuclear p65 DNA-binding activity (n = 21; F). Line of best fit indicates significant correlation with maternal BMI.
The inflammasome complex components caspase-1 and IL-1β were measured by Western blotting (Fig. 3A). While pro-caspase-1 (Fig. 3B) and caspase-1 (Fig. 3C) expression did not correlate with maternal BMI, IL-1β (Fig. 3D) expression was elevated with increasing maternal BMI.
FIG. 3.

Activation of placental inflammasome complex in relation to maternal BMI. A) Representative Western blots of pro-caspase-1, caspase-1, and IL-1β protein expression in homogenates of placentas from pregnancies with varying maternal BMI (n = 32). Scatter plots demonstrate relationship between maternal BMI and placental pro-caspase-1 (B), caspase-1 (C), and IL-1β (D) expression. Line of best fit indicates significant correlation with maternal BMI.
Birth Weight and Placental Inflammatory Pathways
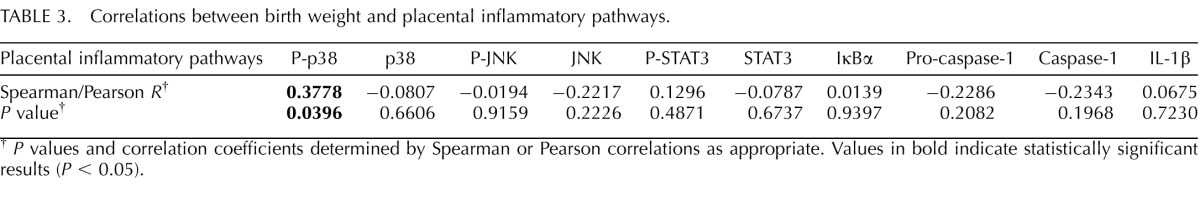
Placental p38-MAPK phosphorylation was positively correlated with birth weight (Table 3). In contrast, phosphorylated or total expression of the other inflammatory proteins (JNK, STAT3, NFκB, caspase-1, and IL-1β) in the placenta did not vary with birth weight.
TABLE 3.
Correlations between birth weight and placental inflammatory pathways.
P values and correlation coefficients determined by Spearman or Pearson correlations as appropriate. Values in bold indicate statistically significant results (P < 0.05).
Activation of Inflammatory Pathways p38-MAPK and STAT3 by Maternal Cytokines—In Vitro Study in Cultured PHTs
In order to determine the link between elevated maternal plasma hormones and cytokines detected with increasing BMI, and the activation of placental inflammatory pathways p38-MAPK and STAT3, we treated isolated PHTs with insulin, MCP-1, or TNFα at high physiological concentrations observed in this study. Treatment of PHTs with insulin did not affect the phosphorylation or the total expression of p38-MAPK or STAT3 (Fig. 4). However, p38-MAPK phosphorylation was stimulated by both MCP-1 and TNFα (Fig. 5B), whereas STAT3 phosphorylation was stimulated by TNFα only (Fig. 5E). TNFα also increased total STAT3 expression (Fig. 5F).
FIG. 4.

Influence of insulin on p38-MAPK and STAT3 phosphorylation in cultured PHTs. Representative Western blots of phosphorylated p38-MAPK (T182/Y180) and p38 MAPK (A), and phosphorylated STAT3 (Y705) and STAT3 expression (D) in PHT cells treated with insulin (5.8 ng/ml) or vehicle control. Histograms illustrate relative protein expression of phosphorylated p38-MAPK (T182/Y180) (B), p38-MAPK (C), phosphorylated STAT3 (Y705) (E), and STAT3 (F). Mean + SEM, n = 4. Cnt, control; Ins, insulin.
FIG. 5.

Influence of MCP-1 and TNFα on p38-MAPK and STAT3 phosphorylation in cultured PHTs. Representative Western blots of phosphorylated p38-MAPK (T182/Y180) and p38 MAPK (A), and phosphorylated STAT3 (Y705) and STAT3 (D) expression in PHT cells treated with MCP-1 (100 pg/ml), TNFα (10 pg/ml), or vehicle control (Cnt). Histograms illustrate relative protein expression of phosphorylated p38-MAPK (T182/Y180) (B), p38-MAPK (C), phosphorylated STAT3 (Y705) (E), and STAT3 (F). Mean + SEM, n = 4. ** P < 0.01, *** P < 0.001 versus control.
DISCUSSION
To our knowledge, this is the first study to examine the impact of maternal BMI on inflammatory signals in the mother, placenta, and fetus in the same cohort. The most important finding is that maternal BMI is associated with activation of placental p38-MAPK and STAT3 signaling, without changes in the classical inflammatory pathways NFκB, JNK, and caspase-1, or fetal systemic inflammatory profile. This finding suggests that inflammation associated with maternal BMI may impact the fetus by altering placental function rather than by fetal exposure to elevated levels of proinflammatory cytokines.
We observed associations between maternal BMI and proinflammatory cytokines TNFα and MCP-1 levels in maternal plasma. Both TNFα and MCP-1 are major proinflammatory cytokines with biological roles in insulin resistance [25, 26]. Accordingly, these findings are consistent with previous studies indicating that obese women are more insulin resistant compared to normal-BMI mothers [3]. Although we did not investigate the origin of these cytokines, previous studies have shown that maternal adipose tissue, which expands greatly in obesity, is a significant source of circulating cytokines [8]. Furthermore, both MCP-1 and TNFα mRNA expression were shown to be increased by 2–5 fold in both the placenta and adipose tissue of obese pregnant women compared to normal-weight women [8, 10], which is consistent with a role for both maternal adipose tissue and the placenta as contributors to maternal systemic inflammation.
While there was evidence of inflammation in maternal plasma, fetal/cord plasma levels of the inflammatory mediators investigated in this study were not influenced by maternal BMI. However, fetal plasma leptin concentrations were positively correlated with maternal BMI, whereas fetal insulin was independent of maternal BMI. Our data are supported by other clinical studies, which also demonstrate a lack of systemic inflammation in fetal plasma in response to maternal obesity [20]. Although our data do not support a role for systemic inflammation in the fetus, it does not exclude a role for local inflammatory processes in tissues such as the fetal adipose.
Placental STAT3 and p38-MAPK pathways were activated with increasing maternal BMI. This finding is consistent with a recent report indicating increased p38-MAPK signaling in placentas of obese compared to lean mothers [27]. The p38-MAPK signaling activates a number of transcription factors, including c-fos, c-jun, and ATF, which regulate proinflammatory gene expression [28], whereas STAT3 is a transcription factor that may either directly regulate gene expression or act indirectly by interacting with other transcription factors [29]. Previous studies have established increased cytokine expression in placentas of obese mothers [9, 10], but whether the activation of placental p38-MAPK or STAT3 in association with high BMI contributes to elevated placental cytokine expression remains to be determined.
Placental p38-MAPK signaling was also associated with birth weight. Fetal growth is highly dependent on placental nutrient supply, and previous studies have established associations between placental nutrient transporters and maternal BMI as well as birth weight [30]. Furthermore, p38-MAPK is involved in the regulation of glucose [31, 32] and amino acid transporters in a variety of cell types [33, 34]. Regulation of placental nutrient transport by p38-MAPK signaling has not been reported, but p38-MAPK has been shown to regulate glucose and amino acid transport in other tissues [32, 34], and our preliminary studies indicate a role for p38-MAPK in the regulation of insulin-dependent system-A and system-L amino acid transport in cultured primary trophoblasts [35]. Likewise, STAT3 activity in primary trophoblasts also regulates amino acid transport [36, 37]. However, placental STAT3 phosphorylation was not correlated with birth weight in this study.
Contrary to our hypothesis, JNK-MAPK and NFκB signaling was not associated with maternal BMI or birth weight. This is in contrast to previous reports by Saben et al. of increased JNK phosphorylation in placentas of obese mothers compared to lean mothers [27]. A possible explanation for the differences in the results may be our selection of placentas only from nonlaboring women, whereas Saben et al. included both laboring and nonlaboring women, and therefore the effects of labor cannot be excluded, especially because recent studies indicate alterations in placental inflammatory signaling pathways with labor [38].
Caspase-1 is a component of the inflammasome complex that regulates the posttranslational maturation of IL-1β. Upon stimulation by inflammatory signals, pro-caspase-1 (p50) is cleaved into caspase-1 (p20), which, in turn, processes IL-1β into the mature and biologically active form. Whereas the placental expression of pro-caspase-1 or caspase-1 was not significantly associated with maternal BMI, placental IL-1β expression was significantly correlated with BMI. This indicates that the increase in placental IL-1β expression with maternal BMI is likely to be regulated by processes other than caspase-1, perhaps through p38-MAPK or STAT3 activity, which were associated with maternal BMI.
It was interesting to note that the pathways typically associated with inflammation, such as NFκB, JNK-MAPK, and caspase-1, were not altered by maternal BMI. These pathways promote insulin resistance in many tissues [39, 40], including the placenta [22]. Moreover, placental insulin signaling has previously been associated with fetal growth, with increased activity reported in obese women giving birth to large babies [30], and reduced signaling in growth-restricted infants [41]. Since the obese women in this study had elevated fasting insulin and delivered larger babies, it is possible that there is greater insulin signaling in the placentas of the obese mothers compared to normal-BMI women. Hence, the lack of placental inflammatory signaling by NFκB, JNK-MAPK, and caspase-1 may have contributed to increased fetal growth.
Although p38-MAPK and STAT3 are implicated in the innate immune response, these pathways are also involved in growth factor signaling [29, 42]. Therefore, the increased placental p38-MAPK and STAT3 may be in response to elevated insulin and leptin as well as inflammatory signals. This is supported by previous reports of increased insulin signaling in the placentas of mothers whose pregnancies were complicated by obesity [30] or gestational diabetes [43]. To investigate a possible link between elevated maternal insulin and cytokines MCP-1 and TNFα, and activation of placental p38-MAPK and STAT3 pathways, we utilized a well-established model of cultured PHT cells from term placentas. Treatment with insulin did not affect p38-MAPK or STAT3 phosphorylation; however, p38-MAPK was activated by both MCP-1 and TNFα, whereas STAT3 phosphorylation was stimulated by TNFα only. Furthermore, leptin has established effects on activation of STAT3 in many tissues, including the placenta [44]. Therefore, increased maternal levels of proinflammatory cytokines TNFα, MCP-1, as well as leptin, may contribute to the increased placental p38-MAPK and STAT3 signaling with maternal BMI. Moreover, circulating nonesterified fatty acids [13] and free fatty acids, such as oleic and palmitic acid [45], which are increased with maternal BMI, activate both p38-MAPK and STAT3 in primary trophoblasts and placental cell lines [13, 27]. Hence, the increased placental activation of p38-MAPK and STAT3 may be a consequence of elevated maternal circulating cytokines, hormones such as leptin, and hyperlipidemia associated with increasing maternal BMI.
One of the strengths of our study is the well-defined study population. We specifically excluded additional clinical factors that may influence the maternal inflammatory profile, such as pre-eclampsia, prepregnancy and gestational diabetes, and hypertension, and further limited our recruitment to nonlaboring women who delivered by Caesarean section and delivered a normal-size infant. Moreover, BMI calculations were based on accurate measurement of weight and height at the clinic, and not self-reported values, which have been shown to lead to miscategorization [46]. The population in our study was predominantly Hispanic women, which reflects the demography of the San Antonio population. However, ethnicity did not influence our results, since re-analysis of the data after separation according to ethnicity exhibited similar results. Furthermore, analysis after subdivision of the data based on fetal sex did not change the pattern of the overall result. Despite the modest sample size, several clinical characteristics of the study population were consistent with previous reports in larger populations. For example, high pre/early pregnancy BMI was associated with higher birth weights [4] and decreased gestational weight gain [47], as well as maternal hyperinsulinemia and hyperleptinemia [3].
In summary, we report that the placental inflammatory pathways p38-MAPK and STAT3 are activated with increasing maternal BMI, which may be due to elevated maternal circulating cytokines TNFα and MCP-1, leptin, and hyperlipidemia. TNFα and MCP-1 activated p38-MAPK and/or STAT3 phosphorylation in cultured PHT cells, further illustrating the possibility of maternal cytokines in influencing placental inflammation. Moreover, placental p38-MAPK activity was correlated with birth weight, possibly indicating a link between placental p38-MAPK pathway, nutrient transport, and fetal growth. Conversely, the placental inflammatory pathways JNK-MAPK, NFκB, and caspase-1, which are commonly associated with insulin resistance, were not altered by maternal BMI or birth weight. These findings support a role for distinct inflammatory pathways in the regulation of fetal growth through diverse effects on placental function. Although the maternal cytokines were elevated with BMI, there were no changes in fetal cytokine levels. Taken together, our data suggests that maternal BMI is associated with systemic maternal inflammation and activation of distinct placental inflammatory pathways, but these effects are not transmitted as systemic inflammation in the fetus. Future studies are warranted to further elucidate the exact role of p38-MAPK and STAT3 pathways in placental function and fetal development.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Ms. Evelyn Miller, patients and staff at Labor and Delivery, University Hospital (San Antonio) for making collection of placental tissue possible, and to Ms. Elizabeth Dudley for excellent technical assistance.
Footnotes
Supported by National Institutes of Health grant DK89989 to T.L.P. and grants from the Swedish Research Council and Swedish Society of Endocrinology to S.L.
These authors contributed equally to this work.
REFERENCES
- Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- Galtier-Dereure F, Boegner C, Bringer J. Obesity and pregnancy: complications and cost. Am J Clin Nutr. 2000;71:1242S–1248S. doi: 10.1093/ajcn/71.5.1242s. [DOI] [PubMed] [Google Scholar]
- Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32:1076–1080. doi: 10.2337/dc08-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Han S, Zhu J, Sun X, Ji C, Guo X. Pre-pregnancy body mass index in relation to infant birth weight and offspring overweight/obesity: a systematic review and meta-analysis. PLoS One. 2013;8:e61627. doi: 10.1371/journal.pone.0061627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake AJ, Reynolds RM. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction. 2010;140:387–398. doi: 10.1530/REP-10-0077. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison FC, Roberts KA, Barr SM, Norman JE. Obesity, pregnancy, inflammation, and vascular function. Reproduction. 2010;140:373–385. doi: 10.1530/REP-10-0074. [DOI] [PubMed] [Google Scholar]
- Basu S, Haghiac M, Surace P, Challier JC, Guerre-Millo M, Singh K, Waters T, Minium J, Presley L, Catalano PM, Hauguel-de Mouzon S. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring) 2011;19:476–482. doi: 10.1038/oby.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29:274–281. doi: 10.1016/j.placenta.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC. Placental structure and inflammation in pregnancies associated with obesity. Placenta. 2011;32:247–254. doi: 10.1016/j.placenta.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab. 2007;92:969–975. doi: 10.1210/jc.2006-2083. [DOI] [PubMed] [Google Scholar]
- Basu S, Leahy P, Challier JC, Minium J, Catalano P, Hauguel-de Mouzon S. Molecular phenotype of monocytes at the maternal-fetal interface Am J Obstet Gynecol 2011. 205 265: e261 e268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lager S, Gaccioli F, Ramirez VI, Jones HN, Jansson T, Powell TL. Oleic acid stimulates system A amino acid transport in primary human trophoblast cells mediated by Toll-like receptor 4. J Lipid Res. 2013;54:725–733. doi: 10.1194/jlr.M033050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathmaperuma AN, Mana P, Cheung SN, Kugathas K, Josiah A, Koina ME, Broomfield A, Delghingaro-Augusto V, Ellwood DA, Dahlstrom JE, Nolan CJ. Fatty acids alter glycerolipid metabolism and induce lipid droplet formation, syncytialisation and cytokine production in human trophoblasts with minimal glucose effect or interaction. Placenta. 2010;31:230–239. doi: 10.1016/j.placenta.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Roberts VH, Smith J, McLea SA, Heizer AB, Richardson JL, Myatt L. Effect of increasing maternal body mass index on oxidative and nitrative stress in the human placenta. Placenta. 2009;30:169–175. doi: 10.1016/j.placenta.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye IL, Waddell BJ, Mark PJ, Keelan JA. Oxysterols exert proinflammatory effects in placental trophoblasts via TLR4-dependent, cholesterol-sensitive activation of NF-kappaB. Mol Hum Reprod. 2012;18:341–353. doi: 10.1093/molehr/gas001. [DOI] [PubMed] [Google Scholar]
- Koga K, Mor G. Expression and function of toll-like receptors at the maternal-fetal interface. Reprod Sci. 2008;15:231–242. doi: 10.1177/1933719108316391. [DOI] [PubMed] [Google Scholar]
- Abrahams VM. The role of the Nod-like receptor family in trophoblast innate immune responses. J Reprod Immunol. 2011;88:112–117. doi: 10.1016/j.jri.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Keelan JA. Pharmacological inhibition of inflammatory pathways for the prevention of preterm birth. J Reprod Immunol. 2011;88:176–184. doi: 10.1016/j.jri.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Radaelli T, Uvena-Celebrezze J, Minium J, Huston-Presley L, Catalano P, Hauguel-de Mouzon S. Maternal interleukin-6: marker of fetal growth and adiposity. J Soc Gynecol Investig. 2006;13:53–57. doi: 10.1016/j.jsgi.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Olsen IE, Groveman SA, Lawson ML, Clark RH, Zemel BS. New intrauterine growth curves based on United States data. Pediatrics. 2010;125:e214–224. doi: 10.1542/peds.2009-0913. [DOI] [PubMed] [Google Scholar]
- Aye IL, Jansson T, Powell TL. Interleukin-1beta inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol Cell Endocrinol. 2013;381:46–55. doi: 10.1016/j.mce.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF., III. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986;118:1567–1582. doi: 10.1210/endo-118-4-1567. [DOI] [PubMed] [Google Scholar]
- Phelps RL, Metzger BE, Freinkel N. Carbohydrate metabolism in pregnancy. XVII. Diurnal profiles of plasma glucose, insulin, free fatty acids, triglycerides, cholesterol, and individual amino acids in late normal pregnancy. Am J Obstet Gynecol. 1981;140:730–736. [PubMed] [Google Scholar]
- Borst SE. The role of TNF-alpha in insulin resistance. Endocrine. 2004;23:177–182. doi: 10.1385/ENDO:23:2-3:177. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saben J, Zhong Y, Gomez-Acevedo H, Thakali KM, Borengasser SJ, Andres A, Shankar K. Early growth response protein-1 mediates lipotoxicity-associated placental inflammation: role in maternal obesity. Am J Physiol Endocrinol Metab. 2013;305:E1–E14. doi: 10.1152/ajpendo.00076.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ. A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010;8:47. doi: 10.1186/1741-7007-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T, Powell TL. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98:105–113. doi: 10.1210/jc.2012-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MA, Moylan JS, Smith JD, Goodyear LJ, Reid MB. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J Physiol. 2009;587:3363–3373. doi: 10.1113/jphysiol.2008.165639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho RC, Alcazar O, Fujii N, Hirshman MF, Goodyear LJ. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2004;286:R342–R349. doi: 10.1152/ajpregu.00563.2003. [DOI] [PubMed] [Google Scholar]
- Ruiz E, Siow RC, Bartlett SR, Jenner AM, Sato H, Bannai S, Mann GE. Vitamin C inhibits diethylmaleate-induced L-cystine transport in human vascular smooth muscle cells. Free Radic Biol Med. 2003;34:103–110. doi: 10.1016/s0891-5849(02)01192-9. [DOI] [PubMed] [Google Scholar]
- Tsai RY, Tai YH, Tzeng JI, Lin SL, Shen CH, Yang CP, Hsin ST, Wang CB, Wong CS. Ultra-low dose naloxone restores the antinociceptive effect of morphine in pertussis toxin-treated rats and prevents glutamate transporter downregulation by suppressing the p38 mitogen-activated protein kinase signaling pathway. Neuroscience. 2009;159:1244–1256. doi: 10.1016/j.neuroscience.2009.01.058. [DOI] [PubMed] [Google Scholar]
- Aye IL, Gao X, Weintraub ST, Jansson T, Powell TL. Adiponectin inhibits insulin function in primary trophoblasts by PPARalpha-mediated ceramide synthesis. Mol Endocrinol. 2014;28:512–524. doi: 10.1210/me.2013-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol. 2009;297:C1228–C1235. doi: 10.1152/ajpcell.00195.2009. [DOI] [PubMed] [Google Scholar]
- Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino acid transport in human primary trophoblast cells. Diabetes. 2010;59:1161–1170. doi: 10.2337/db09-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cindrova-Davies T, Yung HW, Johns J, Spasic-Boskovic O, Korolchuk S, Jauniaux E, Burton GJ, Charnock-Jones DS. Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. Am J Pathol. 2007;171:1168–1179. doi: 10.2353/ajpath.2007.070528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanti JF, Ceppo F, Jager J, Berthou F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front Endocrinol (Lausanne) 2012;3:181. doi: 10.3389/fendo.2012.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviola L, Perrini S, Belsanti G, Natalicchio A, Montrone C, Leonardini A, Vimercati A, Scioscia M, Selvaggi L, Giorgino R, Greco P, Giorgino F. Intrauterine growth restriction in humans is associated with abnormalities in placental insulin-like growth factor signaling. Endocrinology. 2005;146:1498–1505. doi: 10.1210/en.2004-1332. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Alonso A, Del Rey CG, Navarro A, Tolivia J, Gonzalez CG. Effects of gestational diabetes mellitus on proteins implicated in insulin signaling in human placenta. Gynecol Endocrinol. 2006;22:526–535. doi: 10.1080/09513590600921374. [DOI] [PubMed] [Google Scholar]
- von Versen-Hoynck F, Rajakumar A, Parrott MS, Powers RW. Leptin affects system A amino acid transport activity in the human placenta: evidence for STAT3 dependent mechanisms. Placenta. 2009;30:361–367. doi: 10.1016/j.placenta.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Scholl TO, Leskiw M, Savaille J, Stein TP. Differences in maternal circulating fatty acid composition and dietary fat intake in women with gestational diabetes mellitus or mild gestational hyperglycemia. Diabetes Care. 2010;33:2049–2054. doi: 10.2337/dc10-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar LM, Siega-Riz AM, Simhan HN, Diesel JC, Abrams B. The impact of exposure misclassification on associations between prepregnancy BMI and adverse pregnancy outcomes. Obesity (Silver Spring) 2010;18:2184–2190. doi: 10.1038/oby.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt KJ, Alanis MC, Johnson ER, Mayorga ME, Korte JE. Maternal pre-pregnancy weight and gestational weight gain and their association with birthweight with a focus on racial differences. Matern Child Health J. 2013;17:85–94. doi: 10.1007/s10995-012-0950-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.