

Background: The initial step of the signal recognition particle (SRP) targeting pathway requires binding to actively translating ribosomes.
Results: SRP-ribosome binding kinetics and affinities are affected by nascent chain length.
Conclusion: SRP targeting is likely modulated at the initial ribosome-binding step, which is influenced by translation of nascent chain.
Significance: Understanding signal recognition particle-ribosome binding provides insights into the constraints of co-translational targeting.
Keywords: Fluorescence Resonance Energy Transfer (FRET), Protein Targeting, Ribosome Function, Signal Recognition Particle (SRP), Single-Molecule Biophysics
Abstract
The signal recognition particle (SRP) directs ribosome-nascent chain complexes (RNCs) displaying signal sequences to protein translocation channels in the plasma membrane of prokaryotes and endoplasmic reticulum of eukaryotes. It was initially proposed that SRP binds the signal sequence when it emerges from an RNC and that successful binding becomes impaired as translation extends the nascent chain, moving the signal sequence away from SRP on the ribosomal surface. Later studies drew this simple model into question, proposing that SRP binding is unaffected by nascent chain length. Here, we reinvestigate this issue using two novel and independent fluorescence resonance energy transfer assays. We show that the arrival and dissociation rates of SRP binding to RNCs vary according to nascent chain length, resulting in the highest affinity shortly after a functional signal sequence emerges from the ribosome. Moreover, we show that SRP binds RNCs in multiple and interconverting conformations, and that conversely, RNCs exist in two conformations distinguished by SRP interaction kinetics.
Introduction
The signal recognition particle (SRP)7 is a universally conserved RNA-protein complex responsible for the co-translational targeting of membrane and secretory proteins. In eukaryotes, it targets newly synthesized proteins to the endoplasmic reticulum membrane, whereas in prokaryotes, it targets them to the plasma membrane (1). During the initial step of targeting, SRP binds to a ribosome translating a nascent chain, referred to as a ribosome-nascent chain complex (RNC). If the RNC displays a signal sequence, RNC-bound SRP also binds the SRP receptor (SR) at the target membrane. The membrane-localized RNC is then transferred to the translocon, a protein translocation channel through which the nascent chain passes across or into the target membrane (1).
Eukaryotic SRP is composed of a 300-nucleotide RNA and six protein subunits. The simpler Escherichia coli SRP is composed of a 114-nucleotide RNA (4.5S RNA) homologous to a conserved domain of the eukaryotic SRP RNA, and a single protein subunit (Ffh), a homolog of the eukaryotic SRP54 subunit. This protein component has two domains: a methionine-rich domain that can directly bind to signal sequences (2, 3) and an N-terminal four-helix bundle and GTPase domain that can interact with SR, catalyze the hydrolysis of GTP, and contact the ribosome (4–6). The conservation of these components is such that that E. coli SRP can efficiently replace eukaryotic SRP in in vitro-targeting reactions (7, 8).
SRP is unable to target RNCs with nascent chains encoding an N-terminal signal sequence when the chains become longer than ∼140 amino acids (aa) (9, 10). This limit implies the need for an SRP monitoring state with transient RNC-binding kinetics that allows SRP to determine the presence of a signal sequence as early on in translation as possible (11). The limit also plays an important role in the kinetic proof-reading model for SRP substrate selection, which argues that only correct signal sequences allow the completion of all targeting steps before the nascent chain grows too long (12). Finally, it is also used to explain the sensitivity of SRP targeting to translation elongation rates (13).
The first proposed explanation for the nascent chain limit on SRP targeting was that as nascent chains grow past a certain length, the signal sequence becomes unavailable to SRP, thus impairing SRP-RNC binding and overall targeting (9). This simple model was later supplanted by proposals posing that it is not SRP-RNC binding that is affected by nascent chain length but instead later targeting steps (10). These two incompatible proposals exist side by side because three decades of study have not converged on a clear answer about the effect of chain length on SRP-RNC binding. Initial work showed that it does have an effect (9), but later studies disagreed (10, 11, 14). Here, we apply two fluorescence resonance energy transfer (FRET) assays to show definitively that SRP binding to RNCs displaying a signal sequence is sensitive to chain length.
EXPERIMENTAL PROCEDURES
Reagent Cloning, Expression, and Purification
All proteins used in this study were derived from E. coli strain MC4100 and expressed in E. coli. All chemicals used are from Sigma or Fisher Scientific unless otherwise indicated. The Ffh expression construct and purification protocol have been described previously (15, 16). The construct encoding ribosomal protein L29 (gene rpmC) with an N-terminal His6 and factor Xa-cleavable tag was engineered by first amplifying genomic DNA using PCR with primers containing 5′-NdeI and 3′-XhoI restriction sites. This insert was then cloned into the pET16b vector (Novagen). The open reading frame for lepB (gene lepB) was prepared for in vitro translation by amplifying DNA with a 5′-primer, including a T7 transcriptase promoter and a ribosome-binding site, according to the PURE translation system (New England Biolabs) protocol and cloned into the pCR2.1-TOPO vector according to the TOPO TA cloning kit (Invitrogen) manufacturer protocol. Ffh(Q72C) and L29(Q38C) single-cysteine mutants, as well as the lepB signal sequence mutant (peptide sequence, N-MFAEIKVIATPVTGIRWCV-C, underlined residues changed from WT as described in Ref. 17) were engineered using the QuikChange mutagenesis kit (Agilent). L29 was expressed in BL21(DE3) cells (Invitrogen) grown at 37 °C up to an A600 of 0.6 and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 16 h at 25 °C. Cells were then resuspended in resuspension buffer (50 mm K-HEPES, pH 8, 500 mm NaCl, 2 mm DTT, 10% glycerol, and 20 mm imidazole) and lysed at 1000 bar pressure (100 MPa) using an Emulsiflex (Avastin). Cell lysate was then clarified with a 16,000 rpm centrifugation step in an SS-34 rotor (Sorvall) for 30 min. The cleared supernatant was loaded onto nickel-nitrilotriacetic acid beads (Qiagen) pre-equilibrated in wash buffer (50 mm K-HEPES, pH 8, 500 mm NaCl, 2 mm DTT, 10% glycerol, and 50 mm imidazole). Bound protein was eluted from the nickel beads in a single step with elution buffer (50 mm K-HEPES, pH 8, 500 mm NaCl, 2 mm DTT, 10% glycerol, 300 mm imidazole). Eluted protein was buffer-exchanged into factor Xa cleavage buffer (20 mm Tris-HCl, pH 8, at 25 °C, 100 mm NaCl, 2 mm CaCl2, 5 mm β-mercaptoethanol, and 10% glycerol) and digested with 20 units of factor Xa (New England Biolabs) per 30 nmol of protein. Cleaved L29 was buffer-exchanged into L29 labeling buffer (50 mm Tris-HCl, pH 7, at 25 °C, 250 mm NaCl, and 10% glycerol) and labeled as described (see below). LepB-truncated mRNA was transcribed from PCR amplification products using the MEGAShortscript transcription kit (Invitrogen). The mRNA used for single-molecule experiments was further processed by annealing with an equal amount of a synthetic 3-biotinylated DNA oligonucleotide (Integrated DNA Technologies) complementary to the 5′-end of the mRNA (52 °C melting temperature). The E. coli ΔL29 strain was made by replacing the endogenous L29 gene in strain MC4100 with a kanamycin resistance gene according to previously published protocols (18). The deletion was confirmed using diagnostic PCR. 70S ribosomes were purified from the ΔL29 strain using previously described methods (19). 30S ribosomal subunit helix 44 mutant and 50S ribosomal subunit helix 101 mutant ribosomal subunits were expressed and purified as described previously (20–22).
Reagent Labeling with Fluorescent Probes
4.5S RNA was labeled on its 3′-end with Cy5-hydrazide (GE Healthcare) using a previously published protocol (23). The labeling was more than 95% efficient. Ffh (Q72C) was labeled with Cy5-maleimide (GE Healthcare) by buffer exchanging into Ffh labeling buffer (50 mm Tris-HCl, pH 7, at 25 °C, 250 mm NaCl, 2 mm magnesium acetate, 10% glycerol, and 0.5 mm tris(2-carboxyethyl)phosphine) (Soltec Ventures) and treating with 2 mm tris(2-carboxyethyl)phosphine) to reduce the disulfide bonds. The proteins were then incubated with 5-fold excess dye at 25 °C for 30 min. The reaction was stopped by adding 5 mm DTT. Excess dye was removed by gel filtration using three consecutive Illustra NAP-10 desalting columns (GE Healthcare). The efficiency of labeling reaction was ≥ 95%. Labeled Ffh and labeled or unlabeled 4.5S RNA were reconstituted into SRP by mixing Ffh with 1.5–2 fold excess 4.5S RNA in SRP reconstitution buffer (50 mm K-HEPES, pH 7.5, at 25 °C, 150 mm potassium acetate, 5 mm magnesium diacetate, 10% glycerol, and 2 mm DTT) incubating for 10 min at 4 °C and then purifying using an S200 size-exclusion column (GE Healthcare) on an ÄKTA chromatography system (GE Healthcare). In all cases, there were two clear and distinct peaks, with one corresponding to reconstituted SRP and the other corresponding to excess 4.5S RNA. The reconstituted SRP was aliquoted for single use, flash frozen in SRP reconstitution buffer, and stored at −80 °C. L29 (Q38C) was labeled with Cy3 or Cy3B-maleimide using the same method described above for the Ffh proteins, except that the labeling buffer contained no magnesium diacetate or tris(2-carboxyethyl)phosphine). The labeling efficiency was ≥90%. 70S ribosomes derived from the ΔL29 strain were reconstituted with Cy3-labeled L29 by mixing the 70S ribosomes with 1.5–2 fold excess of labeled L29 in L29 reconstitution buffer (10 mm Tris-HCl, pH 7.5, at 4 °C, 300 mm NH4Cl, 10 mm magnesium acetate, and 2 mm DTT) and incubating at 25 °C for 10 min. The 70S-L29 reconstitution mix was then layered on top of a 40% sucrose cushion in L29 wash buffer (10 mm Tris-HCl, pH 7.5, at 4 °C, 400 mm NH4Cl, 10 mm magnesium diacetate and 2 mm DTT) and centrifuged at 100,000 rpm in a 100.4 TLA rotor for 4 h at 4 °C. The ribosome pellets were resuspended in ribosome storage buffer (10 mm Tris-HCl, pH 7.5, at 4 °C, 60 mm NH4Cl, 10 mm magnesium diacetate 2 mm DTT), flash frozen, and stored at −80 °C. 30S helix 44 mutant ribosomal subunits were labeled by hybridization to Cy3B oligonucleotides and 50S helix 101 mutant ribosomal subunits by hybridization to Black Hole Quencher (BHQ)-oligonucleotides according to previously described protocols (20–22). The GTPase activity of all CyDye-labeled SRP mutants in the presence of SR was tested as described previously (15, 16).
Ensemble Ribosome Translation and SRP-ribosome Interaction Measurements
Reconstituted ΔL29 ribosomes were incubated with lepB mRNA in the in vitro translation PURE Δribosome system (New England Biolabs) according to the manufacturer's instructions. In reactions where translation efficiency was measured, [35S]methionine was included (4.34 fmol/reaction with a specific activity of 1175 Ci/mmol) (PerkinElmer Life Science). After 30 min to 1 h of translation, the RNCs were layered onto a 40% sucrose cushion in L29 wash buffer and centrifuged at 100,000 rpm in a 100.4 TLA rotor (Beckman Coulter) for 4 h at 4 °C. The pellets were then resuspended in ribosome storage buffer and either aliquoted and flash frozen for single use in future experiments, loaded onto SDS-polyacrylamide gels for visualization of translation products, or assayed with an LS6500 scintillation counter (Beckman Coulter). To measure the extent of nascent chains associated with RNCs, translation was performed in the presence of [35S]methionine, and the RNCs were purified as described above. The fraction of RNC complexes bound to nascent chains was then established by comparing the concentration of nascent chains as determined by [35S]methionine scintillation counts with the ribosome concentration as determined by absorbance at 260 nm. For the range of nascent chain lengths used in this study >95% of ribosomes had an associated nascent chain. For RNC-SRP interaction measurements, 200 pm Cy3B-labeled reconstituted ΔL29 RNCs stalled after translation were mixed with increasing amounts of Cy5-labeled SRP. All reactions were incubated at 25 °C for at least 20 min, at which point all FRET values measured were stable over time, indicating equilibrium had been reached. All reactions were carried out in reaction buffer (50 mm K-HEPES, pH 7.5, 150 mm potassium acetate, 5 mm magnesium acetate, 2 mm DTT, 10% glycerol, and 100 μm 5′-guanylyl imidodiphosphate. To measure FRET, the sample was excited with 525 nm light, and the donor fluorescence was measured at 573 nm, and the acceptor fluorescence was measured at 670 nm using a Fluorolog-3 spectrofluorometer (HoribaJobin Yvon). FRET efficiency was calculated using Equation 1,
 |
where FD is the donor fluorescence, and FA is the acceptor fluorescence observed at each concentration of SRP. For each concentration of SRP, FA was calculated by subtracting the acceptor fluorescence of labeled SRP alone from the acceptor fluorescence of labeled SRP in the presence of RNCs. The obtained data were then fit to quadratic Equation 2 to find SFRET and KD as described previously (24),
 |
Once an SFRET value of 0.017 was found for 75-aa and 95-aa SRP-RNC binding, the same SFRET was used for all the other fittings.
Single-molecule SRP Delivery Experiments
For each experiment, labeled RNCs were incubated with truncated 5′-biotinylated lepB mRNA using the PURE ΔRibosome system. After translating for 30 min at 37 °C, the stalled RNCs were diluted in a polymix buffer with no reducing agents (50 mm Tris acetate, pH 7.5 at 25 °C, 100 mm KCl, 5 mm ammonium acetate, 5 mm magnesium acetate, 0.5 mm Ca(acetate)2, 0.1 mm EDTA, 5 mm putrescine HCl, and 1 mm spermidine) (21) and immobilized on a neutravidin-derivatized quartz slide, or on a zero mode waveguide (ZMW, Biopac) chip (Pacific Biosciences). The immobilized RNCs were washed with polymix wash buffer containing 1 mm Trolox, an oxygen-scavenging system (2.5 mm 3,4-dihydroxybenzoic acid, 250 nm protocatechuate dioxygenase (25)), and 4 mm GTP. 15 nm Cy5-labeled SRP was then delivered to slides with the immobilized RNCs in polymix wash buffer using a controlled syringe pump. In the ZMW experiments, 100 nm Cy5-labeled SRP was delivered instead of 15 nm. All experiments were collected at 100-ms time resolution using previously described total internal reflection fluorescence microscopy (TIRFM) and ZMW setups (22, 26, 27). All experiments using the nucleotide-labeled assay, (except the one shown in Fig. 4B, in which absolute EFRET values were determined) included a BHQ2-label on helix 101 of the 50S ribosomal subunit. The BHQ quencher was used in previous work (22) and was included in this study because it qualitatively improved the assay signal without interfering with the ability to detect SRP-RNC single-molecule FRET (smFRET) events. Because absolute EFRET values were not determined in these experiments, we used dual illumination to excite both dyes and measured Cy3B and Cy5 fluorescence over time. In this way, we scored all SRP-binding events, even if FRET did not occur. Most Cy5 arrival events displayed distinct anti-correlation with the Cy3B signal, indicative of FRET (“FRET events”), whereas a smaller population of Cy5 arrival events did not (“non-FRET events”). We never observed interconversion between FRET and non-FRET events, and both were significantly above background binding to slides with no RNCs (data not shown). We excluded non-FRET events from further analysis for the following reasons: 1) under our experimental conditions, we could not distinguish individual non-FRET events from background binding; 2) their lack of FRET indicated that, if they reflected RNC binding, it must have occurred at a position on the RNC that we could not validate with our assay; and 3) we also observed non-FRET events using Cy5-labeled 4.5S RNA lacking Ffh, raising additional doubts about whether these events reflect meaningful on-pathway interactions. For the experiment shown in Fig. 4B, the 50S subunit did not include the BHQ2 label so that absolute EFRET values could be accurately determined. All experiments were analyzed with custom MATLAB (Mathworks) scripts described previously (22, 25, 26). Transition state data were analyzed using the vbFRET software described previously (28). Cumulative distributions derived from the data analysis were fit to either a single or double exponential equation using maximum likelihood parameter estimations. The single exponential equation (Equation 3) used was as follows,
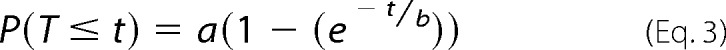 |
where P(T ≤ t) is the probability of event T being less than or equal to time t and a is the proportion of events with an average time b. The double exponential equation (Equation 4) was as follows,
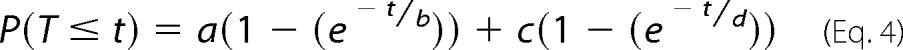 |
where c is the proportion of events with an average time d.
FIGURE 4.

Nucleotide-labeled smFRET assay to detect SRP-RNC interactions. All results shown use the nucleotide-labeled assay. A, single-molecule trace of Cy5-labeled SRP delivered at time = 0 to slides with immobilized Cy3B-labeled 75-aa RNC visualized using TIRFM. The fluorescence intensity of the Cy3B (green) and Cy5 (red) signals is shown. FRET events, arrival time, and residence times are labeled. AU, arbitrary units. B, histogram plot with normal fit of observed EFRET values when SRP was delivered to 75-aa RNC. For the experiment shown in this panel, the 50S subunits were unlabeled (see “Experimental Procedures”). C, bar plot of observed SRP-RNC FRET events and their dependence on aa length of nascent chains on stalled RNC, nature of the signal sequence (WT or mutant (MUT) (see “Experimental Procedures” for sequence)), and composition of SRP (WT or 4.5S RNA alone (RNA). Ø indicates that no events were measured. D, cumulative distribution of lifetimes of the dye on SRP when SRP was directly immobilized on the slide surface. E and F, cumulative distributions of arrival times (E) and residence times (F) of SRP binding to 75-aa RNC. Gray and red lines indicate fits to single and double exponentials, respectively. The percentage and average (AVG.; ± 95% confidence interval of fit) arrival and residence times to the double exponential for each type of event are indicated (n ≥ 419 binding events).
RESULTS
Protein-labeled FRET Assay to Detect SRP-RNC Interactions
To probe SRP-RNC interactions, we developed two sensitive FRET binding assays. In the first assay, both SRP and ribosomes were labeled on their protein components, and we will refer to it as the “protein-labeled” assay. Ribosomes were labeled on a single site in L29, a 63-aa 50S ribosomal subunit protein, in close proximity to the SRP binding site on the ribosome (5, 6). Because L29 is non-essential, we could purify ribosomes lacking L29 (“ΔL29 ribosomes”) from an E. coli strain in which its corresponding gene was deleted. We next reconstituted the ΔL29 ribosomes with a separately expressed and purified L29 variant fluorescently labeled in position 38, which was changed from a glutamine to a cysteine (Fig. 1A).
FIGURE 1.

Protein-labeled FRET assay to detect SRP-RNC interactions. A, L29 ribosome reconstitution scheme. B, L29 ribosomes were separated into 30S and 50S subunits in a 1 mm magnesium acetate 10–40% sucrose gradient. Top panel shows A260 nm of collected gradient fractions. Bottom panel shows fluorescence visualization of Cy3-labeled L29 in fractions analyzed with denaturing PAGE. C, incorporation of Cy3-labeled L29 into ΔL29 but not WT ribosomes as determined after ribosomes were incubated with L29 and pelleted through a 40% sucrose cushion of L29 wash buffer. The pelleted ribosomes were assayed for absorbance at 260 and 550 nm (average of four separate L29 ribosome preps, error bars indicate 1 S.D.). D, translation products of WT or L29 ribosomes using truncated mRNA for 35-aa and 75-aa lepB mRNA after a 2-h translation in PURE system. Stalled RNCs were isolated after spinning over a 40% sucrose 400 mm NH4Cl cushion. Half of the stalled RNCs were visualized via autoradiography of [35S]Met incorporation after denaturing PAGE. The other half of the stalled RNCs were quantified using a scintillation counter to compare translation efficiency. E, SR-dependent GTPase activity of different Cy-labeled SRP constructs as compared with unlabeled WT SRP. SRP GTPase activities derived from fits are as follows: 0.99 ± 0.07 s−1 (± S.E.) for WT, 0.51 ± 0.06 s−1 for protein-labeled SRP, 1.05 ± 0.03 s−1 for nucleotide-labeled SRP.
Labeled L29 was selectively incorporated into the 50S subunit of ΔL29 ribosomes, approaching 1:1 stoichiometry (Fig. 1, B and C). Wild-type (WT) ribosomes, which already contained a copy of L29, did not incorporate the labeled L29 (Fig. 1C). Reconstituted ribosomes functioned as well as WT ribosomes in in vitro translation assays (Fig. 1D). We used these reconstituted ribosomes, labeled with the FRET donor dye Cy3 or Cy3B, to prepare stalled RNCs by translating different length N-terminal portions of leader peptidase (lepB), an in vivo-validated SRP substrate with an N-terminal signal sequence (29). To this end, we used 3′-truncated mRNAs lacking a stop codon and, in agreement with previous work (11, 14, 30), found that ribosomes reaching the truncated end remained stably associated with the mRNA and peptidyl-tRNA, producing stalled RNCs (Fig. 1D). Separately, we labeled the SRP N-terminal GTPase domain with FRET acceptor dye Cy5 on position 72, which was changed from a glutamine to a cysteine. The labeled SRP showed a GTPase activity in the presence of SRP receptor that was within 2-fold of WT (0.51 ± 0.06 s−1 versus 0.99 ± 0.07 s−1, respectively) (Fig. 1E).
Ensemble Equilibrium Measurements Reveal That SRP-RNC Binding Is Sensitive to Nascent Chain Length
The labeled proteins allowed measurement of SRP-ribosome affinity using FRET. A reproducible FRET signal was observed when RNCs stalled with different length nascent chains were equilibrated with increasing concentrations of SRP (Fig. 2A). This allowed us to measure SRP-RNC binding affinities (dissociation constants (Kd)) (Fig. 2, A and B). We found that SRP binding to RNCs with a 35-aa-long nascent chain, in which the signal sequence has not yet emerged from the ribosome (17), displayed a relatively low affinity of Kd = 1.38 ± 0.23 nm. RNCs bearing a 55-aa nascent chain, in which the signal sequence is exposed outside the ribosome, displayed a ∼10-fold higher affinity (Kd = 0.132 ± 0.012 nm). RNCs bearing 75-aa and 95-aa nascent chains displayed affinities that were an additional ∼2-fold higher (Kd = 0.057 ± 0.010, and 0.060 ± 0.013 nm, respectively). By contrast, as the nascent chain continued to grow to 115-aa and 135-aa the affinity decreased by ∼3- and ∼5-fold (Kd = 0.163 ± 0.022 and 0.299 ± 0.057 nm, respectively) (Fig. 2B).
FIGURE 2.

Ensemble equilibrium measurements reveal that SRP-RNC binding is sensitive to nascent chain length. All results shown use the protein-labeled assay. A, Kd measurements of SRP-RNC binding when the RNC was stalled with different aa length lepB nascent chains. Cy5-labeled SRP was incubated with Cy3B-labeled RNCs. Each point is the average of triplicate experiments with error bars indicating S.D. Data were fitted to quadratic binding curves with saturation EFRET set at 0.17, yielding Kd values of 1.38 ± 0.23 nm (± S.E.) for 35-aa RNC, 0.132 ± 0.012 nm for 55 aa, 0.057 ± 0.010 nm for 75 aa, 0.060 ± 0.013 nm for 95 aa, 0.163 ± 0.022 nm for 105-aa RNC, and 0.299 ± 0.057 nm for 135 aa. B, summary of Kd values determined in A. Note that the y axis is split to accommodate values >0.35 nm.
SRP Binds RNCs in Distinct and Interconverting Conformations
In the binding assays presented above, at SRP concentrations higher than 750 nm, the background fluorescence associated with the label on SRP made FRET measurements highly variable. This results in uncertainty of the saturation SRP binding concentrations and saturation FRET values, especially for RNCs associated with nascent chains shorter than 55 aa and longer than 115 aa. However, for SRP binding to 75- and 95-aa RNCs, the equilibrium-binding assays yielded curves in which the saturating FRET efficiency (EFRET) could be reliably determined to be ∼0.017. Because two previous cryo-electron microscopy (cryo-EM) structures of RNC-bound E. coli SRP (5, 6) predict that the labeled residues on the RNC and SRP should be in close proximity, we expected much higher saturating EFRET values (in the range of 0.7–0.9).
To resolve this incongruity, we tested whether the SRP conformation captured in these cryo-EM structures, and used to predict high EFRET values, was just one of many that SRP might assume. This speculation was motivated by kinetic work suggesting multiple RNC-bound SRP conformations (11), and additional cryo-EM and x-ray crystallography structural studies showing alternate SRP conformations that would be more consistent with our observed EFRET values (31–33).
To detect RNC-bound SRP conformations, we analyzed SRP-RNC interactions using the protein-label assay in smFRET experiments. We immobilized 75-aa RNCs with a 3′-biotin-linked oligonucleotide complementary to the 5′-end of the mRNA on a neutravidin-coated and polyethylene glycol-passivated slide surface. We then detected fluorescence from individually labeled RNCs using TIRFM, as described previously (21, 26). This immobilization scheme selects for biosynthetically active RNCs, thereby increasing the likelihood that the immobilized RNCs contain a nascent chain. Upon delivery of labeled SRP to the slides, we detected individual SRP-RNC binding events by FRET (Fig. 3A). Indeed, we observed three major EFRET states (Fig. 3B). One state had a low EFRET of 0.15. A second state had a high EFRET of 0.99. A third state (or perhaps an ensemble of additional states) was broadly distributed between the two peaks. Because of rapid photobleaching of the Cy5 dye on SRP (lifetime ∼ 0.3 s), these data are likely to be biased toward the initial conformation of SRP, when it first binds to the RNCs.
FIGURE 3.

SRP arrives to its RNC in distinct and interconverting conformations. All results shown use the protein-labeled assay, panels A and B used TIRFM, and panels (C–F) used ZMWs. A, example single-molecule trace of Cy5-labeled SRP delivered at time = 0 to slides with immobilized Cy3-labeled 75-aa RNC. The top panel shows the fluorescence intensity of the Cy3 (green) and Cy5 (red) signal. The bottom panel shows the FRET efficiency of the two signals. Dots indicate FRET-binding events, and the line indicates a Cy3-blinking event. The trace shown presented more than the ∼2 event/trace average. AU indicates arbitrary units. B, histogram of the average EFRET values of binding events observed in traces similar to the one shown in A. Lines indicate the normal fits of low EFRET events (red), intermediate EFRET events (green), and high EFRET events (blue). The percentage of total events and mean (± one S.D.) EFRET value for each are indicated (n = 465). C and D, single-molecule traces of Cy5-labeled SRP delivered at time = 0 to slides with immobilized Cy3-labeled 75-aa RNC. Triangles show transitions between different FRET states. E, histogram of the average EFRET values of the distinct FRET states assigned in SRP-RNC binding events with FRET transitions. Lines indicate the normal fits of the low EFRET events (red) intermediate EFRET events (green) and high EFRET events (blue). The percentage of total events and mean EFRET value (± one S.D.) for each type of binding are indicated (n = 218. 42 binding events showed transitions, each of those events had ∼3 transitions yielding ∼5 distinct states per binding event). F, EFRET transition density plot showing the average EFRET of the distinct FRET states from the experiment described in C–E. INT., intermediate EFRET events.
To determine whether SRP can undergo conformational changes once it is bound to RNCs, we repeated the smFRET experiments described above using ZMWs instead of TIRFM slides. ZMWs provide optical confinement in nanoscale wells, yielding an environment for improved dye lifetimes, which affords prolonged observation times (27, 34). This approach extended the Cy5 dye lifetime of RNC-bound SRP by ∼2-fold (from ∼0.3 s to 0.60 ± 0.04 s), allowing us to detect transitions between different EFRET states. We observed FRET signal transitions in ∼6% of binding events (42 events with transitions of 700 total observed events). This subpopulation of FRET binding events had signal lifetimes 10-fold longer than the average (6.3 ± 0.5 s versus ∼0.6 s), suggesting that, even under the improved ZMW conditions, the short lifetimes of most FRET events limited the number of transitions observed.
Using previously described variational Bayesian algorithms (28) alongside visual inspection, we assigned distinct FRET states within single SRP-RNC-binding events (Fig. 3, C and D). A histogram of the average EFRET values of the assigned FRET states within single binding events revealed three distinct EFRET populations (Fig. 3E, which, in contrast to Fig. 3B, only includes data from binding events with transitions). The states are consistent with those observed using TIRFM described above: a low 0.18 EFRET state, a high 0.79 EFRET, and a broad intermediate 0.42 EFRET state. A transition density plot of the average EFRET values of the FRET states before and after each transition revealed that the most prevalent transitions occurred between the low and intermediate EFRET states, followed by transitions between the high and intermediate EFRET states (Fig. 3F). Transitions between the low and high EFRET states were the least common.
These results suggest that SRP arrives at its RNC in distinct but interconverting conformations that include a novel low EFRET conformation in addition to the expected high EFRET conformation. It is therefore likely that the low saturation EFRET observed in solution measurements, which is the average of all states, convoluted with dye photophysical behavior, resulted from the low EFRET SRP conformation predominating under equilibrium conditions.
Nucleotide-labeled smFRET Assay to Detect SRP-RNC Interactions
The protein-labeled FRET affinity measurements discussed so far showed that SRP-RNC binding varied 5-fold as the length of nascent chains displaying a signal sequence changed. To confirm these results, we developed a second independent FRET assay that would provide a unique window of sensitivity to Kd values in the subnanomolar range and be amenable to single particle measurements. In this assay, we labeled both SRP and ribosomes on their RNA components. We will refer to it as the “nucleotide-labeled” assay. Ribosomes were labeled by annealing a Cy3B-labeled oligonucleotide to a loop engineered into the 16S ribosomal RNA. Similarly, there was a Black Hole quencher label on the 23S ribosomal RNA (22). The Black Hole quencher label was included because we found that it qualitatively improved the Cy3B signal. SRP was labeled by chemical attachment of Cy5 to the 3′-end of 4.5S RNA (35), which did not affect its GTPase activity relative to WT (Fig. 1E).
When we delivered labeled SRP to TIRFM slides with immobilized 75-aa RNCs, we observed SRP-RNC binding as the onset of a FRET signal (Fig. 4A). These FRET events displayed all of the expected hallmarks of a bona fide SRP-RNC interaction: (i) the measured EFRET value was consistent with the dye distance predicted by cryo-EM SRP-RNC structures (Fig. 4B) (5, 6). (ii) They required the presence of Ffh on Cy5-labeled 4.5S RNA (Fig. 4C). (iii) The FRET events required the presence of a signal sequence on RNCs (no events were observed with 35-aa RNC in which the signal sequence was concealed, 51 events were observed with 45-aa RNC in which the signal sequence was partially exposed, and 383 events were observed with 55-aa RNC in which the signal sequence was completely exposed) (Fig. 4C). Finally, (iv) the FRET events required that the signal sequence displayed on RNCs be functional (no events were observed with 75-aa RNC displaying a mutant signal sequence shown previously not to interact with SRP (17), whereas 509 events were observed with the WT signal sequence) (Fig. 4C).
In the nucleotide-labeled assay, the FRET donor dye is in a fixed position on the ribosome and the position of the FRET acceptor dye on the 3′-end of 4.5S RNA is not expected to change between different SRP conformations on the ribosome. Therefore, we expected that the FRET signal should be identical for all of the SRP conformations we observed earlier in the protein-labeled assay, as they only differ in the position of the Ffh N-terminal GTPase domain. Consistent with these expectations, smFRET measurements of the SRP-RNC binding EFRET values yielded a single-peak normal distribution (EFRET = 0.16) (Fig. 4B). Additionally, the Cy3B dye allowed for measurements over 4-min time courses, and the Cy5 dye on SRP lasted an average of 83 s when immobilized on the slide surface (Fig. 4D). A direct comparison of the Cy5 dyes on SRP in the protein- and nucleotide-labeling assays using constant excitation light intensities in the TIRFM system indicates that the large differences in average dye lifetimes (∼0.3 and 83 s, respectively) are most likely due to differences in the dye environment and not differences between TIRFM and ZMW excitation light intensities. Ultimately, the long lived signals in the nucleotide-labeled assay allowed for sensitive analyses of a wide time scale of SRP-RNC binding events.
We analyzed the kinetics of SRP-RNC binding events by determining two parameters: arrival times and residence times (Fig. 4, A, E, and F). We defined arrival times as the lag time between SRP delivery to the slide and the onset of the first FRET event. The arrival times between FRET events following the first one were not included in the analysis. This reduced distortion from cases in which the dye on an RNC-bound SRP molecule photobleached before that SRP molecule left and a second labeled SRP molecule bound the same RNC. We defined residence times as the duration of the FRET signal. For all nascent chain lengths that gave rise to FRET signals, we observed that the arrival times fit a double, but not a single, exponential distribution (Fig. 4E), demonstrating that differences in the immobilized RNCs led to distinct SRP average arrival times. The fast arrival times, which were ∼60% of those observed, tended to be ∼10-fold faster than the slow arrival times (3.2 ± 0.1 s and 37.2 ± 2.6 s, respectively, when bound to a 75-aa RNC). Similarly, the residence time measurements for all nascent chain lengths that gave rise to FRET signals also fit a double, but not a single, exponential distribution (Fig. 4F). These observations suggested that the different RNC-bound SRP conformations we observed using the protein-labeled assay have different off-rates. The long residing events, which were ∼80% of those we observed, tended to be ∼10-fold longer than the short-residing events (11.3 ± 0.4 s and >83 s, respectively, when bound to 75-aa RNC).
smFRET Kinetic Measurements Confirm That SRP-RNC Binding Is Sensitive to Nascent Chain Length
A crude analysis of SRP binding kinetics to RNCs with different peptide lengths using the nucleotide-labeled smFRET assay showed a trend similar to the one observed using the protein-labeled ensemble equilibrium FRET assay. Qualitatively, SRP arrival times to RNCs became faster as the nascent chain neared 75 aa and became slower as the chain increased or decreased in length (Fig. 5A). Similarly, SRP residence times on RNC became longer as the nascent chain neared 75 aa and became shorter decreased in length (Fig. 5B). Together, these two trends indicate that SRP-RNC binding affinity (assuming a simple on-off mechanism) is greatest when the RNC has a nascent chain between 65 and 85 aa in length and becomes weaker when the chain deviates from this range, with the main contributor to the variations in affinities resulting from differences in arrival times.
FIGURE 5.

smFRET kinetic measurements confirm that SRP-RNC binding is sensitive to nascent chain length. All results shown use the nucleotide-labeled assay. A and B, cumulative distributions of arrival times (A) and residence times (B) of SRP binding events to RNCs with different length nascent chains. Insets show a magnification of the curves within the dashed lines. Experiments were performed as described (for Fig. 4A). Dashed lines indicate fits to double exponential distributions (n ≥ 192). C, Kd(app) values for SRP-RNC binding to RNC stalled with different length nascent chains derived from the data in (A and B). Each colored line connects affinities determined by the same pairing of binding rates: fast arrivals with either long (blue), or short (red) residence times and slow arrivals with either long (green), or short (purple) residence times. Error bars indicate error propagated from the ± 95% confidence interval of fits for the data shown in A and B from which the affinities were estimated. D, fold difference of affinities shown in C. Colors are the same as in C. Note that the y axis is split to accommodate fold differences larger than 9.
To describe this trend quantitatively, we fit the SRP-RNC binding events to double exponential distributions. For each chain length studied, we obtained a slow and a fast average arrival time as well as a short and a long average residence time. From each of the average arrival and residence times, we derived binding on- and off-rates, respectively. We then determined apparent Kd (Kd(app)) values from these on- and off-rates. For every nascent chain length, we obtained four Kd(app) values because there are four potential ways to pair the arrival and residence times: fast arrivals paired with either (i) short or (ii) long residence times, and slow arrivals with (iii) short or (iv) long residence times (Fig. 5C). The Kd(app) values determined by pairing slow arrival with short residence times are 1000-fold higher than the Kd measurements obtained in the ensemble measurements using the protein-labeled assay. This observation suggests that this pairing is either non-physiological or exceedingly rare in equilibrium conditions. All other Kd(app) values were 10 to 100-fold higher than those determined using the ensemble measurements, a range that can be explained by: (i) surface immobilization of RNC slowing the on-rate of SRP binding by limiting possible SRP approach vectors and (ii) Cy5 dye photobleaching on SRP producing artificially shorter residence times because the average dye lifetime (∼83 s) is similar to the long residence times (∼47–96 s). Regardless of these caveats, we could still compare how nascent chain length changes the SRP-RNC affinities and found that, independent of what pairing gave rise to the Kd(app) values, SRP has the highest binding affinity for 75-aa RNCs. Chains shorter or longer impair binding by as much as 12- to 43-fold (Fig. 5D). The inability to detect SRP-binding to RNCs with chains shorter than 55 aa argues that the absence of a fully exposed signal sequence results in affinities so low that they are undetectable with the nucleotide-labeled smFRET assay. Similarly, SRP binding to 105-aa RNCs pushed the detection limits of the assay, as indicated by arrival times ∼2.5 longer than the 240 s measurement window imposed on the system by dye photobleaching, indicating that that only a relatively fast-arriving subset of a slow-arriving set of SRP-105-aa RNC binding events could be observed. Thus, SRP binding to RNCs with nascent chains shorter than 55 aa or longer than 105 aa has at least a 12-fold lower affinity than SRP binding to 75-aa RNCs.
DISCUSSION
These nucleotide-labeled smFRET results, together with the protein-labeled assay affinity measurements presented earlier, show conclusively that SRP binding to RNCs displaying an N-terminal signal sequence is sensitive to nascent chain length. This notion disagrees with three previous studies (10, 11, 14). Each of these studies suffered from limitations that could explain why the respective author teams did not detect nascent chain-length sensitivities. One study used multiple, heterogeneous attachment dye attachment sites on the ribosome (11), which makes definitive analyses of fluorescent signals difficult. Another study incorporated unnatural fluorescent amino acids into the signal sequence (10), which is likely to disrupt SRP-RNC binding. The last study used a truncation of SRP RNA (14), which is likely to disrupt SRP conformational dynamics (36, 37). By contrast, our assays used uniform and specific labeling positions that did not alter the signal sequence or SRP RNA. These advantages combined with a sensitive subnanomolar detection range allowed us to detect previously unobserved nascent chain length sensitivity in SRP-RNC affinities.
Can the sensitivity of SRP-RNC binding kinetics and affinities to nascent chain length explain why SRP cannot target RNCs whose chains have grown too long? It can be argued that the SRP concentration in E. coli (estimated to be ∼400 nm (38)) saturates binding at the affinities measured here. Therefore, the >12-fold affinity differences from high pm to low nm Kd values measured would not impact targeting. This argument, however, relies on two crucial assumptions: (i) SRP-RNC binding in vivo reaches equilibrium, and (ii) the estimated 400 nm SRP concentration in the cell corresponds to free SRP. However, both of these assumptions can be questioned, especially in light of a vast body of previous work that showed that most regulation of the SRP targeting reaction is kinetically controlled (12, 39–41). Translation proceeds at the rate of ∼30 amino acids per second. Given the off-rates here and elsewhere (42), any particular nascent chain length does not exist long enough for equilibrium to be reached. Furthermore, we have shown that SRP binds tightly (with less than or equal to nanomolar Kd values) to RNCs with short nascent chains (35–55 aa) and exposed signal sequences. The concentration of ribosomes in E. coli is ∼40 μm, <5% of which are probably translating a short nascent chain or exposing a signal sequence (14). These RNCs, therefore, are essentially equimolar with SRP (at ∼400 nm each) providing a sink for SRP, likely lowering the free SRP concentration significantly. Therefore, because SRP-RNC binding is likely kinetically regulated, the nascent chain length-dependent changes in arrival and residence times may contribute specificity in the targeting reaction.
The basic premise that SRP affinities for RNCs with longer nascent chains are reduced was tested previously. These studies showed that targeting of RNCs bearing chains of >140 aa was significantly increased at elevated SRP concentrations (9). Thus, it is likely that early recognition of short chains is important. Once short-chain RNCs are captured by SRP, their nascent chains may elongate and reach a different optimal length for translocon engagement, consistent with the recent finding that a 135-aa-long nascent chain is more effectively transferred to the translocon than an 85-aa-long chain (42). These arguments underscore the importance to extend in future work the scope of protein targeting assays to include actively translating RNCs.
We also show that SRP binds at least 10-fold more tightly to RNCs when they expose a signal sequence. This is likely due to multiple hydrophobic interactions between the SRP M domain and the signal sequence (3). When the SRP-ribosome interactions are added to these SRP-signal sequence interactions, a higher SRP-RNC affinity results. Surprisingly, previous studies had not seen this affinity increase (11, 14), likely due to the same assay limitations discussed above.
Additionally, we show that SRP binds RNCs exposing a signal sequence in multiple interconverting conformations and that these conformations have distinct kinetic properties. This is in agreement with ensemble kinetic observations showing multiple SRP-RNC binding rates (11). We also show that in solution one of the conformations predominates over the others. This predominant conformation is characterized by a low EFRET value between L29 on the ribosome and the SRP N-terminal GTPase domain, indicating a large (∼100 Å) distance between them. This conformation would most resemble cryo-EM structures of SRP-SR complexes on RNCs (31) rather than cryo-EM structures of SRP alone on RNCs (5, 6). This observation indicates that under our experimental conditions, which allow for conformational dynamics on the ribosome, RNC-bound SRP could be in a conformation that predisposes it for SR binding because it does not require the significant rearrangements necessary for transition from the SRP-alone to SRP-SR conformation. This is consistent with observations that binding RNC accelerates SRP-SR binding (41). We observed a high EFRET conformation consistent with the SRP-alone structures when SRP first arrived to its RNC, indicating that this might be a conformation that then transitions into the low EFRET conformation that predisposed SRP for SR binding.
Finally, our results show SRP arriving to immobilized RNCs with kinetics that fit a double exponential distribution. Because in our experimental conditions RNCs are limiting, these results indicate that RNC heterogeneity is responsible for the different binding kinetics. The source for this heterogeneity is unclear, but previous studies indicate some intriguing possibilities: RNC heterogeneity could arise from different elongation states of the ribosomes, which could not be controlled in our experimental system (43); alternatively, RNC heterogeneity could arise from stalled RNCs that display the signal sequence in conformationally distinct ways, as suggested by recent work showing that the ribosomal surface can bias the conformation and folding of nascent chains (44). We do not favor this latter possibility because it assumes that exchange between these conformations is slow on our experimental time scale, which would require stable, long time scale (30 milliseconds or longer) nascent chain interactions, which would be possible but seems unlikely.
The results presented here show that multiple moving parts have an effect on SRP binding to translating RNCs. Both SRP and RNCs are conformationally diverse, which results in distinct binding kinetics whose role remains to be determined. Additionally, translating the nascent chain continually changes the position of the signal sequence, first allowing it to emerge gradually from the ribosome and then gradually distancing it. This dynamic behavior profoundly affects SRP-RNC binding and is likely to impose limits on SRP action.
Acknowledgments
We thank David Morgan, Geeta Narlikar, Hana El-Samad, Joshua Dunn, and the Walter and Puglisi laboratories for helpful discussion and comments.
This work was supported by the Howard Hughes Medical Institute and National Institutes of Health Grants GM032384 (to T. R. N., M. M. E., S. B. N., N. B., and P. W.), GM51266 (to A. T., J. C., and J. D. P.), and GM099687 (to A. P. and J. D. P.).
- SRP
- signal recognition particle
- SR
- signal recognition particle receptor
- FRET
- fluorescent resonance energy transfer
- RNC
- ribosome-nascent chain complex
- aa
- amino acid
- EFRET
- FRET efficiency
- cryo-EM
- cryo-electron micrography
- smFRET
- single-molecule FRET
- TIRFM
- total internal reflection fluorescent microscopy
- ZMW
- zero mode waveguides.
REFERENCES
- 1. Egea P. F., Stroud R. M., Walter P. (2005) Targeting proteins to membranes: structure of the signal recognition particle. Curr. Opin. Struct. Biol. 15, 213–220 [DOI] [PubMed] [Google Scholar]
- 2. Zopf D., Bernstein H. D., Johnson A. E., Walter P. (1990) The methionine-rich domain of the 54 kd protein subunit of the signal recognition particle contains an RNA binding site and can be crosslinked to a signal sequence. EMBO J. 9, 4511–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janda C. Y., Li J., Oubridge C., Hernández H., Robinson C. V., Nagai K. (2010) Recognition of a signal peptide by the signal recognition particle. Nature 465, 507–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Egea P. F., Shan S. O., Napetschnig J., Savage D. F., Walter P., Stroud R. M. (2004) Substrate twinning activates the signal recognition particle and its receptor. Nature 427, 215–221 [DOI] [PubMed] [Google Scholar]
- 5. Schaffitzel C., Oswald M., Berger I., Ishikawa T., Abrahams J. P., Koerten H. K., Koning R. I., Ban N. (2006) Structure of the E. coli signal recognition particle bound to a translating ribosome. Nature 444, 503–506 [DOI] [PubMed] [Google Scholar]
- 6. Halic M., Blau M., Becker T., Mielke T., Pool M. R., Wild K., Sinning I., Beckmann R. (2006) Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature 444, 507–511 [DOI] [PubMed] [Google Scholar]
- 7. Bernstein H. D., Zopf D., Freymann D. M., Walter P. (1993) Functional substitution of the signal recognition particle 54-kDa subunit by its Escherichia coli homolog. Proc. Natl. Acad. Sci. U.S.A. 90, 5229–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Powers T., Walter P. (1997) Co-translational protein targeting catalyzed by the Escherichia coli signal recognition particle and its receptor. EMBO J. 16, 4880–4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siegel V., Walter P. (1988) The affinity of signal recognition particle for presecretory proteins is dependent on nascent chain length. EMBO J. 7, 1769–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flanagan J. J., Chen J. C., Miao Y., Shao Y., Lin J., Bock P. E., Johnson A. E. (2003) Signal recognition particle binds to ribosome-bound signal sequences with fluorescence-detected subnanomolar affinity that does not diminish as the nascent chain lengthens. J. Biol. Chem. 278, 18628–18637 [DOI] [PubMed] [Google Scholar]
- 11. Holtkamp W., Lee S., Bornemann T., Senyushkina T., Rodnina M. V., Wintermeyer W. (2012) Dynamic switch of the signal recognition particle from scanning to targeting. Nat. Struct. Mol. Biol. 19, 1332–1337 [DOI] [PubMed] [Google Scholar]
- 12. Zhang X., Rashid R., Wang K., Shan S. O. (2010) Sequential checkpoints govern substrate selection during cotranslational protein targeting. Science 328, 757–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang D., Shan S. O. (2012) Translation elongation regulates substrate selection by the signal recognition particle. J. Biol. Chem. 287, 7652–7660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bornemann T., Jöckel J., Rodnina M. V., Wintermeyer W. (2008) Signal sequence-independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat. Struct. Mol. Biol. 15, 494–499 [DOI] [PubMed] [Google Scholar]
- 15. Peluso P., Shan S. O., Nock S., Herschlag D., Walter P. (2001) Role of SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry 40, 15224–15233 [DOI] [PubMed] [Google Scholar]
- 16. Bradshaw N., Walter P. (2007) The signal recognition particle (SRP) RNA links conformational changes in the SRP to protein targeting. Mol. Biol. Cell 18, 2728–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Houben E. N., Zarivach R., Oudega B., Luirink J. (2005) Early encounters of a nascent membrane protein: specificity and timing of contacts inside and outside the ribosome. J. Cell Biol. 170, 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spedding G. (1990) Ribosomes and Protein Synthesis: a Practical Approach (Spedding G., ed.) IRL Press at Oxford University Press, Oxford, England [Google Scholar]
- 20. Dorywalska M., Blanchard S. C., Gonzalez R. L., Kim H. D., Chu S., Puglisi J. D. (2005) Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res. 33, 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marshall R. A., Dorywalska M., Puglisi J. D. (2008) Irreversible chemical steps control intersubunit dynamics during translation. Proc. Natl. Acad. Sci. U.S.A. 105, 15364–15369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J., Tsai A., Petrov A., Puglisi J. D. (2012) Nonfluorescent quenchers to correlate single-molecule conformational and compositional dynamics. J. Am. Chem. Soc. 134, 5734–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buskiewicz I., Peske F., Wieden H. J., Gryczynski I., Rodnina M. V., Wintermeyer W. (2005) Conformations of the signal recognition particle protein Ffh from Escherichia coli as determined by FRET. J. Mol. Biol. 351, 417–430 [DOI] [PubMed] [Google Scholar]
- 24. Lam V. Q., Akopian D., Rome M., Henningsen D., Shan S. O. (2010) Lipid activation of the signal recognition particle receptor provides spatial coordination of protein targeting. J. Cell Biol. 190, 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aitken C. E., Marshall R. A., Puglisi J. D. (2008) An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 94, 1826–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aitken C. E., Puglisi J. D. (2010) Following the intersubunit conformation of the ribosome during translation in real time. Nat. Struct. Mol. Biol. 17, 793–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsai A., Petrov A., Marshall R. A., Korlach J., Uemura S., Puglisi J. D. (2012) Heterogeneous pathways and timing of factor departure during translation initiation. Nature 487, 390–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bronson J. E., Fei J., Hofman J. M., Gonzalez R. L., Jr., Wiggins C. H. (2009) Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 97, 3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Gier J. W., Mansournia P., Valent Q. A., Phillips G. J., Luirink J., von Heijne G. (1996) Assembly of a cytoplasmic membrane protein in Escherichia coli is dependent on the signal recognition particle. FEBS Lett. 399, 307–309 [DOI] [PubMed] [Google Scholar]
- 30. Matsuura T., Yanagida H., Ushioda J., Urabe I., Yomo T. (2007) Nascent chain, mRNA, and ribosome complexes generated by a pure translation system. Biochem. Biophys. Res. Commun. 352, 372–377 [DOI] [PubMed] [Google Scholar]
- 31. Estrozi L. F., Boehringer D., Shan S. O., Ban N., Schaffitzel C. (2011) Cryo-EM structure of the E. coli translating ribosome in complex with SRP and its receptor. Nat. Struct. Mol. Biol. 18, 88–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hainzl T., Huang S., Meriläinen G., Brännström K., Sauer-Eriksson A. E. (2011) Structural basis of signal-sequence recognition by the signal recognition particle. Nat. Struct. Mol. Biol. 18, 389–391 [DOI] [PubMed] [Google Scholar]
- 33. Hainzl T., Huang S., Sauer-Eriksson A. E. (2007) Interaction of signal-recognition particle 54 GTPase domain and signal-recognition particle RNA in the free signal-recognition particle. Proc. Natl. Acad. Sci. U.S.A. 104, 14911–14916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J., Dalal R. V., Petrov A. N., Tsai A., O'Leary S. E., Chapin K., Cheng J., Ewan M., Hsiung P. L., Lundquist P., Turner S. W., Hsu D. R., Puglisi J. D. (2014) High-throughput platform for real-time monitoring of biological processes by multicolor single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 111, 664–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buskiewicz I., Kubarenko A., Peske F., Rodnina M. V., Wintermeyer W. (2005) Domain rearrangement of SRP protein Ffh upon binding 4.5S RNA and the SRP receptor FtsY. RNA 11, 947–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shen K., Arslan S., Akopian D., Ha T., Shan S. O. (2012) Activated GTPase movement on an RNA scaffold drives co-translational protein targeting. Nature 492, 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen K., Wang Y., Hwang Fu Y. H., Zhang Q., Feigon J., Shan S. O. (2013) Molecular mechanism of GTPase activation at the signal recognition particle (SRP) RNA distal end. J. Biol. Chem. 288, 36385–36397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jensen C. G., Pedersen S. (1994) Concentrations of 4.5S RNA and Ffh protein in Escherichia coli: the stability of Ffh protein is dependent on the concentration of 4.5S RNA. J. Bacteriol. 176, 7148–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peluso P., Herschlag D., Nock S., Freymann D. M., Johnson A. E., Walter P. (2000) Role of 4.5S RNA in assembly of the bacterial signal recognition particle with its receptor. Science 288, 1640–1643 [DOI] [PubMed] [Google Scholar]
- 40. Bradshaw N., Neher S. B., Booth D. S., Walter P. (2009) Signal sequences activate the catalytic switch of SRP RNA. Science 323, 127–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang X., Schaffitzel C., Ban N., Shan S. O. (2009) Multiple conformational switches in a GTPase complex control co-translational protein targeting. Proc. Natl. Acad. Sci. U.S.A. 106, 1754–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saraogi I., Akopian D., Shan S. O. (2014) Regulation of cargo recognition, commitment, and unloading drives cotranslational protein targeting. J. Cell Biol. 205, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ogg S. C., Walter P. (1995) SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell 81, 1075–1084 [DOI] [PubMed] [Google Scholar]
- 44. Kaiser C. M., Goldman D. H., Chodera J. D., Tinoco I., Jr., Bustamante C. (2011) The ribosome modulates nascent protein folding. Science 334, 1723–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]