

Background: The isolation of capsular polysaccharides from pathogenic bacteria for vaccine production is cost-intensive.
Results: We describe the cloning, recombinant expression, and functional characterization of three enzymes from Neisseria meningitidis serogroup A that facilitate in vitro synthesis of the capsule polymer.
Conclusion: The study presents a novel basis for efficient vaccine production.
Significance: Economic vaccine production is prerequisite to combat meningococcal diseases.
Keywords: Acetyltransferase, Enzyme Catalysis, Gram-negative Bacteria, Nuclear Magnetic Resonance (NMR), Polysaccharide, Recombinant Protein Expression, Vaccine Development, Neisseria meningitidis Serogroup A, Capsule Polymerase, Epimerase
Abstract
The human pathogen Neisseria meningitidis (Nm) is a leading cause of bacterial meningitis and sepsis globally. A major virulence factor of Nm is the capsular polysaccharide (CPS), which in Nm serogroup A consists of N-acetyl-mannosamine-1-phosphate units linked together by phosphodiester linkages [→6)-α-d-ManNAc-(1→OPO3−→]n. Acetylation in O-3 (to a minor extent in O-4) position results in immunologically active polymer. In the capsule gene cluster (cps) of Nm, region A contains the genetic information for CPSA biosynthesis. Thereby the open reading frames csaA, -B, and -C are thought to encode the UDP-N-acetyl-d-glucosamine-2-epimerase, poly-ManNAc-1-phosphate-transferase, and O-acetyltransferase, respectively. With the aim to use a minimal number of recombinant enzymes to produce immunologically active CPSA, we cloned the genes csaA, csaB, and csaC and functionally characterized the purified recombinant proteins. If recombinant CsaA and CsaB were combined in one reaction tube, priming CPSA-oligosaccharides were efficiently elongated with UDP-GlcNAc as the donor substrate, confirming that CsaA is the functional UDP-N-acetyl-d-glucosamine-2-epimerase and CsaB the functional poly-ManNAc-1-phosphate-transferase. Subsequently, CsaB was shown to transfer ManNAc-1P onto O-6 of the non-reducing end sugar of priming oligosaccharides, to prefer non-O-acetylated over O-acetylated primers, and to efficiently elongate the dimer of ManNAc-1-phosphate. The in vitro synthesized CPSA was purified, O-acetylated with recombinant CsaC, and proven to be identical to the natural CPSA by 1H NMR, 31P NMR, and immunoblotting. If all three enzymes and their substrates were combined in a one-pot reaction, nature identical CPSA was obtained. These data provide the basis for the development of novel vaccine production protocols.
Introduction
NmA4 is the major cause of meningococcal disease in the African meningitis belt. Besides seasonal epidemics that occur with almost annual frequency, NmA has been the cause of severe pandemics in the last century (1, 2). A major virulence factor of Nm is the negatively charged capsular polysaccharide (CPS). CPSA consists of N-acetyl-mannosamine-1-phosphate units linked by phosphodiester linkages to give the polymer [→6)-α-d-ManNAc-(1→OPO3−→]n (3). Of note, the six most virulent Nm serogroups (NmA, -B, -C, -W, -Y, and -X) bear negative CPSs. Negative charge in CPSA and CPSX is due to the phosphodiester group, whereas negative charge in CPSB, -C, -W, and -Y results from the incorporation of sialic acid (2).
These Nm CPSs are immunogenic (except CPSB, which is identical to polysialic acid in the human host) and cause the production of antibodies that are bactericidal in the presence of complement (1, 4). In fact, this early observation has made the use of polysaccharide-protein conjugates the gold standard in the development of vaccines against Nm strains. A number of mono- and tetravalent (the latter comprising serogroups A, C, W, and Y) conjugate vaccines against Nm have been licensed (5).
Crucial to the success of vaccination programs in the sub-Saharan meningitis belt is the provision of safe high quality vaccines. MenAfriVac®, a conjugate vaccine with CPSA coupled to tetanus toxoid as carrier protein, has been specifically designed to address these needs (6). With a cost of under 50 cents per dose (7), mass vaccination campaigns were possible in Burkina Faso, Mali, and Niger and installed herd immunity (8–10) protecting not only vaccinated but also non-vaccinated individuals and young children (11).
Recent progress made with the cloning and functional expression of capsule polymerases (CPs) (12–15) and pioneering studies that demonstrate the suitability of recombinant enzymes for the in vitro production of CPSs (16, 17) have opened new perspectives for the economic and safe production of conjugate vaccines. The goal of our study, therefore, was to isolate the minimal number of enzymes needed for in vitro synthesis of immunologically active CPSA and to pioneer protocols for the use of recombinant enzymes in CPSA production chains. As the sugar building block UDP-ManNAc is commercially not available, it was clear from the start that establishing a successful production chain requires the in situ synthesis of UDP-ManNAc from cheap UDP-GlcNAc. Moreover, as the immunogenicity of CPSA depends on O-acetylation (18), an O-acetyltransferase capable to perform this modification was necessary.
The chromosomal locus cps (for capsular polysaccharide synthesis) contains the genetic information for CPS synthesis, modification, and surface transport. The locus is sub-structured into six regions: A–D, D′, and E (Fig. 1A). The sequences encoded in region A are serogroup-specific and encode among other things the polymerases responsible for CPS synthesis (19). Regions B and C are highly conserved and encode the proteins necessary for export and assembly of the polysaccharide on the cell surface. In NmA, region A comprises four open reading frames (ORF) csaA, -B, -C, and -D (previously designated sacA-D or mynA-D). Using insertion-mutagenesis Swartley et al. (20) demonstrated that each of these genes is involved in the production of the NmA capsule. In a later study the gene product encoded in csaC was shown to be an acetyltransferase with specificity for the O-3 and O-4 positions in ManNAc (21) (Fig. 1B). Based on their nucleotide and predicted amino acid sequence, csaA was presumed to encode a UDP-N-acetyl-d-glucosamine-2-epimerase and csaB to encode a capsule polymerase (20) (Fig. 1B). Additional evidence that the product of csaB is in fact the NmA-specific capsule polymerase arose from the demonstration that the protein is part of the stealth family comprising exclusively d-hexose-1-phosphate transferases (22).
FIGURE 1.

The capsule biosynthesis gene cluster. A, schematic representation of the chromosomal locus (cps) of NmA. Products of genes forming region A are involved in the synthesis of the capsule polysaccharide and are serogroup-specific. For more information, see the Introduction (adapted from Harrison et al. (19)). B, reactions catalyzed by the gene products of csaA, csaB, and csaC (1). The putative UDP-N-acetyl-d-glucosamine-2-epimerase CsaA catalyzes the epimerization of UDP-GlcNAc to UDP-ManNAc (2). The putative capsule polymerase CsaB transfers ManNAc-1P from the resulting UDP-N-acetyl-mannosamine onto the non-reducing end of the growing CPSA (3). CsaC O-acetylates CPSA at O-3 or O-4 in the presence of the acetyl-donor acetyl-CoA.
Here we describe the molecular cloning of the genes csaA, csaB, and csaC from NmA, the production of recombinant proteins, and the characterization of their functional properties. Testing a series of synthetic primer compounds, a ManNAc dimer linked together by phosphodiester linkages and carrying a phosphodiester at the reducing end was found to be the minimal acceptor structure. This artificial primer as well as oligosaccharide primers isolated from natural sources was preferentially used by CsaB if presented in non-O-acetylated form. Using a two-step production protocol, O-acetylated CPSA was synthesized by enzyme-catalyzed reactions and purified to homogeneity. Identity with the natural polymer was confirmed by 1H and 31P NMR and immunoblotting.
EXPERIMENTAL PROCEDURES
General Cloning
The genomic DNA isolated from Nm strain Z2491 was a kind gift from Dr. Heike Claus (Institute for Hygiene and Microbiology, University of Würzburg). The csaB sequence was codon-optimized for use in Escherichia coli BL21(DE3) using the Gene Designer software package (DNA 2.0) (23) and the codon frequency tables published by Welch et al. (24). The mean codon frequency for each amino acid was calculated from the codon frequency tables FreqA and FreqB (24), and the resulting codon frequency table was used as the template for the in silico generation of csaBco. csaBco flanked 5′ by a BamHI site and 3′ by a XhoI site was synthesized from Eurofins MWG Operon. All other csaA-C sequences described herein were amplified by polymerase chain reaction (PCR) using the primers shown in Table 1 and genomic DNA from Nm strain Z2491 or csaBco as template. PCR products were cloned via the restriction sites shown in Table 1 into the corresponding sites of the vector pET22b-Strep (25) driving the expression of recombinant proteins under the control of the T7 promoter. PCR products digested with BglII were cloned into the BamHI site of pET22b-Strep.
TABLE 1.
Primers used in this study
Restriction sites are highlighted in bold.
| Primer pair | Resulting construct |
|---|---|
| GCGGATCCAAAGTCTTAACCGTCTTTGGC | StrepII-CsaA-His6 |
| CCGCTCGAGTCTATTCTTTAATAAAGTTTCTACA | |
| GCAGATCTTTTATACTTAATAACAGAAAATGGC | StrepII-CsaB-His6 |
| CCGCTCGAGTTTCTCAAATGATGATGGTAATG | |
| CCGCTCGAGTTTCTCAAATGATGATGGTAATG | StrepII-Δ69-CsaB-His6 |
| GCAGATCTATGTTAATTCCTATTAATTTTTTTAA | |
| CCGCTCGAGTTTCTCAAATGATGATGGTAATG | Δ69-CsaB-His6 |
| GCATCTCATATGTTAATTCCTATTAATTTTTTTTAATTT | |
| GCATCTCATATGCTGATCCCGATCAATTTCTTT | Δ69-CsaBCo-His6 |
| CCGCTCGAGTTTCTCGAAGGAGCTCGGC | |
| CCGCTCGAGTATATTTTGGATTATGGT | StrepII-CsaC-His6 |
| GCGGATCCTTATCTAATTTAAAAACAGG |
Expression and Purification of Recombinant CsaA, CsaB, and CsaC
Freshly transformed E. coli BL21(DE3) were grown at 15 °C in PowerBroth medium for 18 h. At an optical density of A600 = 1.0 protein expression was induced by the addition of 0.1 mm isopropyl-β-d-1-thiogalactopyranoside and allowed to proceed for a period of 20 h. In test expressions, 0.2 ml of culture-volume were pelleted with 16,000 × g for 1 min. Cell pellets were lysed with 0.1 ml of lysis buffer (50 mm Tris, pH 8.0, 2 mm EDTA, 0.1 mg/ml lysozyme). The lysis was intensified by 3 cycles of sonication (Branson sonifier 450, 100% amplitude) interrupted by 3 min of cooling on ice. Soluble and insoluble fractions were separated by centrifugation (16,000 × g, 30 min, 4 °C), and the supernatant was mixed (1:1) with Laemmli buffer and used for PAGE as described below.
For protein purification, pellets from 125 ml of expression culture were pelleted by centrifugation (6000 × g, 10 min, 4 °C). After a washing step with PBS, cells were resuspended in 7.5 ml of binding buffer (50 mm Tris, pH 8.0, 300 mm NaCl) complemented with 40 μg/ml bestatin (Sigma), 1 μg/ml pepstatin (AppliChem), 100 μm PMSF (Stratagene) and sonicated (Branson Digital Sonifier, 50% amplitude, 8 × 30s, interrupted by cooling on ice). After centrifugation at 27,000 × g for 30 min, the soluble fractions were directly loaded onto HisTrap columns (GE Healthcare) to enrich the recombinant proteins by immobilized metal ion affinity chromatography. Columns were washed with binding buffer (50 mm Tris, pH 8.0, 300 mm NaCl), and proteins were eluted in step gradients using 10, 30, 50, and 100% elution buffer (binding buffer containing 500 mm imidazole). Fractions containing recombinant protein were pooled, and the buffer was changed to storage buffer (50 mm Tris, pH 8.0, 50 mm NaCl for CsaA/CsaB; 50 mm Hepes, pH 7.05, 100 mm NaCl, 5 mm MgCl2, and 1 mm EDTA for CsaC) using the HiPrep 26/10 desalting column (GE Healthcare). Isolated proteins were concentrated using Amicon Ultra centrifugal devices (Millipore 30 MWCO). After separation into aliquots, samples were snap-frozen in liquid nitrogen and stored at −80 °C.
SDS-PAGE and Immunoblotting
SDS-PAGE was performed under reducing conditions using 2.5% (v/v) β-mercaptoethanol and 1.5% (w/v) SDS. Proteins were stained using Roti-Blue (Carl Roth GmbH) according to the manufacturer's guidelines. For Western blot analysis samples and standard proteins were blotted onto PVDF membranes (Millipore). His-tagged proteins were detected with 0.5 μg/ml anti-penta-His antibody (Qiagen) and goat anti-mouse IR680 or goat anti-mouse IR800 antibody (LI-COR) as second antibody. Second antibodies were used in a 1:20,000 dilution.
Preparation of CPSA Oligosaccharides
CPSA oligosaccharide samples with an averaged degree of polymerization (avDP) of 6 and 15, respectively, were generated by acidic hydrolysis of long CPSA chains isolated from bacterial cultures (CPSAn). Solutions containing 2.5 mg/ml CPSA in sodium acetate buffer (50 mm sodium acetate, pH 4.8) were incubated at 73 °C for 6 h, and 2 pool fractions (avDP 6 and 15, respectively) were purified by anionic exchange chromatography (Q-Sepharose column, GE Healthcare) using a sodium chloride gradient. The avDP and the dispersion of saccharide chains was determined by 31P NMR and high performance anionic exchange chromatography-pulsed amperometric detection (HPAEC-PAD) analysis following an established protocol (26). If used in enzymatic reactions, hydrolyzed CPSA (CPSAhyd) was dephosphorylated (CPSAhyd-deP) using acid phosphatase (Sigma) according to the manufacturer's guidelines.
Chemical Synthesis of CsaB Acceptors
A short summary of the synthesis of ManNAc and ManNAc derivatives as well as of ManNAc disaccharide units linked together by phosphodiester linkages is provided in the supplemental schemes S1 and S2. A manuscript describing the detailed chemical synthesis and the characterization of these compounds is under preparation.5
Activity Testing of CsaA/CsaB by Use of a Radioactive Assay System
CsaA/CsaB activity was analyzed using an adaptation of a radioactive incorporation assay previously described for the N-acetylglucosamine-1-phosphate transferase from NmX (17). Briefly, assays were carried out with 5 μl of the soluble fractions of bacterial lysates expressing either recombinant CsaB or CsaA (see Fig. 2C) or with purified and epitope-tagged proteins (112 pmol of StrepII-CsaA-His6; 88 pmol of Δ69CsaBco-His6) in a total volume of 25 μl of assay buffer (50 mm Tris pH 8.0 or various pH for determination of the pH optimum). Divalent cations were added from stock solutions. The reaction was primed with 5 ng of avDP15 and started by the addition of 0.05 μmol of UDP-GlcNAc (Calbiochem) containing 0.05 μCi of UDP-[14C]GlcNAc (American Radiolabeled Chemicals). Samples were incubated at 37 °C, and 5-μl aliquots were spotted onto Whatman 3MM Chr paper after 0, 5, 10, and 30 min. After descending paper chromatography, the chromatographically immobile 14C-labled CPSA was quantified by scintillation counting.
FIGURE 2.

Production of recombinant enzymes. A, Coomassie-stained SDS-PAGE of purified StrepII-CsaA-His6 (left panel) and StrepII-CsaC-His6 (right panel). B, to select the construct most suited for the production of active recombinant CsaB, the wild type and a codon-optimized (CsaBco) version of the CsaB sequence were cloned (full-length or after N-terminal truncation, Δ69) to produce proteins with tags on both (StrepII and His6) or only one end (His6), as indicated. Transformed bacteria were lysed, separated into soluble (s) and insoluble (i) fractions, and fractions were separately run on PAGE. After transfer onto nitrocellulose, the blot was developed with an anti-penta-His antibody. m, marker. C, soluble fractions were used to measure CsaB activity in a radioactive incorporation assay. D, Coomassie-stained gel demonstrating the purification result for Δ69-CsaBco-His6 (IMAC, immobilized metal ion affinity chromatography; SEC, size exclusion chromatography).
Activity Testing of CsaA/CsaB by Use of a Multienzyme Spectrophotometric Assay
1.2 μm CsaA and 1 μm CsaB were assayed in the presence of 0.25 mm UDP-GlcNAc (Calbiochem), 20 mm MgCl2, and 50 mm Tris, pH 8.0, in a total volume of 100 μl. The consumption of UDP-GlcNAc was coupled to nicotinamide adenine dinucleotide (NADH) consumption using the following enzymes/substrates: 0.25 mm adenosine triphosphate (ATP, Roche Applied Science), 1 mm phosphoenolpyruvate (ABCR), 0.3 mm NADH (Roche Applied Science), 9–15 units/ml pyruvate kinase, 13.5–21 units/ml lactic dehydrogenase (PK/LDH mix Sigma), and 0.05 mg/ml nucleoside monophosphate kinase (Roche Applied Science). Absorption was measured at 340 nm every 10 s for 30 min using a Biotek EL 808 96-well plate reader.
Physicochemical Analysis of CPSAiv
To produce sufficient CPSA (CPSAiv) for PAGE and NMR analyses, 0.84 nmol (1.2 μm final) of StrepII-CsaA-His6 and 37.5 pmol (50 nm final) of Δ69CsaBco-His6 in reaction buffer (50 mm Tris, pH 8.0, 20 mm MgCl2) were incubated with 5 mm UDP-GlcNAc and 6.8 μg of CPSAhyd of avDP6 after de-O-acetylation and dephosphorylation (CPSAhyd(deOAc)-deP). The total reaction volume was adjusted to 750 μl. In control 1 shown in Fig. 6B, StrepII-CsaA-His6 was also used at 50 nm. Reactions as well as control samples (containing reactants as indicated in Fig. 6) were incubated overnight at 37 °C. 5 μl of each sample were then used for separation on high percentage (25%) PAGE and visualized by a combined Alcian blue/silver staining procedure (27).
FIGURE 6.

In vitro synthesis of CPSA. A, products synthesized in the CsaA/CsaB reaction in the presence of UDP-GlcNAc and CPSAhyd(deOAc) of avDP6 were analyzed by high percentage PAGE and a combined Alcian blue/silver staining. Long chains were produced in the presence of all reactants (reaction) and, although in small amounts, also in control 1 where no priming oligosaccharides were added. The production of long CPSA chains in control 1 argues for the capacity of CsaB to start the polymerization de novo. B, 31P NMR analyses carried out with the reaction, and controls 1 and 4 show signals characteristic for the phosphodiester linkages of CPSA and byproducts of the reaction. C, the 1H NMR analysis of control 4 demonstrates that CsaA catalyzes the UDP-GlcNAc/UDP-ManNAc epimerization via the intermediate 2-acetamidoglucal. D, HPLC analysis of reaction products obtained with variant CsaA:CsaB ratios, as indicated. This experiment clearly showed that UDP formation is prevented if the CsaB concentration is equal to or higher than the concentration of CsaA. UMP, UDP, and UDP-GlcNAc/UDP-ManNAc were detected at 280 nm and CPSA at 214 nm.
The residual sample was freeze-dried, solubilized in 0.75 ml of deuterium oxide (D2O, 99.9% atom D; Aldrich) to give a concentration of 0.5–1 mg/ml saccharide and used for product characterization by NMR. All the 1H and 31P NMR experiments were recorded as previously described (17).
HPLC-anion exchange chromatography (AEC) was performed on a Prominence UFLC-XR (Shimadzu) equipped with a CarboPac PA-100 column (2 × 250 mm, Dionex). Samples were separated as described by Keys et al. (28) with the minor adjustment that H2O and 1 m NaCl were used as mobile phases M1 andM2, respectively. 5 μl of the samples were loaded for the detection of nucleotides at 280 nm and 50 μl for the detection of CPSA at 214 nm. Products were separated using an elution gradient consisting of a −2 curved gradient from 0 to 30% M2 over 4 min followed by a linear gradient from 30 to 84% M2 over 33 min. Enzyme concentrations were used as indicated in Fig. 6. All other reactants were used in the amounts described above.
Analysis of 2-Acetamidoglucal
Assignments of 1H NMR spectrum were in agreement with those reported in literature (29). 1H NMR (D2O, 400 MHz): δ = 6.68 (d, 1 H, J1,2 1.0, H-1), 4.25 (dd, H 1, J3,4 6.5 Hz, H-3), 3.99 (dt, 1 H, J4,5 8.4, J5,6a = J5,6b 4.2 Hz, 6.5 Hz, H-5), 3.86 (d, 2 H, H-6), 3.77 (dd, 1 H, H-4), 2.05 (s, 3 H, CH3CO). Significant signals from 13C NMR (D2O, 100 MHz): δ = 141.47 (C-1), 78.70 (C-5), 68.70 (C-3), 68.36 (C-4), 59.84 (C-6), 21.84 (2 × CH3CO).
In Vitro Synthesis, Purification, and Immunological Analysis of CPSAiv and CPSAiv(OAc)
To generate CPSAiv, 10 nmol of CsaA and 16 nmol of CsaB were incubated overnight at 37 °C in reaction buffer (50 mm Tris, pH 8.0, 20 mm MgCl2) with 10 mm UDP-GlcNAc in a total volume of 9 ml. The reaction was primed with 1 μg of CPSAhyd(deOAc)-deP of avDP6. Acetylation of 1 mg of CPSAiv was performed for 4 h at 37 °C in the presence of 1.2 nmol of CsaC and 14 mm acetyl-CoA (Sigma) in a total volume of 0.5 ml of acetylation buffer (25 mm Tris, pH 7.5, 50 mm NaCl). Both CPSAiv and CPSAiv(OAc) were purified via AEC using a Mono Q HR5/5 column (GE Healthcare) at a flow rate of 1 ml/min and a linear sodium chloride gradient. CPS containing fractions eluting at 540 mm NaCl were pooled, dialyzed (ZelluTrans, Roth, 1 kDa MWCO) against water, and freeze-dried for further analysis. For dot blot analyses, small aliquots of the purified CPSAiv and CPSAiv(OAc) were spotted onto nitrocellulose (Whatman) and incubated with mAb 932 specifically directed against CPSAOAc (mAb 935 was generated in the laboratory of Prof. Dr. D. Bitter-Suermann, Hannover Medical School, Institute for Medical Microbiology, and was kindly provided for this study) in a 1:10,000 dilution. Dot blots were developed with goat anti-mouse IR800 antibody (LI-COR) in a 1:20,000 dilution.
RESULTS
Cloning and Expression of csaA, csaB, and csaC; Production of Recombinant Proteins
Because previous analyses carried out on these genes provided strong evidence that csaA, csaB and csaC encode the UDP-GlcNAc-epimerase, the poly-ManNAc-1-phosphate-transferase (20), and the O-acetyltransferase (21), respectively, primers were constructed (see “Experimental Procedures”) to amplify these ORFs. The genomic DNA isolated from Nm strain Z2491 was used as a template. Obtained PCR products were cloned into the pET22b-Strep vector (25), allowing the expression of recombinant proteins with N-terminal StrepII- and/or C-terminal His6 tag. After transformation into BL21(DE3) and induction of protein expression (see “Experimental Procedures”), the distribution of recombinant proteins between the soluble (s) and insoluble (i) fraction of bacterial lysates was analyzed by Western blotting against the affinity tags. The recombinant epitope tagged forms of CsaA and CsaC appeared mostly in the soluble fraction and could be purified directly from the bacterial lysates. CsaA was purified by immobilized metal ion affinity chromatography followed by a desalting step and yielded 40 mg of protein/liter expression culture. Although some additional faint bands were visible in Coomassie-stained SDS-PAGE (Fig. 2A), a protein fraction highly enriched in CsaA was obtained. CsaC was purified following the protocol described by Gudlavalleti et al. (21) and yielded 96 mg of homogenously pure protein/liter of culture (Fig. 2A).
Similarly, StrepII-CsaB-His6, encoding the putative poly-ManNAc-1-phosphate-transferase, was well expressed, and the excess of the construct appeared in the soluble fraction. However, the major product revealed with the anti-penta-His antibody in Western blot migrated with an apparent molecular mass of 50 kDa, strongly deviating from the calculated molecular mass of 67 kDa (Fig. 2B, left lanes). Because faint signals with molecular masses of >50 kDa were additionally displayed with the anti-penta-His antibody, we concluded that StrepII-CsaB-His6 is either prone to N-terminal degradation or translated from an alternative start codon. Consequently, we reinvestigated the NmA genome with bioinformatics techniques. Indeed, two of the used gene prediction softwares (GeNmark and GeNmarkS; Refs. 30 and 31) retrieved an additional ATG (starting with position 183528 of the NmA genome (NC_003116.1)). In PRODIGAL (32), the prediction for this second start codon was comparable with the published start codon (base no. 183321; Ref. 33).
To investigate if translation from the alternative ATG leads to a stable protein, the corresponding truncation Δ69CsaB was cloned with (StrepII-Δ69-CsaB-His6) and without (Δ69-CsaB-His6) the N-terminal StrepII-tag. Test expressions in BL21(DE3) demonstrated the occurrence of proteins of the expected molecular masses, but in repeated experiments the level of expressed protein was significantly lower than for the full-length construct. Moreover, to our surprise, the construct cloned with free N terminus (Δ69-CsaB-His6) was routinely higher expressed than the StrepII-tagged construct (Fig. 2B). Because rare codons that exist in the CsaB sequence may negatively impact protein expression, csaB was codon-optimized using the Gene Designer (DNA 2.0) software (23) and the codon frequency tables published by Welch et al. (24). Expression of the resulting optimized gene (see supplemental data) was tested with the constructs StrepII-CsaBco-His6 and Δ69-CsaBco-His6. Although increased degradation and concomitantly reduced expression was seen for StrepII-CsaBco-His6, Δ69-CsaBco-His6 was well expressed, and no degradation was detectable in Western blot with the anti-penta-His antibody (Fig. 2B).
Consecutively, enzymatic activity within the soluble fractions of the bacterial lysates was determined with a radioactive incorporation assay previously developed for the poly-GlcNAc-1-phosphate-transferase from NmX (17). The fractions containing the recombinant CsaB variants were tested in the presence of CsaA and UDP-[14C]GlcNAc. In accordance with the levels of expressed protein (Fig. 2B), StrepII-CsaB-His6 and Δ69-CsaBco-His6 showed identical activity profiles (Fig. 2C). Based on these results the protein variant Δ69-CsaBco-His6 was chosen for further experiments. The protein was purified from the soluble fraction of transformed BL21(DE3) by immobilized metal ion affinity chromatography and size exclusion chromatography, yielding 60 mg of highly pure protein from 1 liter of bacterial culture (Fig. 2D).
Optimization of Test Conditions and Characterization of CsaB Substrates
As the donor sugar (UDP-ManNAc) used by CsaB must be produced in situ in the epimerase reaction catalyzed by CsaA, the optimization of test conditions needed the presence of both enzymes. As for CPs of other Nm strains, a hydrolysate of CPSA (CPSAhyd) was used to prime the reaction in the presence of CsaA and its substrate UDP-[14C]GlcNAc. Initial studies carried out to evaluate pH and salt conditions showed the best activity values in the presence of 10–20 mm MgCl2 and a pH between 8.0 and 8.5. Replacement of Mg2+ by Ca2+ or Mn2+ inactivated the enzyme. Although these results were similar to what we had seen with CsxA (CP of NmX), the CsaA/CsaB reaction, in contrast to the CsxA reaction, did not show sensitivity to DTT (up to 2 mm were tested).
The natural CPSA is O-acetylated in positions 3 and to a minor extent also in position 4 of ManNAc (21, 34, 35). We, therefore, questioned whether Δ69-CsaBco-His6 recognizes and elongates acetylated and non-acetylated CPSAhyd with the same efficiency. Hydrolysis of CPSA was carried out before and after base treatment to obtain O-acetylated (CPSAhyd(OAc)) and de-O-acetylated (CPSAhyd(deOAc)) shorter saccharide chains. Knowing that CPSA hydrolysis results in a large distribution of saccharide chain lengths (ranging in size between DP 1 and 70), AEC was used to separate two fractions, the avDP6 (comprising DP1-DP10) and avDP15 (comprising DP10–DP70). Both fractions were used to prime the enzyme reactions as indicated in the subsequent experimental steps.
To quantitatively assess the enzyme reactions, we adapted a spectrophotometric assay previously designed to analyze the CP from NmB (12). In the multienzyme assay shown in Fig. 3, the Δ69-CsaBco-His6 catalyzed product formation is coupled to NADH consumption, which can be continuously monitored at 340 nm. Using this assay the activity of Δ69-CsaBco-His6 was determined with CPSAhyd fractions (CPSAhyd(OAc) and CPSAhyd(deOAc)) of avDP6 and avDP15 (Fig. 4). Moreover, because it was demonstrated that the hydroxyl groups at position 6 (C6-OH) on the non-reducing end sugar in CPSAhyd is blocked by phosphomonoesters (36), both fractions were additionally tested after treatment with acid phosphatase (deP) to remove this group. Independent of the size of the primers used to start the reaction, the native acetylated oligomers (CPSAhyd(OAc) of avDP6 and avDP15) were found to be poor acceptors, but activity increased considerably after removal of acetyl groups and even further after release of the capping phosphate residue, making CPSAhyd(deOAc)-deP the most efficient acceptors. The size of the priming polymers was not significant for enzymatic activity (Fig. 4; compare avDP6 and avDP15). The obtained results allowed the conclusion that the chain elongation by CsaB proceeds via the non-reducing end and by transfer of ManNAc-1P onto C6-OH groups. Furthermore, because CPSAhyd(deOAc) was a better acceptor than CPSAhyd(OAc), it is likely that O-acetylation takes place after polymer synthesis.
FIGURE 3.
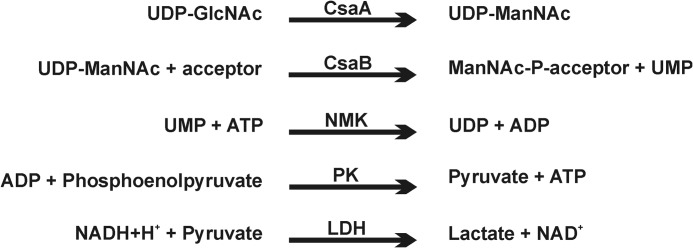
Schematic representation of the multi-enzyme assay used to continuously follow CsaB activity. NMK, nucleoside monophosphate kinase; PK, pyruvate kinase; LDH, lactic dehydrogenase.
FIGURE 4.

Acceptor recognition by CsaB. A, the chemical properties of the primers used to test the acceptor preference of Δ69-CsaBco-His6 are displayed. B, Δ69-CsaBco-His6 activity was followed using the spectrophotometric assay in the presence of CPSAhyd of avDP6 and avDP15 in either native O-acetylated form (CPSAhyd(OAc)) or after de-O-acetylation (CPSAhyd(deOAc)). Because earlier studies showed that the non-reducing ends in CPSAhyd are phosphorylated, samples were additionally tested before and after phosphatase treatment (deP). Samples designated with (deOAc)-deP were the subject of de-acetylation and de-phosphorylation.
Because no information on the minimal length of the priming acceptor for CsaB could be derived from the CPSAhyd fractions, we used well characterized synthetic compounds to interrogate this question. The compounds synthesized are shown in Fig. 5 and varied not only in length but also with respect to O-acetylation (compounds 2, 4, 6 were 3-O-acetylated) and reducing end modifications (37–39). In compounds 1, 2, 5, and 6 the reducing ends were occupied by an n-decyl-phosphate ester, whereas a methyl group (OMe) was present in compounds 3 and 4. The Δ69-CsaBco-His6 activity did not go beyond background (no acceptor) with compounds 1-4 but, intriguingly, steeply increased with disaccharides carrying an n-decyl-phosphate ester at the reducing end (compounds 5 and 6) (Fig. 5). With the non-O-acetylated compound 5, activity values similar to those obtained with the optimized acceptor CPSAhyd(deOAc)-deP were measured. In line with the above data (Fig. 4), O-acetylation of compound 5 (resulting in compound 6) reduced the quality of the acceptor. Based on these data, the minimal acceptor recognized by Δ69-CsaBco-His6 could be defined as the dimer of ManNAc units linked together by phosphodiester linkages. The presence of a phosphodiester at the reducing end seems obligatory, because compounds ending with OMe groups do not work as acceptor substrates.
FIGURE 5.

Determination of the minimal CsaB acceptor. Derivatives of ManNAc ending at the reducing end with a methyl group (compounds 3, 4) or a phospho-n-decyl-ester (compounds 1, 2, 5, 6) were used to prime the Δ69-CsaBco-His6 reaction in the continuous spectrophotometric assay. The dimer of ManNAc-1P carrying a phosphodiester at the reducing end was identified as minimal acceptor. Importantly, similar to the natural oligosaccharides the acceptor quality dropped by roughly 20% if the synthetic acceptor was O-acetylated (compare compounds 5 and 6).
In Vitro Synthesis of CPSA Chains
To analyze if long CPSA chains can be produced with the recombinant enzymes, test reactions were carried out in the presence of StrepII-CsaA-His6, Δ69-CsaBco-His6, and CPSAhyd(deOAc)-deP of avDP6 as the priming oligosaccharide. Reactions were started by the addition of UDP-GlcNAc. Control reactions (Fig. 6A, controls 1–4), in which components were omitted as indicated, were carried out in parallel. After overnight incubation, samples were loaded onto high percentage PAGE and developed by Alcian blue/silver staining. CPSAhyd(deOAc)-deP of avDP15 was loaded as the size marker. In the presence of CsaA and CsaB the added oligosaccharide primers were efficiently elongated to long polymer chains (Fig. 6A). 31P NMR showed the phosphodiester signal, which is characteristic for CPSA as well as the signal that indicates the second reaction product UMP (Fig. 6B, reaction). Unexpected were the signals observed at −5.8 ppm and −9.5 ppm, which indicated the formation of UDP. As these signals were most prominent in control 4 (Fig. 6B), with only StrepII-CsaA-His6 and UDP-GlcNAc present, we speculated that UDP is a side product of the epimerase reaction. A 1H NMR analysis carried out with control 4 revealed a signal that, in perfect agreement with published data (29), represented the anomeric proton of 2-acetamidoglucal (Fig. 6C). 13C,1H heteronuclear multiple quantum coherence (see “Experimental Procedures”) NMR analyses allowed us for the first time to assign a 13C spectrum of this intermediate. The complete absence of signals for the anomeric proton of ManNAc (both α and β) confirmed that UDP is an intermediate of the CsaA reaction and not produced by hydrolysis of UDP-ManNAc.
Another interesting observation was the de novo start of CPSA chains by Δ69-CsaBco-His6 if incubated with equimolar CsaA concentrations (50 nm, see “Experimental Procedures”). The in vitro produced CPSA chains (CPSAiv) could be doubtlessly identified with both PAGE and 31P NMR (Fig. 6, A and B, control 1). However, when CsaA was used in excess of CsaB, none of the methods detected any CPSA signal, and the majority of the donor substrate was converted to UDP and 2-acetamidoglucal (data not shown). Based on these data, it can be concluded that the de novo activity is strongly disadvantaged compared with the elongation of oligosaccharide primers. Although the same type of de novo synthesis was shown for the CP of NmX (17), it came as a surprise in the case of CsaB, as the monosaccharide compounds 1 and 2 were not elongated (see Fig. 5).
To reduce UDP-ManNAc hydrolysis and simultaneously guarantee complete incorporation of ManNAc-1P into the product, the ratio CsaA:CsaB was varied as indicated in Fig. 6D. The reactions were carried out as described above with CPSAhyd(deOAc)-deP of avDP6 as primer. After overnight incubation, products were separated by HPLC and recorded at 214 nm (CPSAiv) and 280 nm (UDP and UMP). As long as the concentration of CsaB was equal to or higher than the concentration of CsaA, no hydrolysis of UDP-ManNAc was detectable (Fig. 6D), not even if the enzymes were present in only 50 nm concentration and the donor sugar was not completely consumed. These data convincingly show that in vitro CPSA production with high efficiency is possible with the recombinant enzymes.
With the intention to obtain bio-identical CPSA in milligram amounts, the CsaA/CsaB reaction was up-scaled. Long polymers were purified by AEC using a protocol similar to the one described in Fiebig et al. (17). CPSAiv eluted at 540 mm NaCl clearly separated from all other reaction components (Fig. 7A). 1H NMR spectra, comparatively recorded for CPSAiv and natural CPSA (CPSAn) treated with alkaline to remove the acetyl-group (CPSAn(deOAc)), were in congruence (Fig. 7D). 1 mg of the purified CPSAiv was then used for acetylation with recombinant CsaC. To enable acetylation at both O-3 and O-4, the acetyl donor acetyl-CoA was added in a 2-fold molar excess of ManNAc. After a 4-h reaction step, CPSAiv(OAc) was purified by a second AEC step. The single peak that eluted at 540 mm NaCl (Fig. 7B) contained material that in dot blot analysis was recognized by mAb 932, specifically directed against the O-acetylated form of CPSA (Fig. 7C). Based on the signal intensity of the H-2 in 1H NMR, 88% of the available O-3 groups were acetylated. This value is in perfect agreement with the value obtained for the reference CPSAn (Fig. 7D). However, whereas CPSAn to a minor extent also carried O-4 acetyl groups, this modification was not detectable in CPSAiv(OAc).
FIGURE 7.

Purification and characterization of in vitro synthesized CPSA. A, in vitro synthesized CPSA (CPSAiv) was separated from all contaminating reaction products using anion exchange chromatography with the indicated sodium chloride gradient. B, purified CPSAiv after O-acetylation (CPSAiv(OAc)) was re-purified under the same conditions resulting in a well separated product peak, which in panel C dot blot analyses were recognized by mAb 932. D, corresponding 1H NMR analysis of the produced CPSAiv in comparison to CPSAn (from natural source). Slight variations in chemical shifts between CPSAn and CPSAiv are due to pH variations resulting from different purification protocols.
To determine on the analytical scale if acetylated, bio-identical polymer can be produced in a one-step reaction starting with one substrate and just using the polymerase, we incubated CsaB in the presence of 3-O-acetylated UDP-ManNAc (UDP-3OAcManNAc) and analyzed the reaction products by PAGE and 31P NMR. However, no product signals could be detected by any of the methods (Fig. 8, A and B) and 31P NMR revealed that UDP-3OAcManNAc was not used by CsaB.
FIGURE 8.

A one-pot reaction for the synthesis of immunologically active O-acetylated CPSA. A, UDP-3OAcManNAc was chemically synthesized to test if O-acetylated CPSA could be obtained in a one-step reaction. Analysis of the reaction by high percentage PAGE followed by combined Alcian blue/silver staining did not reveal any product signals (left lane), whereas long CPSA was obtained in the control reaction (right lane). B, 31P NMR analysis of the samples revealed that UDP-3OAcManNAc (top spectrum) was not consumed by CsaB, whereas product signals were detected in the control (bottom spectrum). C, in vitro synthesis of O-acetylated and non-O-acetylated CPSA using all enzymes CsaA-C in a one-pot reaction. To control product formation, substrates and enzymes were added as indicated. After a 2-h incubation, products were displayed on high percentage PAGE by a combined Alcian blue/silver staining. Long chains were synthesized in all reactions containing CsaA, CsaB, and UDP-GlcNAc. D, only products obtained in the reaction where CsaC and acetyl-CoA were present could be detected with mAb 932 specifically directed against the CPSAOAc.
Finally we explored if O-acetylated CPSA can be produced in a one-pot reaction. Therefore, the reaction mixture containing CsaA, CsaB, and UDP-GlcNAc was supplemented with recombinant CsaC and acetyl-CoA. Moreover, control reactions with single compounds missing (see Fig. 8, C and D) were carried out in parallel. After overnight incubation, products were analyzed by Alcian blue/silver-stained high percentage PAGE (Fig. 8C) and immunoblotting with mAb 932 (Fig. 8D). In the presence of all components, a product recognized by mAb 932 was produced (Fig. 8D), indicating that CsaC can acetylate the produced CPSA in situ. The control reactions carried out in this experiment provided clear evidence for the functional nature of CsaA and CsaC being UDP-GlcNAc/UDP-ManNAc epimerase and O-acetyltransferase, respectively. Last, the similarity (size and concentration) of reaction products identified in lanes 1, 4, and 6 (Fig. 8C) indicated that suitable conditions were established for all enzymes in the one-pot-reaction scheme.
DISCUSSION
Of all pathogenic Nm serogroups, NmA has caused the most disastrous epidemics in sub-Saharan Africa. The prevalence of this pathogen provoked an unprecedented endeavor to develop a highly effective and economic vaccine, MenAfriVac® (7). With costs of less than 50 cents per dose, MenAfriVac® enabled mass vaccination campaigns in Burkina Faso, Mali, and Niger (8–10), which installed herd immunity, leading to protection not only for vaccinated but also for non-vaccinated individuals and in particular of young children (11).
All NmA vaccines licensed today are glycoconjugate vaccines coupled to carrier proteins, with CPSA polysaccharides isolated from large scale NmA cultures or oligosaccharides having shorter chain length obtained by acidic hydrolysis (40). To avoid the significant cost and biohazard in association with large scale NmA cultures and pyrogen-free production of polysaccharides, the enzyme-catalyzed in vitro synthesis of CPSA (CPSAiv) would provide an attractive alternative. Toward this goal, we describe in this study the molecular cloning and functional expression of the three enzymes (UDP-GlcNAc-2-epimerase, CsaA; poly-ManNAc-1-phosphate-transferase, CsaB; O-acetyltransferase, CsaC) that are part of the capsular biosynthesis complex in NmA and represent the minimal number of enzymes needed to produce immunologically active CPSAiv(OAc) in vitro starting from economic precursors. Using the well characterized BL21(DE3) strain as expression host, C-terminal His6-tagged and N-terminal StrepII-tagged versions of CsaA and CsaC could be purified in high quality and remarkable quantity (CsaA and CsaC, 40 and 96 mg/liter bacterial culture, respectively). In CsaB a second start codon was identified encoding methionine 70. In the overexpression system the use of this second start codon (construct Δ69-CsaB-His6) generated a stable protein that could be purified in remarkable amounts (60 mg/liter bacterial culture) after its DNA sequence had been optimized for codon usage in BL21(DE3). Whether this second start codon is actually used in the natural environment remains an open question.
When the CsaA/CsaB reaction was carried out under suboptimal conditions (CsaA concentrations were higher than CsaB concentrations), UDP and 2-acetamidoglucal were formed as side products. A similar finding was made by Sala et al. (41), who showed that the E. coli UDP-N-acetylglucosamine-2-epimerase, if present at high concentrations, releases the two intermediates of the epimerization reaction (UDP and 2-acetamidoglucal) into solution. Because E. coli UDP-N-acetylglucosamine-2-epimerase and CsaA share significant sequence similarity, it is reasonable to believe that CsaA uses the same catalytic mechanism. This CsaA side reaction could be completely suppressed if CsaA concentrations were equal or lower than the CsaB concentrations.
Using a two-step protocol (in test reactions even a one-pot reaction; see Fig. 8) O-acetylated CPSA (CPSAiv(OAc)) could be produced in vitro in high purity and at medium scale (1 mg). Similar to the CPSA isolated from natural source (CPSAn) (42), the CPSAiv(OAc) fraction was of high molecular weight, showed 31P NMR and 1H NMR profiles consistent with CPSAn, and was recognized by mAb 932, a standard reagent in the characterization of immunologically active CPSA. Small differences were seen in the acetylation patterns. While the CPSAn reference contained some O-4-acetylation, this modification was not detectable in CPSAiv(OAc). Although this difference at first sight may suggest the existence of a second enzyme with preference for the 4-O position, this interpretation is highly unlikely since csaC-knockouts are completely devoid of O-acetylation (21). Interesting, rather diverse values exist in literature for the relative occurrence of 3-O-Ac, 4-O-Ac, and free hydroxyl groups in CPSAn (70:0:30 (34), 87:8:5 (35), 40:27:33 (21)). To resolve the question of the importance of O-4-acetylation for the immunogenic quality of in vitro produced CPSA, further experimental work is needed.
In the current study we provide clear evidence that the acceptor quality of CPSAhyd increases after removal of the O-acetyl-groups. Consequently, also, the synthetic ManNAc-1P dimer carrying 3-O-Ac-groups (compound 6) was a less suited primer than the respective compound 5 without O-acetyl-groups. Asking if UDP-activated and 3-O-acetylated ManNAc (UDP-3OAcManNAc) may be a donor substrate for CsaB, we chemically synthesized this compound using a modification of a literature procedure for the preparation of UDP-GlcNAc (43) (see the supplemental scheme 3). Remarkably, no insertion of the modified compound was seen, strongly arguing for a highly selective recognition of the donor substrate by CsaB and further emphasizing the hypothesis that O-acetylation in NmA takes place on the synthesized polysaccharide. Of relevance in this context are the previous demonstrations that O-acetylation of sialic acid residues in the CPSs of NmC and E. coli K1 takes place after the polymer has reached a certain length and could not be detected on the donor substrate (44, 45).
In 1999 a study by Ravenscroft et al. (36) demonstrated that acidic hydrolysis of CPSA results in fragments that are capped by phosphate at the non-reducing end. We show in the current study that removal of this capping phosphate steeply increases CsaB activity values (Fig. 4) and thus provide clear evidence that chain elongation proceeds by transfer of ManNAc-1P residues to the non-reducing end of the priming oligosaccharide. In addition, the use of synthetic priming compounds demonstrated that the minimal acceptor for CsaB is the ManNAc-phosphate dimer and that the phosphate group at the reducing end can be extended with rather large chemical groups (n-decyl ester in the compounds tested in this study). Particularly this latter finding is of biotechnological relevance because it provides the perspective that CPSA chains can be built with priming oligosaccharides that carry functional groups ready for conjugation to carrier proteins (5).
Similar to the capsule polymerase of NmX (17), CsaB is also capable of initiating polymerization in the absence of any acceptor. Remarkably, this de novo activity was not altered in the presence of the artificial compounds 1-4. More experimental work is needed to interpret these findings, but based on these results it is tempting to speculate that the de novo reaction involves two UDP-ManNAc residues bound to acceptor and donor site in the enzyme.
In summary, we present data in this study that provide a new basis for the development of efficient and economic protocols (even one-pot-reaction protocols) for the synthesis of highly pure and immunologically active CPSAiv(OAc). This means a large step forward in the combat of epidemics caused by one of the predominant neisserial serogroups NmA.
Supplementary Material
Acknowledgments
We thank Drs. Heike Claus and Ulrich Vogel, Institute for Hygiene and Microbiology, University of Würzburg, for providing mAb 932 and the genomic DNA of Nm strain Z2491 and Dr. Paolo Costantino (Novartis Vaccines and Diagnostics, Research, Siena) as well as Dr. Timothy G. Keys and Dr. Martina Mühlenhoff (both Hannover Medical School) for helpful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) in the framework of DFG Research Unit 548 (Ge801/10-1).

This article contains supplemental data and Schemes S1–S3.
D. V. Yashunsky, A. J. Black, and A. V. Nikolaev, manuscript in preparation.
- NmA
- Neisseria meningitidis serogroup A
- AEC
- anionic exchange chromatography
- DP
- degree of polymerization
- avDP
- averaged DP
- CP
- capsule polymerase
- CPS
- capsular polysaccharide
- CPSA
- CPS of NmA
- CPSAhyd
- hydrolyzed CPSA
- deP
- dephosphorylated
- CsaA
- UDP-GlcNAc-2-epimerase
- CsaB
- poly-ManNAc-1-phosphate-transferase
- CsaC
- O-acetyltransferase
- 1P
- 1-phosphate.
REFERENCES
- 1. Stephens D. S., Greenwood B., Brandtzaeg P. (2007) Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 369, 2196–2210 [DOI] [PubMed] [Google Scholar]
- 2. Stephens D. (2009) Biology and pathogenesis of the evolutionarily successful, obligate human bacterium Neisseria meningitidis. Vaccine 27, B71–B77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu T. Y., Gotschlich E. C., Jonssen E. K., Wysocki J. R. (1971) Studies on the meningococcal polysaccharides. I. Composition and chemical properties of the group A polysaccharide. J. Biol. Chem. 246, 2849–2858 [PubMed] [Google Scholar]
- 4. Gotschlich E. C., Goldschneider I., Artenstein M. S. (1969) Human immunity to the meningococcus. IV. Immunogenicity of group A and group C meningococcal polysaccharides in human volunteers. J. Exp. Med. 129, 1367–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Costantino P., Rappuoli R., Berti F. (2011) The design of semi-synthetic and synthetic glycoconjugate vaccines. Expert Opin. Drug Discov. 6, 1045–1066 [DOI] [PubMed] [Google Scholar]
- 6. Frasch C. E., Preziosi M. P., LaForce F. M. (2012) Development of a group A meningococcal conjugate vaccine, MenAfriVac(TM). Hum. Vaccin. Immunother. 8, 715–724 [DOI] [PubMed] [Google Scholar]
- 7. Roberts L. (2010) Vaccine introduction. The beginning of the end for Africa's devastating meningitis outbreaks? Science 330, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 8. Djingarey M. H., Barry R., Bonkoungou M., Tiendrebeogo S., Sebgo R., Kandolo D., Lingani C., Preziosi M. P., Zuber P. L., Perea W., Hugonnet S., Dellepiane de Rey Tolve N., Tevi-Benissan C., Clark T. A., Mayer L. W., Novak R., Messonier N. E., Berlier M., Toboe D., Nshimirimana D., Mihigo R., Aguado T., Diomandé F., Kristiansen P. A, Caugant D. A, Laforce F. M. (2012) Effectively introducing a new meningococcal A conjugate vaccine in Africa: the Burkina Faso experience. Vaccine 30, B40–B45 [DOI] [PubMed] [Google Scholar]
- 9. Caini S., Beck N. S., Yacouba H., Maiga I., Chaibou I., Hinsa I., Adakal A., Issoufou A., Kim S. H., Pezzoli L. (2013) From Agadez to Zinder: estimating coverage of the MenAfriVacTM conjugate vaccine against meningococcal serogroup A in Niger, September 2010-January 2012. Vaccine 31, 1597–1603 [DOI] [PubMed] [Google Scholar]
- 10. LaForce F., Okwo-Bele J. M. (2011) Eliminating epidemic Group A meningococcal meningitis in Africa through a new vaccine. Health Aff. (Millwood) 30, 1049–1057 [DOI] [PubMed] [Google Scholar]
- 11. Kristiansen P. A., Diomandé F., Ba A. K., Sanou I., Ouédraogo A. S., Ouédraogo R., Sangaré L., Kandolo D., Aké F., Saga I. M., Clark T. A., Misegades L., Martin S. W., Thomas J. D., Tiendrebeogo S. R., Hassan-King M., Djingarey M. H., Messonnier N. E., Préziosi M. P., Laforce F. M., Caugant D. A. (2013) Impact of the serogroup A meningococcal conjugate vaccine, MenAfriVac, on carriage and herd immunity. Clin. Infect. Dis. 56, 354–363 [DOI] [PubMed] [Google Scholar]
- 12. Freiberger F., Claus H., Günzel A., Oltmann-Norden I., Vionnet J., Mühlenhoff M., Vogel U., Vann W. F., Gerardy-Schahn R., Stummeyer K. (2007) Biochemical characterization of a Neisseria meningitidis polysialyltransferase reveals novel functional motifs in bacterial sialyltransferases. Mol. Microbiol. 65, 1258–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romanow A., Haselhorst T., Stummeyer K., Claus H., Bethe A., Mühlenhoff M., Vogel U., von Itzstein M., Gerardy-Schahn R. (2013) Biochemical and biophysical characterization of the sialyl-/hexosyltransferase synthesizing the meningococcal serogroup W135 heteropolysaccharide capsule. J. Biol. Chem. 288, 11718–11730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peterson D. C., Arakere G., Vionnet J., McCarthy P. C., Vann W. F. (2011) Characterization and acceptor preference of a soluble meningococcal group C polysialyltransferase. J. Bacteriol. 193, 1576–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muindi K. M., McCarthy P. C., Wang T., Vionnet J., Battistel M., Jankowska E., Vann W. F. (2014) Characterization of the meningococcal serogroup X capsule N-acetylglucosamine-1-phosphotransferase. Glycobiology 24, 139–149 [DOI] [PubMed] [Google Scholar]
- 16. McCarthy P. C., Saksena R., Peterson D. C., Lee C. H., An Y., Cipollo J. F., Vann W. F. (2013) Chemoenzymatic synthesis of immunogenic meningococcal group C polysialic acid-tetanus Hc fragment glycoconjugates. Glycoconj. J. 30, 857–870 [DOI] [PubMed] [Google Scholar]
- 17. Fiebig T., Berti F., Freiberger F., Pinto V., Claus H., Romano M. R., Proietti D., Brogioni B., Stummeyer K., Berger M., Vogel U., Costantino P., Gerardy-Schahn R. (2014) Functional expression of the capsule polymerase of Neisseria meningitidis serogroup X: A new perspective for vaccine development. Glycobiology 24, 150–158 [DOI] [PubMed] [Google Scholar]
- 18. Berry D. S., Lynn F., Lee C. H., Frasch C. E., Bash M. C. (2002) Effect of O-acetylation of Neisseria meningitidis serogroup A capsular polysaccharide on development of functional immune responses. Infect. Immun. 70, 3707–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harrison O. B., Claus H., Jiang Y., Bennett J. S., Bratcher H. B., Jolley K. A., Corton C., Care R., Poolman J. T., Zollinger W. D., Frasch C. E., Stephens D. S., Feavers I., Frosch M., Parkhill J., Vogel U., Quail M. A., Bentley S. D., Maiden M. C. (2013) Description and nomenclature of Neisseria meningitidis capsule locus. Emerg. Infect. Dis. 19, 566–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swartley J. S., Liu L. J., Miller Y. K., Martin L. E., Edupuganti S., Stephens D. S. (1998) Characterization of the gene cassette required for biosynthesis of the (α1→6)-linked N-acetyl-d-mannosamine-1-phosphate capsule of serogroup A Neisseria meningitidis. J. Bacteriol. 180, 1533–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gudlavalleti S. K., Datta A. K., Tzeng Y. L., Noble C., Carlson R. W., Stephens D. S. (2004) The Neisseria meningitidis serogroup A capsular polysaccharide O-3 and O-4 acetyltransferase. J. Biol. Chem. 279, 42765–42773 [DOI] [PubMed] [Google Scholar]
- 22. Sperisen P., Schmid C. D., Bucher P., Zilian O. (2005) Stealth proteins: in silico identification of a novel protein family rendering bacterial pathogens invisible to host immune defense. PLoS Comput. Biol. 1, e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Villalobos A., Ness J. E., Gustafsson C., Minshull J., Govindarajan S. (2006) Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC Bioinformatics 7, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Welch M., Govindarajan S., Ness J. E., Villalobos A., Gurney A., Minshull J., Gustafsson C. (2009) Design parameters to control synthetic gene expression in Escherichia coli. PloS ONE 4, e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwarzer D., Stummeyer K., Haselhorst T., Freiberger F., Rode B., Grove M., Scheper T., von Itzstein M., Mühlenhoff M., Gerardy-Schahn R. (2009) Proteolytic release of the intramolecular chaperone domain confers processivity to endosialidase F. J. Biol. Chem. 284, 9465–9474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berti F., Romano M. R., Micoli F., Pinto V., Cappelletti E., Gavini M., Proietti D., Pluschke G., MacLennan C. A., Costantino P. (2012) Relative stability of meningococcal serogroup A and X polysaccharides. Vaccine 30, 6409–6415 [DOI] [PubMed] [Google Scholar]
- 27. Min H., Cowman M. (1986) Combined alcian blue and silver staining of glycosaminoglycans in polyacrylamide gels: application to electrophoretic analysis of molecular weight distribution. Anal. Biochem. 155, 275–285 [DOI] [PubMed] [Google Scholar]
- 28. Keys T. G., Freiberger F., Ehrit J., Krueger J., Eggers K., Buettner F. F., Gerardy-Schahn R. (2012) A universal fluorescent acceptor for high-performance liquid chromatography analysis of pro- and eukaryotic polysialyltransferases. Anal. Biochem. 427, 107–115 [DOI] [PubMed] [Google Scholar]
- 29. Lai E. C., Withers S. G. (1994) Stereochemistry and kinetics of the hydration of 2-acetamido-d-glucal by β-N-acetylhexosaminidases. Biochemistry 33, 14743–14749 [DOI] [PubMed] [Google Scholar]
- 30. Borodovsky M., McIninch J. (1993) GENMARK: Parallel gene recognition for both DNA strands. Comput. Chem. 17, 123–133 [Google Scholar]
- 31. Besemer J., Lomsadze A., Borodovsky M. (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hyatt D., Chen G. L., Locascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parkhill J., Achtman M., James K. D., Bentley S. D., Churcher C., Klee S. R., Morelli G., Basham D., Brown D., Chillingworth T., Davies R. M., Davis P., Devlin K., Feltwell T., Hamlin N., Holroyd S., Jagels K., Leather S., Moule S., Mungall K., Quail M. A., Rajandream M. A., Rutherford K. M., Simmonds M., Skelton J., Whitehead S., Spratt B. G., Barrell B. G. (2000) Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature 404, 502–506 [DOI] [PubMed] [Google Scholar]
- 34. Bundle D. R., Smith I. C., Jennings H. J. (1974) Determination of the structure and conformation of bacterial polysaccharides by carbon 13 nuclear magnetic resonance. Studies on the group-specific antigens of Neisseria meningitidis serogroups A and X. J. Biol. Chem. 249, 2275–2281 [PubMed] [Google Scholar]
- 35. Lemercinier X., Jones C. (1996) Full 1H NMR assignment and detailed O-acetylation patterns of capsular polysaccharides from Neisseria meningitidis used in vaccine production. Carbohydr. Res. 296, 83–96 [DOI] [PubMed] [Google Scholar]
- 36. Ravenscroft N., Averani G., Bartoloni A., Berti S., Bigio M., Carinci V., Costantino P., D'Ascenzi S., Giannozzi A., Norelli F., Pennatini C., Proietti D., Ceccarini C., Cescutti P. (1999) Size determination of bacterial capsular oligosaccharides used to prepare conjugate vaccines. Vaccine 17, 2802–2816 [DOI] [PubMed] [Google Scholar]
- 37. Black A., Nikolaev A. V. (2010) Synthesis of structures corresponding to the capsular polysaccharide of Neisseria meningitidis group A. 4th Baltic Meeting on Microbial Carbohydrates, September 19–22, 2010, p. 56 (Abstract 5), Hyytiälä Forestry Field Station, Finland [Google Scholar]
- 38. Black A., Yashunsky D. V., Sizova O. V., Nikolaev A. V. (2011) Towards synthetic glycoconjugates based on the structure of Neisseria meningitidis group A capsular polysaccharide. 16th European Carbohydrate Symposium, July 3–7, 2011, Abstract p. 61 (OL-02), Sorrento, Italy [Google Scholar]
- 39. Nikolaev A. V. (2011) From phosphosaccharide chemistry to potential anti-parasite and anti-bacterial carbohydrate vaccines. Carbohydrate Gordon Research Conference, June 19–24, 2011, Abstracts p. 4, Colby College, Waterville, ME [Google Scholar]
- 40. Bardotti A., Averani G., Berti F., Berti S., Carinci V., D'Ascenzi S., Fabbri B., Giannini S., Giannozzi A., Magagnoli C., Proietti D., Norelli F., Rappuoli R., Ricci S., Costantino P. (2008) Physicochemical characterisation of glycoconjugate vaccines for prevention of meningococcal diseases. Vaccine 26, 2284–2296 [DOI] [PubMed] [Google Scholar]
- 41. Sala R., Morgan P., Tanner M. (1996) Enzymatic formation and release of a stable glycal intermediate: the mechanism of the reaction catalyzed by UDP-N-acetylglucosamine 2-epimerase. J. Am. Chem. Soc. 118, 3033–3034 [Google Scholar]
- 42. Gotschlich E. C., Liu T. Y., Artenstein M. S. (1969) Human immunity to the meningococcus. 3. Preparation and immunochemical properties of the group A, group B, and group C meningococcal polysaccharides. J. Exp. Med. 129, 1349–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang R., Moquist P., Finney N. (2004) Chemical synthesis of UDP-4-O-methyl-GlcNAc, a potential chain terminator of chitin synthesis. Carbohydr. Res. 339, 1531–1536 [DOI] [PubMed] [Google Scholar]
- 44. Bergfeld A. K., Claus H., Vogel U., Mühlenhoff M. (2007) Biochemical characterization of the polysialic acid-specific O-acetyltransferase NeuO of Escherichia coli K1. J. Biol. Chem. 282, 22217–22227 [DOI] [PubMed] [Google Scholar]
- 45. Bergfeld A. K., Claus H., Lorenzen N. K., Spielmann F., Vogel U., Mühlenhoff M. (2009) The polysialic acid-specific O-acetyltransferase OatC from Neisseria meningitidis serogroup C evolved apart from other bacterial sialate O-acetyltransferases. J. Biol. Chem. 284, 6–16 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.