

Background: Ligand-specific receptor signaling is often referred to as functional selectivity or biased agonism.
Results: Single amino acid substitution on β2-adrenoreceptor (Y308F) converts a ligand-specific signaling from Gs-biased to promiscuous Gs and Gi dual signaling.
Conclusion: Specific ligand-receptor interaction results in receptor conformation(s) sufficient to convey biased signaling.
Significance: Our work reveals a molecular mechanism for biased agonism.
Keywords: Adrenergic Receptor, Cardiovascular, G Protein-coupled Receptor (GPCR), Molecular Docking, Molecular Pharmacology, Signal Transduction, Site-directed Mutagenesis, Cardiomyocyte Contraction, Functional Selectivity
Abstract
Interaction of a given G protein-coupled receptor to multiple different G proteins is a widespread phenomenon. For instance, β2-adrenoceptor (β2-AR) couples dually to Gs and Gi proteins. Previous studies have shown that cAMP-dependent protein kinase (PKA)-mediated phosphorylation of β2-AR causes a switch in receptor coupling from Gs to Gi. More recent studies have demonstrated that phosphorylation of β2-AR by G protein-coupled receptor kinases, particularly GRK2, markedly enhances the Gi coupling. We have previously shown that although most β2-AR agonists cause both Gs and Gi activation, (R,R′)-fenoterol preferentially activates β2-AR-Gs signaling. However, the structural basis for this functional selectivity remains elusive. Here, using docking simulation and site-directed mutagenesis, we defined Tyr-308 as the key amino acid residue on β2-AR essential for Gs-biased signaling. Following stimulation with a β2-AR-Gs-biased agonist (R,R′)-4′-aminofenoterol, the Gi disruptor pertussis toxin produced no effects on the receptor-mediated ERK phosphorylation in HEK293 cells nor on the contractile response in cardiomyocytes expressing the wild-type β2-AR. Interestingly, Y308F substitution on β2-AR enabled (R,R′)-4′-aminofenoterol to activate Gi and to produce these responses in a pertussis toxin-sensitive manner without altering β2-AR phosphorylation by PKA or G protein-coupled receptor kinases. These results indicate that, in addition to the phosphorylation status, the intrinsic structural feature of β2-AR plays a crucial role in the receptor coupling selectivity to G proteins. We conclude that specific interactions between the ligand and the Tyr-308 residue of β2-AR stabilize receptor conformations favoring the receptor-Gs protein coupling and subsequently result in Gs-biased agonism.
Introduction
Increasing evidence has accumulated over the past decade indicating that a G protein-coupled receptor (GPCR)3 does not respond similarly to all agonist ligands. Ligands can initiate multiple cascades of intracellular reactions that can be mediated by G proteins or be G protein-independent. The term biased agonism initially referred to the ability of a ligand to selectively activate either a G protein-mediated event, such as stimulation of adenylyl cyclase, or activation of the G protein-independent noncanonical β-arrestin-dependent signal transduction pathway (1). The emerging paradigm of biased agonism or functional selectivity suggests that binding of one ligand can stabilize receptor conformation(s) preferentially favoring recognition by a given set of signaling proteins or pathways on the intracellular side, although another ligand stabilizes a receptor state that is preferred by a different set of signaling proteins. In this manner, ligands can trigger qualitatively distinct signaling events in the cell (1–8).
β-Adrenergic receptors (β-AR) are Gs-coupled GPCRs and in fact β1-AR couples only to Gs. For β2-AR, the prototypical member of the GPCR family, studies (9, 10) have shown that agonist binding can activate both Gs and Gi. Our previous study has shown that fenoterol (Fen) is unique among the β2-AR agonists in terms of ligand-induced receptor-G protein coupling selectivity (11). Although most β2-AR agonists produce contractile responses in cardiomyocytes that can be sensitized by the Gi disruptor pertussis toxin (PTX), indicating dual Gs and Gi coupling, the inotropic effect of Fen is PTX-insensitive, suggesting that Fen preferentially promotes β2-AR-Gs coupling. Fen contains two chiral centers in its molecule and may exist as four stereoisomers (Fig. 1). The stereoisomers of Fen and a series of Fen analogs have been synthesized (12–14). The role of ligand chirality in G protein-coupling selectivity has recently been demonstrated using these Fen derivatives (15). Specifically, we have shown that (R,R′)-Fen and (R,R′)-4′-methoxyfenoterol ((R,R′)-methoxyFen) preferentially activated Gs signaling, as evidenced by the lack of PTX sensitivity of their contractile responses in cardiomyocytes and their inability to activate Gi-dependent ERK signaling in HEK-293 cells. In contrast, the corresponding (S,R′)-isomers exhibited robust PTX sensitivity in these responses suggesting that they activated both Gs and Gi.
FIGURE 1.

Structures of Fen and its derivatives used in the study. The following terms are used: fenoterol (Fen), 4′-methoxyfenoterol (methoxyFen); 4′-aminofenoterol (aminoFen); phenylfenoterol (PhFen); 1-naphthylfenoterol (1-NapFen); ethylfenoterol (EtFen); 2-naphthylfenoterol (2-NapFen); and 4′-methoxy-1-naphthylfenoterol (MNFen).
The mechanism for the differential G protein coupling of β-ARs has been the major focus of various studies. It has been suggested that phosphorylation of the β2-AR by cAMP-dependent protein kinase (PKA) or G protein-coupled receptor kinases (GRKs) promotes the receptors to couple to Gi proteins (16–20). However, some evidence argues against this perception (21, 22). Thus, the molecular basis for inducing β2-AR's coupling selectivity to different G proteins remains largely elusive.
Our recent simulation studies employing data obtained with [3H]CGP-12177 as the marker ligand have identified hydrogen bond (HB) formation between the tyrosine 308 residue (Tyr-308 or Y7.35 in Ballesteros-Weinstein numbering) in transmembrane (TM) 7 of β2-AR and a HB acceptor at the 4′-position of (R,R′)-Fen and (R,R′)-methoxyFen (13, 23). These preliminary results suggest that the Tyr-308 residue is essential for agonist-induced β2-AR preferential coupling to Gs protein. In this study, using site-directed mutagenesis, receptor pharmacology, and cardiomyocyte activation in conjunction with computer simulation, we demonstrated that (R,R′)-4′-aminofenoterol ((R,R′)-aminoFen) (Fig. 1) targets the WT β2-AR but not the mutant receptor, β2-AR Y308F, to Gs-biased signaling. These results experimentally verify our computation-based predictions, and here we present the first evidence that interactions with an individual residue in a conformation of the β2-AR can induce or stabilize a conformation that leads to selective coupling to a G protein subunit.
EXPERIMENTAL PROCEDURES
β2-AR Model Construction and Docking Methodology
Docking of ligands to β2-AR models was performed as recently described (23). In brief, a crystallographic model of the β2-AR co-crystallized with an inverse agonist carazolol (PDB entry 2RH1) was used as a docking target in simulations. The model was modified by swapping the tyrosine 308 residue into phenylalanine using Yasara to obtain the model representing the Y308F mutant. Molegro Virtual Docker software (MVD version 2010.4.0.0, Aarhus, Denmark) was used for docking simulations within the binding cavity of the target model using MolDock SE algorithm as a search engine.
Compounds and Reagents
Fen analogs used in this study (Fig. 1) were synthesized as described previously (13, 14). Cell culture reagents were purchased from Invitrogen. Zinterol was obtained from Tocris Bioscience (Bristol, UK). Forskolin, ICI-118,551, 3-isobutyl-1-methylxanthine, (−)-isoproterenol (ISO), PTX, and other reagents were purchased from Sigma.
Animals
Male Sprague-Dawley rats (200–250 g) were purchased from Charles River. β2-AR knock-out mice were generous gifts from Dr. Brian Kobilka (Stanford University Medical Center, Palo Alto, CA). Animals were housed and studied in conformance with the “Guide for the Care and Use of Laboratory Animals, Eighth Edition” from the National Institutes of Health (NOT-OD-12–020, released 2011), with institutional Animal Care and Use Committee approval.
Generation of Stable Cell Lines and Recombinant Adenoviruses
HEK293A cells were obtained from Invitrogen and maintained in DMEM supplemented with 10% FBS at 37 °C in a humidified 5% CO2 incubator. The plasmids encoding for the human β2-AR and β2-AR Y308F mutant were kindly provided by Dr. Brian Kobilka and Dr. Hitoshi Kurose (Kyushu University, Fukuoka, Japan), respectively. Each of these coding sequences of β2-AR was subcloned into pcDNA3.1− vector (Invitrogen). HEK cells were transfected with the resultant plasmids using Lipofectamine 2000 reagent (Invitrogen), and stably transfected clones were selected against G418 (0.8 mg/ml). When stable expression was achieved, the cells were cultured in the presence of 0.3 mg/ml G418. Adenoviruses for human β2-AR (adeno-β2-AR) and green fluorescent protein (adeno-GFP) have been described previously (24, 25). Adenoviral expression vector carrying the β2-AR Y308F coding sequence was generated by subcloning. Viral particles were purified from transfected HEK cells using standard viral amplification and CsCl purification methods. Viral titers were determined in dilution assays by an immunocytochemical technique using an antibody raised against β2-AR (sc-569, Santa Cruz Biotechnology, Santa Cruz, CA).
Cardiomyocyte Isolation, Adenoviral Gene Transfer, and Contractility Measurement
Cardiomyocytes were isolated from male β2-AR knock-out mice (2–4 months old) or Sprague-Dawley rats using a standard enzymatic technique (9, 24). Mouse cardiomyocytes were seeded on laminin-coated coverslips and infected with the adenoviruses at a multiplicity of infection of 100 (24). The cells were subsequently cultured for 24 h in minimal essential medium (M1018, Sigma) supplemented with forskolin (1 μm) and 2,3-butanedione monoxime (10 mm). Contractility of single cardiomyocytes was measured as described previously (9). In brief, cardiomyocytes were perfused with a buffer containing (in mm) 137 NaCl, 4.9 KCl, 1.2 MgCl2, 1 NaH2PO4, 1 CaCl2, 20 glucose, and 20 HEPES (pH 7.4) and electrically paced (0.5 Hz for rat cardiomyocytes or 1 Hz for mouse cardiomyocytes) at ambient temperature on a microscopic stage. Cell length was monitored by an optical edge-tracking method using an instrument setup manufactured by IonOptix (Milton, MA). Measurements were made under steady-state conditions before and after exposure of the myocytes to a single dose of the agonist. In a subset of experiments, aliquots of cells were incubated with PTX (0.75 μg/ml at 37 °C for >3 h) to block Gi signaling, as described previously (9).
cAMP Accumulation Assay
Cells cultured on poly-d-lysine-coated 12-well plates were treated with PTX (0.3 μg/ml) or vehicle overnight and incubated for 10 min with the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (1 mm) in HEPES-buffered Hanks' balanced salt solution prior to stimulation. Cells were then treated with the agonist or control vehicle for 10 min. Reactions were stopped by the addition of HCl. cAMP contents in the clarified cellular extracts were determined with an enzyme immunoassay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's protocol. cAMP contents were normalized with total cellular protein. Protein contents were determined by the method of Lowry (Bio-Rad).
Immunoblotting
Whole-cell lysates in lysis buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitor mixture (Roche Diagnostics) and phosphatase inhibitor mixture (EMD Millipore, Billerica, MA) were centrifuged at 15,000 × g for 10 min. Clarified cell lysates (20 μg) for β2-AR detection were further denatured and treated with 125 units of peptide:N-glycosidase (New England Biolabs) for 2 h at 37 °C. The samples were denatured in Laemmli sample buffer and resolved by SDS-PAGE. Phosphorylation of ERK and β2-AR was detected by immunoblotting using the same antibodies as described previously (15).
Radioligand Binding Assay
Receptor density was determined on membranes derived from HEK stable cell lines expressing WT or mutant forms of β2-AR as described previously (14). The Bmax and Kd values were determined by nonlinear regression analysis using Prism 4 (GraphPad Software, San Diego).
Statistical Analysis
Results are expressed as means ± S.E. Unless described otherwise, unpaired Student's t test was performed to compare the means between two groups and one-way analysis of variance for multiple group comparison followed by post hoc analysis with Bonferroni's t test. Statistical analysis and curve-fitting of the concentration-response curves were conducted using Prism 4. The curves of the cAMP assays were fitted to the sigmoid curves by nonlinear regression analysis using the four-parameter logistic model without giving any constraints. Curve-fitting of the cardiomyocyte contractility data was conducted using the same algorithms and constraints laid out in our previous study (15).
RESULTS
Role of the Aminoalkyl Substituent of (R,R′)-Fen on Preferential β2-AR-Gs Coupling
To define the structural features of Fen compounds contributing to selective β2-AR-Gs signaling, we have undertaken a structure-activity relationship approach. In this campaign, PTX was used to distinguish the contribution of β2-AR-Gi signaling in the agonist-stimulated inotropic effects of a collection of Fen derivatives (Fig. 1) on a cardiomyocyte contractility model. By inhibiting the Gi signaling with PTX, the regulatory inhibition of adenylyl cyclase on cAMP synthesis would be decreased, and as a result the Gs-stimulated contractile response would be enhanced. Four Fen derivatives ((R,R′)-Fen, (R,R′)-methoxyFen, (R,R′)-aminoFen, and (R,S′)-aminoFen) eliciting a Gi-independent activation of β2-AR (thus PTX-insensitive) were identified (Table 1 and Fig. 2B) with (R,R′)-aminoFen demonstrating the highest Gs selectivity (as assessed by the small difference between the EC50 values of the −PTX and the +PTX groups, Table 1). Their contractility stimulatory effects were mediated through β2-AR because these effects could be antagonized by ICI-118,551, a specific β2-AR antagonist (Fig. 3). (R,R′)-AminoFen was subsequently used as the typical Gs-selective β2-AR agonist in the rest of this investigation.
TABLE 1.
Effect of PTX treatment on the contractile responses to the Fen derivatives
EC50 ± S.E. (nm) for PTX-treated and control groups determined based on concentration-response profiles for the compound-stimulated cardiomyocyte contraction.
| Compound | EC50 (nm) −PTX | EC50 (nm) +PTX | p value | PTX sensitivity |
|---|---|---|---|---|
| (R,R′)-Fen | 73 ± 18a,b | 37 ± 10a | 0.085c | No |
| (R,S′)-Fen | 575 ± 122b | 270 ± 100 | 0.034 | Yes |
| (S,R′)-Fen | 3451 ± 3788a | 830 ± 234a | 0.003 | Yes |
| (R,R′)-MethoxyFen | 186 ± 50a,b | 144 ± 50a | 0.579c | No |
| (R,S′)-MethoxyFen | 506 ± 107b | 182 ± 53 | 0.022 | Yes |
| (S,R′)-MethoxyFen | 3184 ± 1160a | 1161 ± 336a | 0.018 | Yes |
| (R,R′)-PhFen | 547 ± 167 | 124 ± 52 | 0.015 | Yes |
| (R,R′)-AminoFen | 176 ± 78 | 145 ± 64 | 0.641 | No |
| (R,S′)-AminoFen | 605 ± 225 | 311 ± 93 | 0.071 | No |
| (S,R′)-AminoFen | 3083 ± 1286 | 778 ± 169 | 0.004 | Yes |
| (R,R′)-1-NapFen | 82 ± 2 | 12 ± 2 | 0.004 | Yes |
| (R,S′)-1-NapFen | 111 ± 43 | 16 ± 6 | <0.001 | Yes |
| (R,R′)-EtFen | 8551 ± 4992b | 1330 ± 443 | 0.004 | Yes |
| (R,R′)-2-NapFen | 133 ± 25b | 27 ± 7 | <0.001 | Yes |
| (R,R′)-MNFen | 16 ± 7b | 3 ± 2 | 0.028 | Yes |
a EC50 values were recalculated from Ref. 15.
b EC50 values have been reported in Ref. 14 as partial data. Complete sets of data are presented here. Comparisons between the −PTX and +PTX groups and the calculation of the p values were performed in experiments with a parallel design.
c p values were adopted from Ref. 15.
FIGURE 2.
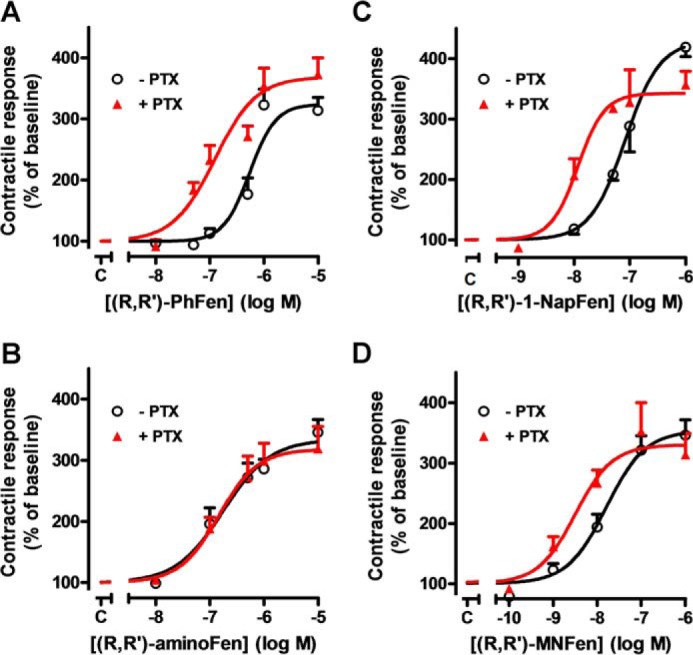
Substitution on the aminoalkyl portion of (R,R′)-Fen determines the PTX sensitivity of the agonist-stimulated contractile response in rat cardiomyocytes. Concentration-response profiles of cardiomyocyte contractility in cells subjected to (R,R′)-PhFen (A), (R,R′)-aminoFen (B), (R,R′)-1-NapFen (C), and (R,R′)-MNFen (D) with (▴) and without (○) PTX treatment (0.75 μg/ml at 37 °C for >3 h). Contractile response to the agonist is expressed as a percentage of the basal contractility (mean ± S.E., n = 9–11 cells from 5 to 9 hearts for each data point).
FIGURE 3.

(R,R′)-AminoFen exhibits β2-AR subtype selectivity in cardiomyocyte contractile response. Single ventricular myocytes from rats were set to pace under perfusion. The contraction amplitude of a cell in response to (R,R′)-aminoFen (0.1 μm) followed by ICI-118,551 (ICI, 0.1 μm) was monitored. Steady-state contractility was recorded. Contractile response is expressed as a percentage of the basal contractility. Data are means ± S.E., n = 4 cells from four hearts. ***, p < 0.001 (by paired t test).
(R,R′)-AminoFen Selectively Activates β2-AR-Gs Signaling in Cardiomyocytes Expressing WT β2-AR but Activates Both Gs and Gi in Cardiomyocytes Expressing the β2-AR Y308F Mutant
Cardiomyocytes express both β1-AR and β2-AR, and robust β2-AR-Gi coupling has been demonstrated in freshly isolated adult mouse cardiomyocytes expressing endogenous β2-AR or human β2-AR at 200-fold over basal level (10). Hence, we employed cardiomyocytes from β2-AR knock-out mice transduced with exogenous β2-AR or its mutants as a physiological model to investigate the role of the β2-AR Tyr-308 residue on ligand-directed G protein selectivity. In our recent study, we have shown that β2-AR in adult rodent cardiomyocytes lost its coupling to Gi after overnight culture, and addition of forskolin in the culture medium could maintain functional dual coupling of β2-AR to Gs and Gi proteins (26). In this investigation, we first confirmed the presence of functional β2-AR-Gi coupling in β2-AR knock-out mouse cardiomyocytes reconstituted with human β2-AR using zinterol, a selective β2-AR agonist (Fig. 4). In another control experiment, cultured cardiomyocytes from β2-AR knock-out mice were infected with adeno-GFP and then subjected to (R,R′)-aminoFen stimulation to study the effect of this compound on stimulating β1-AR. The results in Fig. 5A show that the β1-AR stimulatory effect of (R,R′)-aminoFen was undetectable at 100 nm, was minor at 500 nm (175 ± 26%), and became very substantial (about 350%) at 1 μm. In subsequent contractility studies, we only tested (R,R′)-aminoFen up to 500 nm.
FIGURE 4.

Addition of forskolin reconstitutes functional coupling of β2-AR to Gi protein in cultured β2-AR knock-out mouse cardiomyocytes induced with human β2-AR. Cardiomyocytes from β2-AR knock-out mice were infected with adeno-GFP (white bar) or adeno-β2-AR (black bars) and cultured for 24 h in the presence or absence of forskolin (1 μm) and/or PTX (0.75 μg/ml) as indicated. Cells were transferred to a perfusion chamber, electrically paced, and subjected to stimulation with zinterol (0.2 μm, a concentration without an inotropic effect in freshly isolated cardiomyocytes from WT mice, see Fig. 1A in Ref. 26). Steady-state contractility was measured. Data (mean ± S.E., n = 10–15 cells from 5 to 9 hearts for each data point) are expressed as percentages of the basal contractility. *, p < 0.05. Zinterol (0.2 μm) did not increase contractility in cells infected with adeno-GFP demonstrating no β1-AR stimulatory effect at this concentration. In cells infected with adeno-β2-AR and cultured in the absence of forskolin, the inotropic response produced by zinterol stimulation was the result of a pure β2-AR-Gs-mediated effect because β2-AR and Gi proteins were functionally uncoupled. In cells infected with adeno-β2-AR in the presence of forskolin, the coupling of β2-AR to Gi protein was reestablished. Therefore, the cardiomyocytes were unresponsive to zinterol as if they were freshly isolated WT β2-AR+ cells when β2-AR-Gi coupling was intact. In cells infected with adeno-β2-AR in the presence of forskolin and PTX, the coupling of β2-AR to Gi protein still occurred, but Gi had lost its function and could no longer negatively regulate β2-AR-Gs activation by zinterol.
FIGURE 5.

PTX increases the inotropic effect of (R,R′)-aminoFen in cardiomyocytes expressing β2-AR Y308F mutant but not in cardiomyocytes expressing WT human β2-AR. A, (R,R′)-aminoFen-stimulated contractile responses in β2-AR knock-out mouse cardiomyocyte adenoviral gene transfer GFP. Cardiomyocytes from β2-AR knock-out mice were infected with adeno-GFP and cultured for 24 h. Cells were paced under perfusion and subjected to (R,R′)-aminoFen (100, 500, or 1000 nm). Steady-state contractility before and after agonist stimulation was measured. B, (R,R′)-aminoFen-stimulated contractile responses in β2-AR knock-out mouse cardiomyocytes adenoviral gene transfer WT β2-AR in the presence or absence of PTX treatment. Cardiomyocytes from β2-AR knock-out mice were infected with adeno-β2-AR and cultured with or without PTX (0.75 μg/ml) for 24 h. Cells were paced under perfusion and subjected to (R,R′)-aminoFen (10, 100, or 500 nm). C, (R,R′)-aminoFen-stimulated contractile responses in β2-AR knock-out mouse cardiomyocyte adenoviral gene transfer β2-AR Y308F in the presence or absence of PTX treatment. Contractile responses are expressed as percentages of the basal contractility (mean ± S.E., n = 9–14 cells from 4 to 8 hearts for each data point). Concentration dependence of the (R,R′)-aminoFen-stimulated responses was verified (by two-way analysis of variance, p < 0.0001) for all datasets. *, p < 0.05; ***, p < 0.001 versus corresponding −PTX group.
Next, we investigated the positive inotropic effects of (R,R′)-aminoFen on β2-AR knock-out mouse cardiomyocytes infected with adeno-β2-AR or adeno-β2-AR Y308F and the sensitivities of these responses toward PTX. In cardiomyocytes transduced with the WT β2-AR, the positive inotropic effect of (R,R′)-aminoFen was insensitive to PTX treatment (Fig. 5B). Notably, the (R,R′)-aminoFen-stimulated positive inotropic effect was markedly enhanced by PTX treatment in cells transduced with the β2-AR Y308F mutant (Fig. 5C).
Residue Tyr-308 of β2-AR Is Necessary for Ligand-directed Gs-biased β2-AR Signaling
It has been demonstrated in HEK cells that β2-AR agonists trigger an acute increase in ERK phosphorylation, which peaks at 5 min, and this effect is mediated in part by a Gi-dependent mechanism (16, 27). Furthermore, both Gs- and Gi-mediated β2-AR activation can lead to ERK phosphorylation (16, 22, 27). Cell lines stably expressing WT β2-AR (HEK-β2-AR cells) and β2-AR Y308F mutant (HEK-β2-AR Y308F cells) were established from HEK293A cells. The levels of β2-AR in these cell lines were 4033 ± 826 and 2300 ± 80 fmol/mg protein, respectively, as assayed by radioligand binding, whereas the level of β2-AR in the parental cells was 30–40 fmol/mg (21). Next, we studied the G protein pathways responsible for phospho-ERK (p-ERK) induction by (R,R′)-aminoFen and ISO in these cell lines. The sensitivity of agonist-induced ERK phosphorylation toward PTX was used to indicate Gi activation.
Stimulation with ISO increased p-ERK by about 6-fold in HEK-β2-AR cells (Fig. 6, A and B) and HEK-β2-AR Y308F cells (Fig. 6, C and D). This activation of p-ERK was mediated by a combination of Gs- and Gi-dependent pathways as demonstrated by the decreases in maximal ERK phosphorylation in the PTX-treated groups. In contrast, in HEK-β2-AR cells, (R,R′)-aminoFen induced ERK phosphorylation in a PTX-insensitive manner (Fig. 6, A and B). Importantly, the increase in phosphorylation of ERK in response to (R,R′)-aminoFen exhibited a robust PTX sensitivity in HEK-β2-AR Y308F cells, and this Gi-dependent effect appeared to be positively correlated with the concentration of (R,R′)-aminoFen (Fig. 6, C and D).
FIGURE 6.

Y308F substitution on β2-AR increases the PTX sensitivity of (R,R′)-aminoFen-induced ERK phosphorylation in HEK stable cell lines. Confluent cultures of HEK-β2-AR cells and HEK-β2-AR Y308F cells were deprived of serum overnight. Treatment with PTX (0.3 μg/ml, +) or vehicle (−) was implemented during serum starvation. Cells were then stimulated with ISO (1 μm) or (R,R′)-aminoFen (10−9 to 10−6 m) for 5 min at 37 °C as indicated. ERK phosphorylation was determined by immunoblotting. A, immunoblots of p-ERK and total ERK (as protein loading control) in response to agonist stimulation in HEK-β2-AR cells, and B, averaged data. C, immunoblots of p-ERK and total ERK in response to agonist stimulation in HEK-β2-AR Y308F cells, and D, averaged data. Data are presented as fold increase over −PTX control (means ± S.E. in 3–4 independent experiments). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle controls; #, p < 0.05 versus −PTX group.
We also measured cAMP accumulation in HEK cells stably expressing β2-AR and its Y308F mutant. Although (R,R′)-aminoFen produced a PTX-insensitive cAMP response in HEK-β2-AR cells (Fig. 7A), the concentration-response profile shifted upwards in response to PTX treatment in HEK-β2-AR Y308F cells (Fig. 7B). Efficacy data (Table 2) show that PTX significantly increased the Emax value (from 89 ± 1 to 117 ± 2, p < 0.01) without altering the logEC50 value of the cAMP response of (R,R′)-aminoFen in HEK-β2-AR Y308F cells (from −8.05 ± 0.01 to −8.03 ± 0.02, p = 0.75). We did not observe sensitivity of cAMP response to PTX when HEK-β2-AR cells were stimulated with ISO (data not shown), thus corroborating a previous experiment (17). This may be due to an inherent limitation of the assay. In contrast, PTX caused an increase in the Emax of the ISO-stimulated cAMP response (from 109 ± 4 to 126 ± 5, p < 0.05, Table 2) in HEK-β2-AR Y308F cells, mirroring a similar observation in cells expressing the β2-AR D-4 mutant, a receptor phenotype having a reduced Gs-coupling, and an increased Gi-coupling (17). Taken together, these results demonstrate that the β2-AR-Y308 residue is necessary for the Gs-biased β2-AR signaling and that Y308F mutation fully restored β2-AR-Gi signaling in response to (R,R′)-aminoFen, a Gs-selective β2-AR agonist, in cardiomyocytes and in HEK cells.
FIGURE 7.
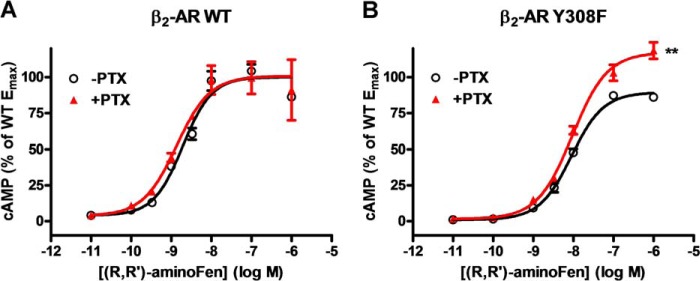
Y308F substitution on β2-AR increases the PTX sensitivity of (R,R′)-aminoFen-stimulated cAMP production in HEK stable cell lines. HEK-β2-AR cells and HEK-β2-AR Y308F cells were cultured in 12-well plates in parallel, and subsets of the cells were treated with PTX (0.3 μg/ml) or vehicle overnight. Agonist stimulation was allowed to proceed for 10 min at 25 °C in the presence of 3-isobutyl-1-methylxanthine (1 mm). Cellular cAMP contents were determined by enzyme immunoassay. HEK-β2-AR cells were subjected to (R,R′)-aminoFen (10−11 to 10−6 m) (A), and HEK-β2-AR Y308F cells were subjected to (R,R′)-aminoFen (B), with (▴) and without (○) PTX treatment. Data (means ± S.E. in three independent experiments performed in triplicate) are expressed as percentages of the Emax response of the β2-AR WT −PTX group. Curve-fitting analysis of the concentration-response curves were conducted using Prism. **, p < 0.01 versus −PTX group.
TABLE 2.
Emax % and logEC50 values of the (R,R′)-aminoFen- and ISO-induced cAMP responses in HEK cell lines stably expressing β2-AR WT or β2-AR Y308F
Calculations of logEC50 and Emax values were based on concentration-response profiles for the compound-stimulated cAMP production. Emax values are expressed as percentages of the Emax response of the β2-AR WT −PTX group. Means ± S.E., n = 3. *, p < 0.05; **, p < 0.01 versus −PTX group.
| Cell lines | (R,R′)-AminoFen |
ISO |
||||||
|---|---|---|---|---|---|---|---|---|
| −PTX |
+PTX |
−PTX |
+PTX |
|||||
| Emax % | log EC50 | Emax % | log EC50 | Emax % | log EC50 | Emax % | log EC50 | |
| β2-AR WT | 100 ± 3 | −8.71 ± 0.05 | 101 ± 2 | −8.87 ± 0.03 | 100 ± 4 | −10.14 ± 0.10 | 97 ± 3 | −10.25 ± 0.07 |
| β2-AR Y308F | 89 ± 1 | −8.05 ± 0.01 | 117 ± 2** | −8.03 ± 0.02 | 109 ± 4 | −9.36 ± 0.06 | 126 ± 5* | −9.33 ± 0.07 |
Effects of Agonist-induced Receptor Phosphorylation on G Protein-coupling Selectivity of β2-AR
It is well established that agonist stimulation of β2-AR leads to phosphorylation of the receptor by PKA and GRK, with important implications on receptor desensitization (28). In addition, phosphorylation of β2-AR at the GRK or the PKA sites has been suggested to be necessary for β2-AR-Gi protein coupling in naive cells (16, 17), cardiomyocytes (18–20), and in vivo hearts (20). Therefore, we investigated the agonist-stimulated receptor phosphorylation in HEK cells expressing either the WT β2-AR or the β2-AR Y308F mutant using phosphosite-specific antibodies (29). Stimulation with ISO (1 μm) for 5 min, a treatment time period reportedly leading to near-maximal receptor phosphorylation responses (30–33), increased the phosphorylation of β2-AR and β2-AR Y308F at Ser-262 (PKA-site) to about 5-fold of basal (Fig. 8, A and B). Similarly, (R,R′)-aminoFen (1 μm) produced the same maximal responses in both HEK-β2-AR cells and HEK-β2-AR Y308F cells (Fig. 8, A and B). Treatment with ISO also increased the phosphorylation of β2-AR at Ser-355,356 (GRK sites) by about 17-fold in these cell lines (Fig. 8, C and D). Because it has been reported that phosphorylation of the β2-AR mediated by GRK depends on a high concentration of agonists (27–31), we subsequently performed the receptor phosphorylation assay at higher concentrations of (R,R′)-aminoFen (namely 100- and 1000-fold of EC50 cAMP concentrations, corresponding to a near-saturating and a saturating concentration of the agonist for receptor stimulation, respectively, refer to Fig. 7, A and B). As shown in Fig. 8, C and D, (R,R′)-aminoFen at the 100-fold EC50 cAMP concentration (R100) produced a significant increase in the phosphorylation of β2-AR at Ser-355,356 (GRK sites) as compared with the vehicle control in HEK cell lines expressing either the WT β2-AR or the β2-AR Y308F mutant. Treatment with (R,R′)-aminoFen at the 1000-fold EC50 cAMP concentration (R1000) caused β2-AR-Ser-355,356 phosphorylation in both HEK-β2-AR cells and HEK-β2-AR Y308F cells indistinguishable in magnitude as compared with the stimulation with ISO (1 μm, a saturating concentration, refer to Table 2) (Fig. 8, C and D).
FIGURE 8.

(R,R′)-AminoFen induces phosphorylation of β2-AR and β2-AR Y308F mutant at the GRK and the PKA sites in HEK stable cell lines. Confluent cultures of HEK-β2-AR cells and HEK-β2-AR Y308F cells were incubated in serum-free medium for 3 h and then stimulated with vehicle control (−), ISO (1 μm), or (R,R′)-aminoFen (R, 1 μm; R100 for WT, 0.2 μm; R100 for Y308F, 1 μm; R1000 for WT, 2 μm; R1000 for Y308F, 10 μm) for 5 min at 37 °C. Phosphorylated β2-AR was detected by phosphosite-specific antibodies against Ser(P)-262 for PKA sites and Ser(P)-355,356 for GRK sites. Total β2-AR was detected after stripping and reprobing the membrane with the β2-AR-CT antibody. A, immunoblots of Ser(P)-262-β2-AR and total β2-AR in response to agonist stimulation, and B, averaged data (normalized to total β2-AR). C, immunoblots of Ser(P)-355,356-β2-AR and total β2-AR in response to agonist stimulation, and D, averaged data. Data are expressed as fold increase over control (means ± S.E. in at least three independent experiments). *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle controls (two-way analysis of variance with post hoc t test). No significant differences were found for all within-group comparisons between WT and Y308F, p > 0.05.
In HEK-β2-AR cells, increased phosphorylation of the β2-AR at the PKA site could be observed 5 min after stimulation with ISO or (R,R′)-aminoFen at 1 μm (Fig. 8, A and B). Similarly, (R,R′)-aminoFen (0.2 and 2 μm) and ISO (1 μm) also increased the phosphorylation of β2-AR at the GRK sites (Fig. 8, C and D). The same treatment with (R,R′)-aminoFen (10−8 to 10−6 m) increased the p-ERK level by about 4-fold via activating Gs but not Gi (Fig. 6, A and B). Treatment with ISO after disrupting the activity of Gi with PTX also increased the p-ERK level by about 4-fold, which could be further increased to 6-fold in the absence of PTX (Fig. 6, A and B). Thus, both (R,R′)-aminoFen and ISO can induce the phosphorylation of ERK and β2-AR via a Gs-dependent pathway, but only ISO can activate ERK through a β2-AR-Gi signaling pathway. These results clearly demonstrate that (R,R′)-aminoFen and ISO produced similar effects in triggering phosphorylation of β2-AR at the PKA sites or the GRK sites, although they exhibited diverse G protein selectivity in HEK stable cell lines expressing the WT β2-AR. In addition, Y308F substitution on the β2-AR caused a qualitative change in the G protein selectivity of (R,R′)-aminoFen from being exclusively Gs-activating (Fig. 6, A and B) to dually Gs/Gi-activating (Fig. 6, C and D) without significantly affecting its activities in eliciting receptor phosphorylation by both PKA and GRKs (p > 0.05, Fig. 8, B and D). Thus, phosphorylation of β2-AR at its PKA or GRK sites is insufficient to trigger the receptor coupling to Gi proteins.
Docking Simulation on β2-AR
To reveal the molecular interactions important for the ligand-directed Gs-biased agonism of β2-AR, a molecular model of (R,R′)-aminoFen was docked to the crystal model of the β2-AR-binding site as well as to a model representing the Y308F mutant receptor using the same procedures as described previously (23). As shown in Fig. 9, A and C, the ligand molecule can be fitted reasonably well into the binding sites of both β2-AR conformations (PDB entry 2RH1 for carazolol-bound and PDB entry 3SN6 for BI-167107- and Gs protein-bound, respectively) with the resorcinol ring of the ligand pointed in the direction of S2035.42 and S2075.46, the β-OH and the secondary amine groups interacted with D1133.32 and N3127.39 residues. Notably, a HB interaction between the 4′-amino group of the ligand and the hydroxyl group of Y3087.35 was found probable in the carazolol-bound β2-AR model (Fig. 9A). When (R,R′)-aminoFen was docked to the modified model of the Y308F mutant receptor (Fig. 9B), the position of the molecule within the binding site was very similar, and the above-mentioned interactions still occurred with the exception of the HB created by the 4′-amino moiety. In effect, the 4′-aminobenzyl ring assumed a slightly different position than during docking to the WT receptor model.
FIGURE 9.

Binding poses of (R,R′)-aminoFen and (R,R′)-MNFen docked to the β2-AR models. A, docking of (R,R′)-aminoFen to the carazolol-bound β2-AR model (PDB entry 2RH1), and B, to the β2-AR Y308F mutant. The 4′-amino group of (R,R′)-aminoFen forms a HB with the Tyr-308 residue of β2-AR (green arrow), and the interaction is lost in the β2-AR Y308F mutant. C, docking of (R,R′)-aminoFen to a BI-167107 and Gs protein-bound conformation of β2-AR (PDB entry 3SN6). The location of the docked molecule highly resembles the orientation of co-crystallized agonist, BI-167107, as both molecules share significant structural similarities. The agonists form a network of analogous HB interactions with the receptor residues Ser-203, Ser-207, Asn-312, and Asp-113. An additional interaction can be observed between the 4′-amino moiety of the ligand and Lys-305 residue. D, docking of (R,R′)-MNFen to a carazolol-bound conformation of β2-AR (PDB entry 2RH1). Aromatic residues in the ligand binding pocket able to form π-π interactions with the naphthyl moiety of the ligand are shown. Ligand molecule is rendered in atom-type color-coded stick mode, and five essential TM helices of target β2-AR model are shown and colored as follows: TM3, magenta; TM4, green; TM5, red; TM6, yellow; and TM7, blue.
DISCUSSION
The current data (Figs. 5–8) suggest that the Gs-selective signaling depends on specific interactions between the agonist and the β2-AR-Y308 residue, and induction of receptor phosphorylation alone does not necessarily lead to a switching of the receptor coupling from Gs to Gi as once proposed (16). These results are consistent with those reported in our previous study on cardiomyocytes (15) in which the stereoisomers of Fen and methoxyFen possessing different G protein selectivity induced similar phosphorylation of β2-AR at the PKA sites. Because (R,R′)-Fen, (R,R′)-methoxyFen, and (R,R′)-aminoFen are full agonists of β2-AR capable of inducing receptor phosphorylation just as ISO (Fig. 8) (15), the only explanation for their preferential Gs selectivity would be their exceptional abilities in stabilizing a receptor conformation favoring receptor-Gs protein interaction. As the emerging paradigm of functional selectivity suggests, ligands can perturb a GPCR to attain “ensembles” of multiple conformations, and each of these conformations is capable of activating a distinct set of signaling events (1–8). Therefore, one possibility is that the binding of the Gs-selective agonists causes the β2-AR to assume or stabilize conformations leading to Gs protein coupling. β2-AR in such conformation(s) regardless of its phosphorylation status interacts strongly with Gs protein, prohibiting receptor-Gi protein interaction from taking place. Furthermore, if the β2-AR-Y308 residue is mutated, the Gs selectivity of these agonists will be lost (Figs. 5 and 6), suggesting that mutation of this residue affects the receptor conformations stabilized by these agonists. The latter conformations, possibly resembling the ISO-bound receptor conformations, are Gi protein-permissive.
Y3087.35 has been implicated for the high affinity binding of β2-AR-selective agonists (34). Importantly, our recent docking simulation study (23) supported the initial determination (13, 14) that in inverse agonist-associated conformations, such as the S1 and S2 conformations described by Kim et al. (35), Y3087.35 may interact directly with the 4′-hydroxyl, 4′-methoxy, or 4′-amino group of the (R,R′)-Fen derivatives through hydrogen bonding. This hypothesis was confirmed in this study (Fig. 9A).
Because the human β2-AR and the rat β2-AR share a high sequence homology particularly at the TM regions encompassing the ligand binding pocket (data not shown), the data derived from the rat cardiomyocyte study (Fig. 2 and Table 1) could be interpreted in the light of the structural insight gained from the human β2-AR models. As shown in Fig. 2, A, C, and D, and Table 1, the R,R′-isomers of Fen derivatives containing the following substituents on the aminoalkyl portion, phenyl (PhFen), 1-naphthylfenoterol, 2-naphthylfenoterol, ethylfenoterol, and 4′-methoxy-1-naphthyl (MNFen), produced PTX-sensitive contractile responses in cardiomyocytes. These results suggest that these compounds activate both Gs and Gi pathways of β2-AR. In contrast, the positive inotropic effects of (R,R′)Fen, (R,R′)-methoxyFen, (R,R′)-aminoFen, and (R,S′)-aminoFen were PTX-insensitive (Table 1 and Fig. 2B), indicating that they selectively activate β2-AR-Gs signaling. Together, these data illustrate the structural features of a Fen compound for ligand-directed selective β2-AR-Gs signaling as follows: (i) a benzyl rather than a naphthyl moiety on its aminoalkyl substituent; (ii) a 4′-oxygen or a 4′-nitrogen moiety on this aromatic substituent; and (iii) a mandatory R-configuration on the chiral center of the β-OH group and a preferred R-configuration on the second chiral center.
The fact that (R,R′)-PhFen, but not (R,R′)-Fen, (R,R′)-methoxyFen, and (R,R′)-aminoFen, produced a PTX-sensitive inotropic effect (Fig. 2, A and B, and Table 1) suggests that the 4′-(N/O) moieties in these (R,R′)-Fen derivatives are indispensable for agonist-induced preferential Gs activation. If either the phenyl hydroxyl group of Tyr-308 or the 4′-(N/O) moiety is lost, the HB between the ligand and the 7.35 residue (Fig. 9A) will not exist, and promiscuous Gs and Gi dual signaling rather than Gs-biased signaling will be induced (Figs. 5C, 6C, and 7B). This indicates that specific interaction between Tyr-308 and the 4′-(N/O)-benzyl moiety promotes preferential receptor-Gs protein coupling. Our simulation study (23) has also shown that (R,R′)-Fen derivatives with naphthyl moieties interact not by hydrogen bonding with Tyr-308 but rather by π-π interactions with the other aromatic residues in the ligand binding pocket (Fig. 9D). The opposite is true for compounds with 4′-(N/O)-benzyl moieties (23). The dominance of the π-π interactions with (R,R′)-MNFen binding, irrespective of the presence of a potentially hydrogen bonding 4′-methoxy moiety, is associated with dual Gs and Gi protein coupling of β2-AR (Fig. 2D). These key features in the ligand-receptor interaction make (R,R′)-MNFen a superior negative model compound as compared with ISO or the (S,R′)-isomers in the study of Gs-biased signaling, and the functional data with (R,R′)-MNFen stimulation (Fig. 2D) also point to the same conclusion. Thus, based on our simulated docking study and experimental evidence, we conclude that HB interactions between the 4′-(N/O)-benzyl moiety of the (R,R′)Fen derivatives and the β2-AR-Y308 residue play an important role on ligand-directed β2-AR-Gs signaling.
The structural features of Fen derivatives for preferential Gs selectivity and receptor subtype selectivity have both similarities and differences. Our initial studies (13, 14) have shown that both stereochemistry and the aminoalkyl substituent play essential roles on the β2-AR subtype selectivity of the Fen compounds. Although an R-configuration, hydrogen bonding with Tyr-308, and π-π interactions with aromatic residues in the ligand binding pocket can all contribute to high ligand binding affinity and increased selectivity to β2-AR (14), only the R-configuration and hydrogen bonding with Tyr-308 correlate with Gs selectivity. The π-π interactions, however, likely have a detrimental effect on Gs selectivity of the Fen derivatives. It is conceivable that the above-mentioned molecular interactions would impact the receptor conformational ensembles stabilized by different β2-AR agonists and subsequently result in the differential G protein-coupling selectivity.
Recent studies have identified H6.55 to be a major determinant of ligand-biased signaling in dopaminergic D2L receptors (36). A follow-up study has further characterized the receptor conformations involved in D2L receptor functional selectivity (37). Briefly, a model of functional selectivity for D2L receptor has been proposed in which TM6 represents a rotatory switch in response to the binding of different functionally selective agonists. In this model, if H6.55 in TM6 rotates toward S5.43 on TM5, the resultant ligand-stimulated receptor conformation will favor the activation of the arachidonic acid pathway. Conversely, if H6.55 rotates toward TM7 and interacts with Y7.35, the ligand-stimulated receptor conformation will lead to a signaling bias toward cAMP/MAPK activation.
A very similar mechanism occurs in β2-AR. Y3087.35 is known to form a HB with a neighboring residue N2936.55 in TM6, and this specific interaction (additionally shown on Fig. 9A) remains intact in all reported crystallographic structures of the receptor. Because the 4′-(N/O) moiety of the Gs-selective ligand involves the hydroxyl group of Tyr-308 in another HB interaction, a competition occurs between the Tyr-308–Asn-293 interaction and Tyr-308–ligand interaction. It is therefore postulated that the naturally occurring Tyr-308–Asn-293 HB is disrupted when the Gs-selective ligand binds to the receptor. The Tyr-308–Asn-293 interaction bridges the upper parts of TM6 and TM7. Breaking this interaction during the receptor conformational transition might be a key phenomenon leading to specific activation of the β2-AR to a form favoring selective Gs protein coupling.
Using acetylcholine M2 receptor as a model, Bock et al. (38) have designed “dualsteric” agonists to study the role of allosteric vestibules on G protein activation. The allosteric vestibule is located at the entrance of the orthosteric binding cavity of many class A GPCRs and has been implicated for ligand binding (39). Acetylcholine is known to activate Gi and Gs signaling of the M2 receptor. The authors have found that dualsteric agonists (such as iper-6-phth and iper-6-naph) exhibited a Gi over Gs signaling bias compared with acetylcholine and their parent compound Iperoxo, an orthosteric muscarinic agonist. Mutagenic studies have identified the M2-W4227.35 residue located at the allosteric vestibule to be critical for both Gs and Gi protein activation, with the gain in dualsteric probe efficacy for Gi activation in the allosteric mutant. Interestingly, W4227.35 and Y1775.32 in extracellular loop 2 of the M2 receptor and the analogous Y3087.35 and F1935.32 in β2-AR that line the passage to the orthosteric binding cavity have been suggested to undergo a conformational rearrangement during receptor activation (39, 40). The authors implied from their findings that spatial rearrangement of this passage is critical for receptor movements required for appropriate unfolding of the intracellular domain region for G protein coupling.
The two previous studies and this study indicate the important role of the 7.35 residue on GPCR conformational transition leading to G protein activation. Notably, here we provide the direct evidence to pinpoint the role of this residue on functional selectivity and illustrate this point with an “extreme” form of signaling bias (in nominal terms of with or without PTX sensitivity rather than in ratiometric terms of a biased factor) in a physiological context of adult cardiomyocytes. Further studies are needed to determine whether this deduction could be generalized in a broader sense, such as to other class A aminergic GPCRs.
Our study design necessitates the investigation of a single aspect of the β2-AR agonists, specifically their differential selectivity to Gs and Gi proteins. From a chemical biology perspective, however, the β2-AR is only one of the many possible in vivo targets of the Fen compounds. Indeed, in complex biological systems such as the adult cardiomyocytes, a compound is likely to produce its effects via interactions with multiple cellular proteins. Therefore, it is unsurprising to find that high concentrations of (R,R′)-aminoFen also stimulate β1-AR (Fig. 5A) given that only 1 out of the 15 amino acids that constitute the ligand binding pocket differs between β1-AR and β2-AR (41). Consistently, our previous binding affinity data (13) have also shown that the subtype selectivity of β2-AR relative to β1-AR in terms of Kiβ1-AR/Kiβ2-AR ratio was 9 for (R,R′)-aminoFen. Interestingly, it is the 7.35 residue (β2-AR-Y308) that is different, and the corresponding residue is a phenylalanine or Phe-359 in β1-AR, incidentally the same mutation characterized in this study. However, mutation on β2-AR to convert the amino acid residues in its ligand binding pocket to that resembling β1-AR produced a dissimilar function of increased receptor-Gi protein coupling in the β2-AR Y308F mutant, yet it is known that β1-AR does not normally couple to Gi (10). Thus, it is not the amino acid residues themselves but rather their different interactions with the ligand and the resultant conformational changes (42) that determine the diverse selectivity of the β-AR subtypes or mutants to different G proteins. As a cautionary note, this interpretation is only confined to the very first step of G protein coupling at the receptor level, without taking into account other intracellular mechanisms such as phosphorylation, internalization, G protein abundance, and subcellular compartmentation.
In addition, cross-talk between different receptor-mediated signaling pathways is very common when a given ligand simultaneously stimulates two or more receptors. We observed this cross-talk of signals between β1-AR and β2-AR in cardiomyocytes stimulated by 500 nm (R,R′)-aminoFen as exemplified by a higher contractile response in the β2-AR Y308F mutant-expressing cells (Fig. 5C) versus the WT β2-AR-expressing cells (Fig. 5B) in the PTX-treated groups. The detailed mechanism of the cross-talk between the β1- and β2-AR signals is beyond the scope of this study, although an elaborated discussion can be found in Zhang et al. (43). This example illustrates that no single assay or approach can adequately elucidate the complex pharmacology of a compound.
In conclusion, this study has identified an amino acid residue in β2-AR necessary for functional selectivity. Mutation of this residue causes a Gs-selective agonist to gain the ability to activate Gi when it binds to the β2-AR. We also provide, for the first time, functional data confirming the identification of the ligand-receptor interactions important for Gs-biased signaling in β2-AR. Advances in structural biological techniques (42, 44–48) will ultimately unravel how these interactions during ligand binding translate into receptor conformation(s) for selective coupling to different G proteins. This investigation has elucidated the molecular basis of Gs-biased agonism in β2-AR, and this is one step closer to structure-based design of signaling pathway-specific drugs.
Acknowledgments
We thank Dr. Brian Kobilka and Dr. Hitoshi Kurose for providing the plasmids encoding for the human β2-AR and β2-AR Y308F mutant. Special thanks to Dr. Richard B. Clark for providing the anti-Ser(P)-262 and anti-Ser(P)-355,356 antibodies.
This work was supported, in whole or in part, by National Institutes of Health Intramural Research Program from NIA (to A. Y. H. W., R. K. P., M. B., and I. W. W.) and by National Institutes of Health Grant N01-AG31009 from NIA. This work was also supported by National Basic Research Program of China Grant 2012CB518000 and National Science Foundation of China Project 31221002 (to R. P. X.), National Major Scientific Research Program of China Grant 2012CB910402, National Scientific Technology Major Project of China Grant 2013ZX09507001 (to A. Y. H. W.), Beijing Key Laboratory of Cardiometabolic Molecular Medicine, Peking University, and Foundation for Polish Science Grant TEAM 2009-4/5 Programs (to K. J.).
- GPCR
- G protein-coupled receptor
- adeno
- adenovirus
- aminoFen
- 4′-aminofenoterol
- β-AR
- β-adrenoceptor
- β1-AR
- β1-adrenoceptor
- β2-AR
- β2-adrenoceptor
- Fen
- fenoterol
- GRK
- G protein-coupled receptor kinase
- HB
- hydrogen bond
- ISO
- (−)-isoproterenol
- methoxyFen
- 4′-methoxyfenoterol
- MNFen
- 4′-methoxy-1-naphthylfenoterol
- PDB
- Protein Data Bank
- PhFen
- phenylfenoterol
- PKA
- cyclic AMP-dependent protein kinase
- PTX
- pertussis toxin
- TM
- transmembrane.
REFERENCES
- 1. Violin J. D., Lefkowitz R. J. (2007) β-Arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 28, 416–422 [DOI] [PubMed] [Google Scholar]
- 2. Kenakin T. (2007) Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol. Sci. 28, 407–415 [DOI] [PubMed] [Google Scholar]
- 3. Mailman R. B. (2007) GPCR functional selectivity has therapeutic impact. Trends Pharmacol. Sci. 28, 390–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Urban J. D., Clarke W. P., Zastrow M. V., Nichols D. E., Kobilka B., Weinstein H. (2007) Functional selectivity and classical concepts of quantitative pharmacology. Pharmacology 320, 1–13 [DOI] [PubMed] [Google Scholar]
- 5. Neubig R. R. (2007) Missing links: mechanisms of protean agonism. Mol. Pharmacol. 71, 1200–1202 [DOI] [PubMed] [Google Scholar]
- 6. Seifert R., Dove S. (2009) Functional selectivity of GPCR ligand stereoisomers: new pharmacological opportunities. Mol. Pharmacol. 75, 13–18 [DOI] [PubMed] [Google Scholar]
- 7. Evans B. A., Sato M., Sarwar M., Hutchinson D. S., Summers R. J. (2010) Ligand-directed signalling at β-adrenoceptors. Br. J. Pharmacol. 159, 1022–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kenakin T., Miller L. J. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiao R. P., Ji X., Lakatta E. G. (1995) Functional coupling of the β2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol. Pharmacol. 47, 322–329 [PubMed] [Google Scholar]
- 10. Xiao R. P., Avdonin P., Zhou Y. Y., Cheng H., Akhter S. A., Eschenhagen T., Lefkowitz R. J., Koch W. J., Lakatta E. G. (1999) Coupling of β2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ. Res. 84, 43–52 [DOI] [PubMed] [Google Scholar]
- 11. Xiao R. P., Zhang S. J., Chakir K., Avdonin P., Zhu W., Bond R. A., Balke C. W., Lakatta E. G., Cheng H. (2003) Enhanced Gi signaling selectively negates β2-AR- but not β1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation 108, 1633–1639 [DOI] [PubMed] [Google Scholar]
- 12. Beigi F., Bertucci C., Zhu W., Chakir K., Wainer I. W., Xiao R. P., Abernethy D. R. (2006) Enantioselective separation and online affinity chromatographic characterization of R,R- and S,S-fenoterol. Chirality 18, 822–827 [DOI] [PubMed] [Google Scholar]
- 13. Jozwiak K., Khalid C., Tanga M. J., Berzetei-Gurske I., Jimenez L., Kozocas J. A., Woo A., Zhu W., Xiao R. P., Abernethy D. R., Wainer I. W. (2007) Comparative molecular field analysis of the binding of the stereoisomers of fenoterol and fenoterol derivatives to the β2-adrenergic receptor. J. Med. Chem. 50, 2903–2915 [DOI] [PubMed] [Google Scholar]
- 14. Jozwiak K., Woo A. Y., Tanga M. J., Toll L., Jimenez L., Kozocas J. A., Plazinska A., Xiao R. P., Wainer I. W. (2010) Comparative molecular field analysis of fenoterol derivatives: a platform towards highly selective and effective β2-adrenergic receptor agonists. Bioorg. Med. Chem. 18, 728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woo A. Y., Wang T. B., Zeng X., Zhu W., Abernethy D. R., Wainer I. W., Xiao R. P. (2009) Stereochemistry of an agonist determines coupling preference of β2-adrenoceptor to different G proteins in cardiomyocytes. Mol. Pharmacol. 75, 158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Daaka Y., Luttrell L. M., Lefkowitz R. J. (1997) Switching of the coupling of the β2-adrenergic receptor to different G proteins by protein kinase A. Nature 390, 88–91 [DOI] [PubMed] [Google Scholar]
- 17. Zamah A. M., Delahunty M., Luttrell L. M., Lefkowitz R. J. (2002) Protein kinase A-mediated phosphorylation of the β2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J. Biol. Chem. 277, 31249–31256 [DOI] [PubMed] [Google Scholar]
- 18. Wang Y., De Arcangelis V., Gao X., Ramani B., Jung Y. S., Xiang Y. (2008) Norepinephrine- and epinephrine-induced distinct β2-adrenoceptor signaling is dictated by GRK2 phosphorylation in cardiomyocytes. J. Biol. Chem. 283, 1799–1807 [DOI] [PubMed] [Google Scholar]
- 19. Liu R., Ramani B., Soto D., De Arcangelis V., Xiang Y. (2009) Agonist dose-dependent phosphorylation by protein kinase A and G protein-coupled receptor kinase regulates β2 adrenoceptor coupling to Gi proteins in cardiomyocytes. J. Biol. Chem. 284, 32279–32287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu W., Petrashevskaya N., Ren S., Zhao A., Chakir K., Gao E., Chuprun J. K., Wang Y., Talan M., Dorn G. W., 2nd, Lakatta E. G., Koch W. J., Feldman A. M., Xiao R. P. (2012) Gi-biased β2AR signaling links GRK2 upregulation to heart failure. Circ. Res. 110, 265–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedman J., Babu B., Clark R. B. (2002) β2-Arenergic receptor lacking the cyclic AMP-dependent protein kinase consensus sites fully activates extracellular signal-regulated kinase 1/2 in human embryonic kidney 293 cells: lack of evidence for Gs/Gi switching. Mol. Pharmacol. 62, 1094–1102 [DOI] [PubMed] [Google Scholar]
- 22. Schmitt J. M., Stork P. J. (2000) β2-Adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein Rap1 and the serine/threonine kinase B-Raf. J. Biol. Chem. 275, 25342–25350 [DOI] [PubMed] [Google Scholar]
- 23. Plazinska A., Kolinski M., Wainer I. W., Jozwiak K. (2013) Molecular interactions between fenoterol stereoisomers and derivatives and the β2-adrenergic receptor binding site studied by docking and molecular dynamics simulations. J. Mol. Model. 19, 4919–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Y. Y., Wang S. Q., Zhu W. Z., Chruscinski A., Kobilka B. K., Ziman B., Wang S., Lakatta E. G., Cheng H., Xiao R. P. (2000) Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am. J. Physiol. Heart Circ. Physiol. 279, H429–H436 [DOI] [PubMed] [Google Scholar]
- 25. Baillie G. S., Sood A., McPhee I., Gall I., Perry S. J., Lefkowitz R. J., Houslay M. D. (2003) β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. U.S.A. 100, 940–945 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Chakir K., Zhu W., Tsang S., Woo A. Y., Yang D., Wang X., Zeng X., Rhee M. H., Mende U., Koitabashi N., Takimoto E., Blumer K. J., Lakatta E. G., Kass D. A., Xiao R. P. (2011) RGS2 is a primary terminator of β2-adrenergic receptor-mediated Gi signaling. J. Mol. Cell. Cardiol. 50, 1000–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lefkowitz R. J., Pierce K. L., Luttrell L. M. (2002) Dancing with different partners: protein kinase a phosphorylation of seven membrane-spanning receptors regulates their G protein-coupling specificity. Mol. Pharmacol. 62, 971–974 [DOI] [PubMed] [Google Scholar]
- 28. Lefkowitz R. J. (1998) G protein-coupled receptors. III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J. Biol. Chem. 273, 18677–18680 [DOI] [PubMed] [Google Scholar]
- 29. Tran T. M., Friedman J., Qunaibi E., Baameur F., Moore R. H., Clark R. B. (2004) Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the β2-adrenergic receptor using phosphoserine-specific antibodies. Mol. Pharmacol. 65, 196–206 [DOI] [PubMed] [Google Scholar]
- 30. January B., Seibold A., Whaley B., Hipkin R. W., Lin D., Schonbrunn A., Barber R., Clark R. B. (1997) β2-Adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J. Biol. Chem. 272, 23871–23879 [DOI] [PubMed] [Google Scholar]
- 31. Iyer V., Tran T. M., Foster E., Dai W., Clark R. B., Knoll B. J. (2006) Differential phosphorylation and dephosphorylation of β2-adrenoceptor sites Ser262 and Ser355,356. Br. J. Pharmacol. 147, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lohse M. J., Benovic J. L., Caron M. G., Lefkowitz R. J. (1990) Multiple pathways of rapid β2-adrenergic receptor desensitization. Delineation with specific inhibitors. J. Biol. Chem. 265, 3202–3211 [PubMed] [Google Scholar]
- 33. Hausdorff W. P., Lohse M. J., Bouvier M., Liggett S. B., Caron M. G., Lefkowitz R. J. (1990) Two kinases mediate agonist-dependent phosphorylation and desensitization of the β2-adrenergic receptor. Symp. Soc. Exp. Biol. 44, 225–240 [PubMed] [Google Scholar]
- 34. Isogaya M., Sugimoto Y., Tanimura R., Tanaka R., Kikkawa H., Nagao T., Kurose H. (1999) Binding pockets of the β1- and β2-adrenergic receptors for subtype-selective agonists. Mol. Pharmacol. 56, 875–885 [DOI] [PubMed] [Google Scholar]
- 35. Kim T. H., Chung K. Y., Manglik A., Hansen A. L., Dror R. O., Mildorf T. J., Shaw D. E., Kobilka B. K., Prosser R. S. (2013) The role of ligands on the equilibria between functional states of a G protein-coupled receptor. J. Am. Chem. Soc. 135, 9465–9474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tschammer N., Bollinger S., Kenakin T., Gmeiner P. (2011) Histidine 6.55 is a major determinant of ligand-biased signaling in dopamine D2L receptor. Mol. Pharmacol. 79, 575–585 [DOI] [PubMed] [Google Scholar]
- 37. Fowler J. C., Bhattacharya S., Urban J. D., Vaidehi N., Mailman R. B. (2012) Receptor conformations involved in dopamine D2L receptor functional selectivity induced by selected transmembrane-5 serine mutations. Mol. Pharmacol. 81, 820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bock A., Merten N., Schrage R., Dallanoce C., Bätz J., Klöckner J., Schmitz J., Matera C., Simon K., Kebig A., Peters L., Müller A., Schrobang-Ley J., Tränkle C., Hoffmann C., De Amici M., Holzgrabe U., Kostenis E., Mohr K. (2012) The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 3, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dror R. O., Pan A. C., Arlow D. H., Borhani D. W., Maragakis P., Shan Y., Xu H., Shaw D. E. (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 108, 13118–13123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jäger D., Schmalenbach C., Prilla S., Schrobang J., Kebig A., Sennwitz M., Heller E., Tränkle C., Holzgrabe U., Höltje H. D., Mohr K. (2007) Allosteric small molecules unveil a role of an extracellular E2/transmembrane helix 7 junction for G protein-coupled receptor activation. J. Biol. Chem. 282, 34968–34976 [DOI] [PubMed] [Google Scholar]
- 41. Kobilka B. K. (2011) Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 32, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ring A. M., Manglik A., Kruse A. C., Enos M. D., Weis W. I., Garcia K. C., Kobilka B. K. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang S. J., Cheng H., Zhou Y. Y., Wang D. J., Zhu W., Ziman B., Spurgoen H., Lefkowitz R. J., Lakatta E. G., Koch W. J., Xiao R. P. (2000) Inhibition of spontaneous β2-adrenergic activation rescues β1-adrenergic contractile response in cardiomyocytes overexpressing β2-adrenoceptor. J. Biol. Chem. 275, 21773–21779 [DOI] [PubMed] [Google Scholar]
- 44. Kahsai A. W., Xiao K., Rajagopal S., Ahn S., Shukla A. K., Sun J., Oas T. G., Lefkowitz R. J. (2011) Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat. Chem. Biol. 7, 692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. West G. M., Chien E. Y., Katritch V., Gatchalian J., Chalmers M. J., Stevens R. C., Griffin P. R. (2011) Ligand-dependent perturbation of the conformational ensemble for the GPCR β2 adrenergic receptor revealed by HDX. Structure 19, 1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu J. J., Horst R., Katritch V., Stevens R. C., Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malik R. U., Ritt M., DeVree B. T., Neubig R. R., Sunahara R. K., Sivaramakrishnan S. (2013) Detection of G protein-selective G protein-coupled receptor (GPCR) conformations in live cells. J. Biol. Chem. 288, 17167–17178 [DOI] [PMC free article] [PubMed] [Google Scholar]