

Background: The consequences of calpain activation after myocardial infarction (MI) are not fully elucidated.
Results: Post-MI remodeling was exacerbated in calpastatin-deficient hearts, and calpain activation disrupted N-cadherin-based cell adhesions.
Conclusion: Unregulated activation of calpains contributes to progression of post-MI remodeling.
Significance: Pharmacological intervention of the calpain-calpastatin system will be a promising strategy in the treatment of post-MI remodeling.
Keywords: Apoptosis, Calcium, Cell Adhesion, Heart Failure, Protease
Abstract
Enzymatic proteolysis by calpains, Ca2+-dependent intracellular cysteine proteases, has been implicated in pathological processes such as cellular degeneration or death. Here, we investigated the role of calpain activation in the hearts subjected to myocardial infarction. We produced myocardial infarction in Cast−/− mice deficient for calpastatin, the specific endogenous inhibitory protein for calpains, and Cast+/+ mice. The activity of cardiac calpains in Cast+/+ mice was not elevated within 1 day but showed a gradual elevation after 7 days following myocardial infarction, which was further pronounced in Cast−/− mice. Although the prevalence of cardiomyocyte death was indistinguishable between Cast−/− and Cast+/+ mice, Cast−/− mice exhibited profound contractile dysfunction and chamber dilatation and showed a significant reduction in survival rate after myocardial infarction as compared with Cast+/+ mice. Notably, immunofluorescence revealed that at 28 days after myocardial infarction, calpains were activated in cardiomyocytes exclusively at the border zone and that Cast−/− mice showed higher intensity and a broader extent of calpain activation at the border zone than Cast+/+ mice. In the border zone of Cast−/− mice, pronounced activation of calpains was associated with a decrease in N-cadherin expression and up-regulation of molecular markers for cardiac hypertrophy and fibrosis. In cultured rat neonatal cardiomyocytes, calpain activation by treatment with ionomycin induced cleavage of N-cadherin and decreased expression levels of β-catenin and connexin 43, which was attenuated by calpain inhibitor. These results thus demonstrate that activation of calpains disassembles cell-cell adhesion at intercalated discs by degrading N-cadherin and thereby promotes left ventricular remodeling after myocardial infarction.
Introduction
The calpains represent a family of Ca2+-dependent cysteine proteases consisting of several ubiquitously expressed and tissue-specific isoforms (1). The main isoforms in mammalian cells are ubiquitous μ-calpain (calpain 1) and m-calpain (calpain 2), which demand micromolar and millimolar levels of Ca2+, respectively, for exhibiting proteolytic activity in vitro (1). The μ- and m-calpains are heterodimers consisting of an isoform-specific catalytic subunit encoded by Capn1 and Capn2, respectively, and a common regulatory subunit encoded by Capns1 (1). In vivo, the proteolytic activity of both μ- and m-calpains is tightly controlled by calpastatin, a major and specific endogenous inhibitory protein of calpains (2). Recent studies have suggested that the calpain-calpastatin system controls fundamental cellular functions such as cytoskeletal remodeling, cell cycle regulation, gene expression, and cell death (1). Thus, the imbalance between calpains and calpastatin can give rise to a wide spectrum of diseases, such as Alzheimer disease, muscular dystrophy, cataract formation, diabetes mellitus, ischemia, and cancers (3).
In cardiac muscle, Ca2+ plays a crucial role in excitation-contraction coupling, and dysregulation of cellular Ca2+ homeostasis, often in the form of Ca2+ overload, leads to myocardial cell injury. Although multiple destructive processes are triggered by Ca2+ overload, the cellular demise ensues largely from Ca2+-induced mitochondrial permeability transition and from Ca2+-induced proteolysis (4). In ex vivo or in vivo hearts, Ca2+ overload after ischemia-reperfusion have caused contractile dysfunction (post-ischemic stunning), which is associated with proteolysis of structural and regulatory proteins such as α-actinin, desmin, α-spectrin, troponin I, Na+/K+-ATPase, and protein kinase Cα (5–10). Furthermore, calpains have been implicated in the execution of myocardial cell death during ischemia-reperfusion. Calpain activation mediates Ca2+ overload-induced proteolysis in these processes, as is evident from observations that pharmacological inhibition of calpains has significantly attenuated myocardial stunning and reduced infarct size after ischemia-reperfusion (5–9, 11–14).
However, the consequences of calpain activation after myocardial infarction (MI)3 remain to be fully elucidated in an in vivo context. During acute ischemia, the intracellular Ca2+ is elevated mainly through Ca2+ entry by reverse mode Na+/Ca2+ exchanger (15, 16), leading to the assumption that calpain activation during an acute phase may exacerbate myocardial cell death. Meanwhile, it has been reported that enzymatic activities as well as mRNA and protein levels of calpains are elevated in a chronic phase (17–21), suggesting that calpain activation may be implicated in the progression of left ventricular (LV) remodeling after MI. Indeed, several reports have demonstrated that calpain inhibitors reduced infarct size and prevented LV dysfunction after MI in animal models (22, 23), but there are significant limitations to these pharmacological approaches. Most of the calpain inhibitors ineluctably exert nonspecific effects on other proteases (such as caspases and cathepsins) or proteasome activity in some degree and fail to reach the effective concentrations in vivo because of low permeability across the cell membrane and measurable toxicity to living cells (24).
In the present study we examined the temporal transition and regional and subcellular distribution of calpain activity in the heart after MI. Furthermore, we produced MI in calpastatin knock-out (Cast−/−) mice and wild-type (Cast+/+) mice to elucidate the consequences of enhanced calpain activation. We found that calpains were activated in the border zone, adjacent to the infarct zone, during the chronic phase and that profound activation of calpains exacerbated LV remodeling after MI in Cast−/− mice. Mechanistically, unregulated activation of calpain induced cleavage of N-cadherin and disrupted the cadherin-based cell adhesions, which potentially led to progression of LV remodeling after MI.
EXPERIMENTAL PROCEDURES
Mice, MI Operation, Blood Pressure Measurement, Echocardiography
Generation of Cast−/− mice has been described previously (25). The systolic and diastolic blood pressures and pulse rates were measured in conscious mice noninvasively by a programmable sphygmomanometer (BP-98A, Softron) using the tail-cuff method. To produce MI, we anesthetized 8–12-week-old mice by intraperitoneal injection of medetomidine hydrochloride (0.3 mg/kg), midazolam (4 mg/kg), and butorphanol (5 mg/kg) (26), and anesthesia was monitored by pinching the toe. We ligated the left descending coronary artery with 10-0 nylon suture at the level of below left atrium, and post-operative analgesia (meloxicam, 5 mg/kg/24 h) was administered subcutaneously for 48 h. For evaluation of cardiac dimensions and contractility, transthoracic echocardiography was performed on conscious mice with Vevo 770 Imaging System using a 25-MHz linear probe (Visual Sonics). At the indicated time points, mice were anesthetized by intraperitoneal injection of an overdose of pentobarbital (200 mg/kg). The hearts were removed and prepared for further histological and molecular analysis. All protocols were approved by the Institutional Animal Care and Use Committee of Chiba University.
Histological Analysis, Immunohistochemistry, TUNEL Assay
For histological analysis, hearts were excised, fixed in 10% neutralized formalin, and embedded in paraffin. Serial sections at 5 μm were deparaffinized and stained with Masson's trichrome for evaluation of fibrosis. We evaluated a ratio of infarct area to left ventricular free wall area for quantification of the infarct area (%). Infarct length (mm) was measured as the midline of the length of infarct that included >50% of the whole thickness of the myocardial wall (27). For immunofluorescence, hearts were excised and immediately embedded in Tissue-Tek OCT cryo-embedding compound (Miles Laboratories). Cryostat sections at 5 μm were fixed in acetone, and primary antibodies were applied overnight at 4 °C. Alexa Fluor 488-conjugated anti-rabbit IgG antibody (Invitrogen) or Cy3-conjugated anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories) was applied to visualize expression of specific proteins. Images were acquired with either a microscope (Eclipse E600; Nikon) using the Radiance 2000 confocal scanning system (Bio-Rad) or an LSM 700 confocal microscope (Carl Zeiss). A TUNEL assay with nuclear staining with Hoechst 33258 was performed using an in situ apoptosis detection kit (Takara Bio), according to the manufacturer's protocol. Images were captured using a Nikon Eclipse E600 microscope equipped with epifluorescence optics and a CCD camera (Axiocam; Carl Zeiss). The images were analyzed for calculation of areas with intense fluorescence using the BZ-Analyzer software in Hybrid Cell Count mode (Keyence).
Cell Culture, Immunocytochemistry, Calpain Activity Assay
Primary cultures of cardiac myocytes were prepared from ventricles of 1-day-old Wistar rats as described previously (28). The experimental protocol was approved by the Animal Study Committee of Osaka University. Briefly, cervical dislocation euthanasia was performed by trained personal before harvesting of the cardiac tissue according to the American Veterinary Medical Association guidelines for the euthanasia of animals. Cardiomyocytes were plated at a field density of 1 × 105 cells/cm2 and cultured in DMEM supplemented with 10% bovine growth serum. Forty-eight hours after plating cells were washed twice in phosphate-buffered saline, pretreated with 10 μm MDL28170 (Sigma) in serum-free DMEM for 1 h, and stimulated with 10 μm ionomycin (Sigma) for 10 min. For immunocytochemistry, the cells were fixed in PBS containing 4% paraformaldehyde for 15 min at room temperature. The cells were stained with primary antibodies and visualized with secondary antibodies: Alexa Fluor 488-conjugated anti-rabbit IgG (H+L) antibody and Alexa Fluor 647-conjugated anti-mouse IgG (H+L) antibody (Invitrogen). Images were acquired with a confocal microscope (LSM 700; Carl Zeiss). The enzymatic activity of calpains was measured using the Calpain-Glo Protease Assay (Promega) according to the manufacture's protocol.
Western Blot Analysis
Protein samples were fractionated with SDS-PAGE and transferred to PVDF membrane (GE Healthcare). The blotted membranes were incubated with primary antibodies followed by horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody (Jackson ImmunoResearch Laboratories). Immunoreactive signals were detected with the ECL Plus Western blotting Detection System (GE Healthcare) and developed onto a film (Hyperfilm ECL; GE Healthcare Biosciences) or visualized using a lumino-image analyzer (ImageQuant LAS 4000 mini; GE Healthcare).
Primary Antibodies
The following antibodies were used: mouse monoclonal anti-N-cadherin antibody (clone 3B9, Invitrogen) raised against the intracellular domain of chicken N-cadherin, rabbit polyclonal anti-N-cadherin antibody (Santa Cruz Biotechnology, Inc.) raised against the extracellular domain of human N-cadherin, rabbit polyclonal anti-β-catenin antibody (Abcam), mouse monoclonal anti-Cx43 antibody (clone CX-1B1, Invitrogen), rabbit polyclonal anti-Cx43 antibody (Millipore), mouse monoclonal anti-sarcomeric α-actinin antibody (clone EA-53, Sigma), rabbit polyclonal anti-collagen 1 antibody (Abcam), mouse monoclonal anti-α-tubulin antibody (clone DM1A, Sigma), rabbit monoclonal anti-GAPDH antibody (clone 14C10, Cell Signaling Technology). A rabbit polyclonal antibody specific to the calpain cleavage site of the N-terminal 135-kDa fragment of αII-spectrin was described previously (25).
Real Time RT-PCR Analysis
Total RNA was extracted by using TRIzol reagent (Invitrogen), and single-stranded cDNA was transcribed by using QuantiTect Reverse Transcription kit (Qiagen) according to the manufacturer's protocol. We conducted quantitative real-time PCR analysis using Light Cycler TaqMan Master Kit (Roche Applied Science) with the target-specific primers and the matching probes designed by the Universal ProbeLibrary System (Roche Applied Science). Amplification conditions were initial denaturation for 10 min at 95 °C followed by 45 cycles of 10 s at 95 °C and 25 s at 60 °C. Individual PCR products were analyzed by melting point analysis. The expression level of a gene was normalized relative to that of Gapdh by using a comparative Ct method. The primer sequences and Universal Probe numbers were designed with the ProbeFinder software as following: Nppa, 5′-cacagatctgatggatttcaaga-3′ and 5′-cctcatcttctaccggcatc-3′, no. 25; Nppb, 5′-gtcagtcgtttgggctgtaac-3′ and 5′- agacccaggcagagtcagaa-3′, no. 71; Acta1, 5′-aatgagcgtttccgttgc-3′ and 5′-atccccgcagactccatac-3′, no. 94; Gapdh, 5′-tgtccgtcgtggatctgac-3′ and 5′-cctgcttcaccaccttcttg-3′, no. 80.
Statistics
The results are expressed as the mean ± S.E. Differences in measured values were analyzed using an unpaired 2-tailed Student's t test for two-group comparison and a 1-way analysis of variance followed by the Bonferroni's method for multi-group comparison. We estimated survival curves after MI by the Kaplan-Meier method and compared the two groups by the log-rank test. Values of p < 0.05 were considered statistically significant.
RESULTS
Calpains Are Activated in the Chronic Phase but Not in the Acute Phase after MI
First, we examined the time course of change in calpain activity after MI in wild-type mice by using an antibody specific to the calpain-cleaved N-terminal 135-kDa fragment of αII-spectrin (25). Although it has been reported that intracellular Ca2+ is elevated immediately after myocardial ischemia (15), the level of spectrin proteolysis was unchanged within 24 h after MI (Fig. 1A). However, at 7 days after MI, spectrin proteolysis was significantly increased and sustained thereafter (Fig. 1A). These results suggest that calpains are activated not in the acute phase but in the subacute and chronic phase after MI.
FIGURE 1.
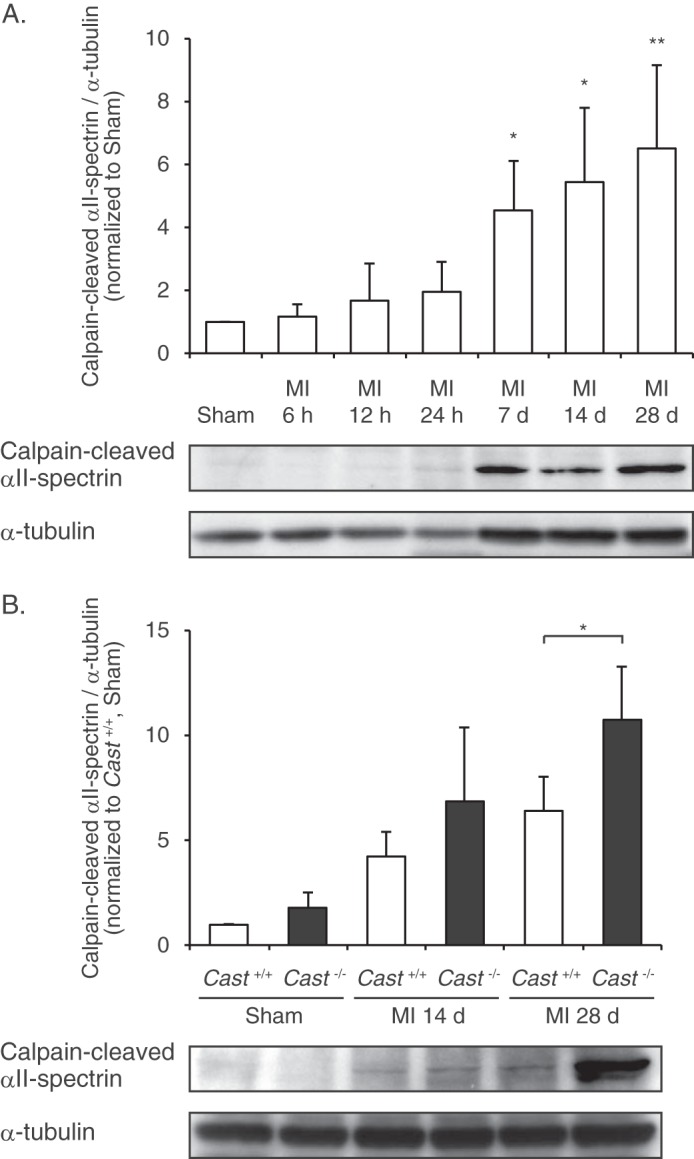
Calpain activity in MI hearts of Cast−/− and Cast+/+ mice. A, time course of calpain activation after MI, determined by immunoblot analysis of calpain-cleaved αII-spectrin in the hearts of wild-type mice (n = 5). α-Tubulin was used as an internal control for the amount of loaded protein. The calpain-cleaved αII-spectrin/α-tubulin ratios were quantified by densitometry and plotted (upper panel). Data are presented as the mean ± S.E. *, p < 0.05; **, p < 0.01 versus Sham. B, comparison of calpain activation after MI between Cast−/− and Cast+/+ mice (n = 3 ∼ 4). The calpain-cleaved αII-spectrin/α-tubulin ratios were quantified by densitometry and plotted (upper panel). Data are presented as the mean ± S.E. *, p < 0.05.
Calpain Activation in the Chronic Phase after MI Is Enhanced in Cast−/− Mice
Next, we compared the activities of cardiac calpains after MI between Cast−/− and Cast+/+ mice. It has been reported that basal activity of calpain is important for protein homeostasis in unstressed hearts (29), but the calpain activity in sham-operated hearts, as assessed by the level of spectrin proteolysis, was indistinguishable between Cast−/− and Cast+/+ mice (Fig. 1B). Essentially, Cast−/− mice showed normal development, fertility, and life span (25). In addition, Cast−/− mice exhibited normal heart rates, blood pressures, and cardiac function under the physiological conditions as assessed by echocardiographic evaluation (Table 1). These results suggest that calpastatin deficiency has little effect on basal activity of calpain under the physiological conditions. However, as compared with Cast+/+ mice, Cast−/− mice showed a significant increase in spectrin proteolysis at 28 days after MI (Fig. 1B), suggesting that calpastatin deficiency exaggerates activation of calpains when calpain activation is induced under stressed conditions.
TABLE 1.
Basal heart rates, blood pressures, and echocardiographic parameters of Cast−/− and Cast+/+ mice
Values are the mean ± S.E. LVEDD, left ventricular end-diastolic dimension; LVESD, left ventricular end-systolic dimension; IVSth, intraventricular septal thickness; PWth, left ventricular posterior wall thickness; FS, fractional shortening; HR, heart rate; SBP, systolic blood pressure; DBP, diastolic blood pressure; bpm, beats per min.
| Parameters | Cast+/+ | Cast−/− | P |
|---|---|---|---|
| Number | 4 | 4 | |
| Age (weeks) | 8.60 ± 0.08 | 8.60 ± 0.20 | 0.54 |
| Heart rate (bpm) | 640 ± 13.6 | 646 ± 32.6 | 0.79 |
| SBP (mm Hg) | 98.9 ± 5.08 | 97.9 ± 5.73 | 0.79 |
| DBP (mm Hg) | 58.2 ± 4.26 | 52.6 ± 3.12 | 0.76 |
| LVEDD (mm) | 3.48 ± 0.03 | 3.46 ± 0.27 | 0.90 |
| LVESD (mm) | 1.71 ± 0.02 | 1.69 ± 0.04 | 0.51 |
| IVSth (mm) | 0.86 ± 0.11 | 0.89 ± 0.04 | 0.62 |
| PWth (mm) | 0.81 ± 0.07 | 0.90 ± 0.05 | 0.13 |
| FS (%) | 50.7 ± 1.28 | 50.9 ± 2.47 | 0.91 |
LV Remodeling after MI Is Enhanced in Cast−/− Mice
At 14 days after MI, histological analysis with Masson's trichrome staining and echocardiographic examination revealed no significant difference in LV geometry and function between Cast−/− and Cast+/+ mice (Fig. 2, A and B). However, at 28 days after MI, Cast−/− mice showed more severe LV dilatation and dysfunction than Cast+/+ mice (Fig. 2, C and D). Because there was no significant difference in infarct size, determined either by area measurement or by length measurement, between Cast−/− and Cast+/+ mice (Fig. 2, E and F), LV remodeling was promoted independently of the infarct size in Cast−/− mice. As a consequence, the survival rate was significantly lower in Cast−/− mice than in Cast+/+ mice (p < 0.05), although the early death within 14 days after MI was comparable (p = 0.058) (Fig. 2G). These results suggest that enhanced activation of calpains in the chronic phase enhances LV remodeling in Cast−/− mice, leading to death possibly due to heart failure.
FIGURE 2.

LV remodeling after MI in Cast−/− and Cast+/+ mice. A, Masson's trichrome staining of Cast−/− and Cast+/+ hearts at 14 days after MI. Scale bars, 2 mm. B, echocardiographic parameters of Cast−/− and Cast+/+ mice at 14 days after MI. C, Masson's trichrome staining of Cast−/− and Cast+/+ hearts at 28 days after MI. Scale bars, 2 mm. D, echocardiographic parameters of Cast−/− and Cast+/+ mice at 28 days after MI. E and F, infarct area (light panels) and infarct length (right panels) of Cast−/− and Cast+/+ hearts at 14 days (E) and 28 days (F) after MI. G, Kaplan-Meier survival curves of Cast+/+ (n = 31) and Cast−/− mice (n = 40) after MI. LVEDD, LV end-diastolic dimension; FS, fractional shortening. Values represent the mean ± S.E. of data from 10 mice in each group. NS, not significant. *, p < 0.05; **, p < 0.01 versus Cast+/+ mice.
Myocardial Cell Death after MI Is Comparable between Cast−/− and Cast+/+ Mice
Calpains have been implicated in the execution of cell death (1). Thus, we compared the prevalence of cardiomyocyte apoptosis in Cast−/− and Cast+/+ mice by TUNEL staining. There was no significant difference in the number of TUNEL-positive cardiomyocytes in the infarct area of Cast−/− and Cast+/+ hearts at 24 h after MI (Fig. 3, A and B). Throughout the period from 3 to 28 days, apoptotic cardiomyocytes were scarcely observed in the infarct or non-infarct area of both genotypes, and the number was not significantly different between Cast−/− and Cast+/+ mice (Fig. 3, C and D). In addition, the infarct area of Cast−/− mice was not significantly different from that of Cast+/+ mice at 28 days after MI (Fig. 2, E and F). These results suggest that calpastatin deficiency has little impact on myocardial cell death both in the acute phase and in the chronic phase after MI.
FIGURE 3.

Cardiomyocyte apoptosis after MI in Cast−/− and Cast+/+ mice. A, TUNEL staining with nuclear staining with Hoechst 33258 in the infarct zone of Cast−/− and Cast+/+ mice at 24 h after MI. Scale bars, 50 μm. B, percentage of TUNEL-positive cardiomyocytes in the infarct zone of Cast−/− and Cast+/+ mice at 24 h after MI was calculated. Values represent the mean ± S.E. (500 cardiomyocytes sampled from 12 visual fields from 3 mice in each group). NS, not significant. C and D, percentage of TUNEL-positive cardiomyocytes in the infarct zone (C) and non-infarct zone (D) of Cast−/− and Cast+/+ mice at 7 days after MI was calculated. Values represent the mean ± S.E. (800 cardiomyocytes sampled from 12 visual fields from 3 mice in each group). NS, not significant.
Calpain Activation Is Associated with a Decrease in N-cadherin Expression in the Border Zone of Cast−/− Hearts
To gain insights into the mechanism of how calpain activation leads to exacerbation of LV remodeling, we utilized immunofluorescence staining and assessed the cellular and subcellular localization of calpain activation in the infarct and border zones. In sham-operated hearts of Cast+/+ mice, calpain-cleaved αII-spectrin was primarily localized in cardiomyocytes (Fig. 4A). Especially in cardiomyocytes, the fluorescence signals showed a characteristic cross-striated pattern that is typically seen with sarcomeric proteins, and intense fluorescence was colocalized with N-cadherin at the intercalated discs (Fig. 4A). A similar localization pattern of spectrin proteolysis with comparable intensity was observed in Cast−/− and Cast+/+ mice (Fig. 4A). At 28 days after MI, the levels of calpain-cleaved αII-spectrin were indistinguishable in the non-infarct zone between Cast−/− and Cast+/+ mice (Fig. 4B). However, we observed exaggeration of calpain-mediated spectrin proteolysis exclusively in cardiomyocytes at the border zone, adjacent to the infarct zone (Fig. 4C). Cast−/− mice showed significantly broader area of cardiomyocytes with intense immunofluorescence for calpain-cleaved αII-spectrin at the border zone than Cast+/+ mice (Fig. 4, C and D). Notably, in cardiomyocytes at the border zone of Cast−/− mice, profound activation of calpains was associated with a considerable decrease in the expression of N-cadherin (Fig. 4C). N-cadherin is a Ca2+-dependent transmembrane glycoprotein that enhances cell adhesion by binding to α-, β-, and γ-catenins at adherens junction (30) and stabilizes gap junction formation by maintaining connexin 43 (Cx43) at intercalated discs (31). In parallel with the decrease in N-cadherin expression, the expression levels of β-catenin and Cx43 were decreased in cardiomyocytes at the border zone of Cast−/− mice (Fig. 4, E and F). Next, we isolated RNA from the border zone of MI hearts for quantification of the expression levels of fetal cardiac genes. Cast−/− mice showed a significant increase in the expressions of Nppa, Nppb, and Acta1 as compared with Cast+/+ mice (Fig. 5A). Furthermore, immunohistochemical analysis revealed that the expression of collagen 1 was significantly increased in the border zone of Cast−/− mice as compared with Cast+/+ mice (Fig. 5B). These results suggest that molecular and cellular remodeling in the border zone was more prominent in Cast−/− mice than in Cast+/+ mice. Collectively, we speculate that unregulated activation of calpains in cardiomyocytes may disassemble the cadherin-mediated structures of adherens junction and gap junction of cardiomyocytes in the border zone and thereby lead to progression of LV remodeling after MI.
FIGURE 4.

Cellular and subcellular localization of calpain activation in the border zone of Cast−/− and Cast+/+ hearts after MI. A, immunofluorescence of sham-operated Cast−/− and Cast+/+ hearts (n = 5, in each group). Calpain-cleaved αII-spectrin and N-cadherin are represented in green and red, respectively. Co-localization of calpain-cleaved αII-spectrin and N-cadherin at the intercalated discs is indicated by the yellow merging of the green and red labels. Scale bars, 50 μm. B, immunofluorescence of the non-infarct zone in Cast−/− and Cast+/+ hearts at 28 days after MI. Calpain-cleaved αII-spectrin and N-cadherin are represented in green and red, respectively. Scale bars, 100 μm. C, immunofluorescence of the border zone in Cast−/− and Cast+/+ hearts at 28 days after MI (n = 5, in each group). Calpain-cleaved αII-spectrin and N-cadherin are represented in green and red, respectively. Scale bars, 100 μm. D, calculated areas of cardiomyocytes with intense immunofluorescence for calpain-cleaved αII-spectrin at the border zone in Cast−/− and Cast+/+ hearts at 28 days after MI (n = 5, in each group). Data are presented as the mean ± S.E. *, p < 0.05. E, immunofluorescence of the border zone in Cast−/− and Cast+/+ hearts at 28 days after MI (n = 5, in each group). β-Catenin and sarcomeric α-actinin are represented in green and red, respectively. Scale bars, 100 μm. F, immunofluorescence of the border zone in Cast−/− and Cast+/+ hearts at 28 days after MI (n = 5, in each group). Cx43 and sarcomeric α-actinin are represented in green and red, respectively. Scale bars, 100 μm. The dotted lines (C, E, and F) indicate the boundary between the border zone where viable cardiomyocytes remain (above the dotted line) and the infarct zone (below the dotted line).
FIGURE 5.

Molecular and histological changes in the border zone of Cast−/− and Cast+/+ hearts after MI. A, the mRNA expressions of Nppa, Nppb, and Acta1 in the border zone of Cast−/− and Cast+/+ hearts at 28 days after MI and in sham-operated Cast−/− and Cast+/+ hearts (n = 3 ∼ 5, in each group). Values represent the mean ± S.E. *, p < 0.05; **, p < 0.01 versus sham-operated Cast+/+ mice. B, immunofluorescence of the border zone in Cast−/− and Cast+/+ hearts at 28 days after MI (n = 5, in each group). Collagen 1 and sarcomeric α-actinin are represented in green and red, respectively. Scale bars, 200 μm. The dotted lines indicate the boundary between the border zone where viable cardiomyocytes remain (above the dotted line) and the infarct zone (below the dotted line).
Calpain Activation Causes N-cadherin Cleavage and Disassembles Cadherin-based Cell Adhesions in Cultured Cardiomyocytes of Neonatal Rats
To examine whether calpain activation was sufficient for disruption of N-cadherin-based cell adhesions, we stimulated cultured cardiomyocytes of neonatal rats with the Ca2+ ionophore ionomycin. Direct measurement of the enzymatic activity of calpains using a luminescent assay revealed that calpain enzymatic activity was significantly enhanced after stimulation with ionomycin, which was significantly, but not completely, repressed by pretreatment with the cell-permeable calpain inhibitor MDL28170 (Fig. 6A). Western blot analysis showed that the expression levels of full-length N-cadherin (140 kDa) were significantly decreased by stimulation with ionomycin, which was attenuated by pretreatment with MDL28170 (Fig. 6, B and C). In parallel with a decrease in the levels of full-length N-cadherin, ionomycin significantly increased the generation of the 106- and 37-kDa fragments, as detected by anti-N-cadherin antibodies raised against the N-terminal extracellular domain and the C-terminal intracellular domain, respectively (Fig. 6B). Similarly, the expression levels of β-catenin and Cx43 were significantly decreased by stimulation with ionomycin, which was attenuated by pretreatment with MDL28170 (Fig. 6, B and C).
FIGURE 6.

Calpain-mediated degradation of N-cadherin and down-regulation of intercalated disc proteins in rat neonatal cardiomyocytes. A, ionomycin-induced calpain activation in rat neonatal cardiomyocytes. Cells were pretreated with MDL28170 (10 μm) and stimulated with ionomycin (10 μm) for 10 min, and calpain activity was determined by a luminescent assay. Experiments were repeated three times in triplicate, and data are shown as -fold induction over vehicle control (mean ± S.E.). Iono, ionomycin; MDL, MDL28170. **, p < 0.01. B, immunoblot (IB) analysis of N-cadherin, β-catenin, and Cx43 in rat neonatal cardiomyocytes. Besides the full-length N-cadherin (arrows), the degraded N-terminal fragments (arrowhead) and C-terminal fragments (arrowhead) were detected by anti-N-cadherin antibody raised against the extracellular domain and intracellular domain of N-cadherin, respectively. C, quantitation of the N-cadherin (N terminus)/GAPDH (n = 6), N-cadherin (C terminus)/GAPDH (n = 7), β-catenin/GAPDH (n = 3), and Cx43/GAPDH (n = 4) are shown as bar graphs (right panel). Data are presented as the mean ± S.E. *, p < 0.05; **, p < 0.01.
Confocal immunocytochemistry revealed reduction in the expression levels of N-cadherin and β-catenin at the intercalated discs when cardiomyocytes were stimulated with ionomycin (Fig. 7A). Meanwhile, Cx43 immunofluorescent signals showed reduction at the intercalated discs and concomitant intracellular redistribution in response to ionomycin stimulation (Fig. 7B). Ionomycin-induced effects on the amount and spatial distribution of these intercalated disc proteins were prevented by pretreatment with MDL28170 (Fig. 7, A and B). These results suggest that calpain activation is sufficient to cause degradation of N-cadherin and to disassemble cadherin-based cell adhesions in rat neonatal cardiomyocytes.
FIGURE 7.
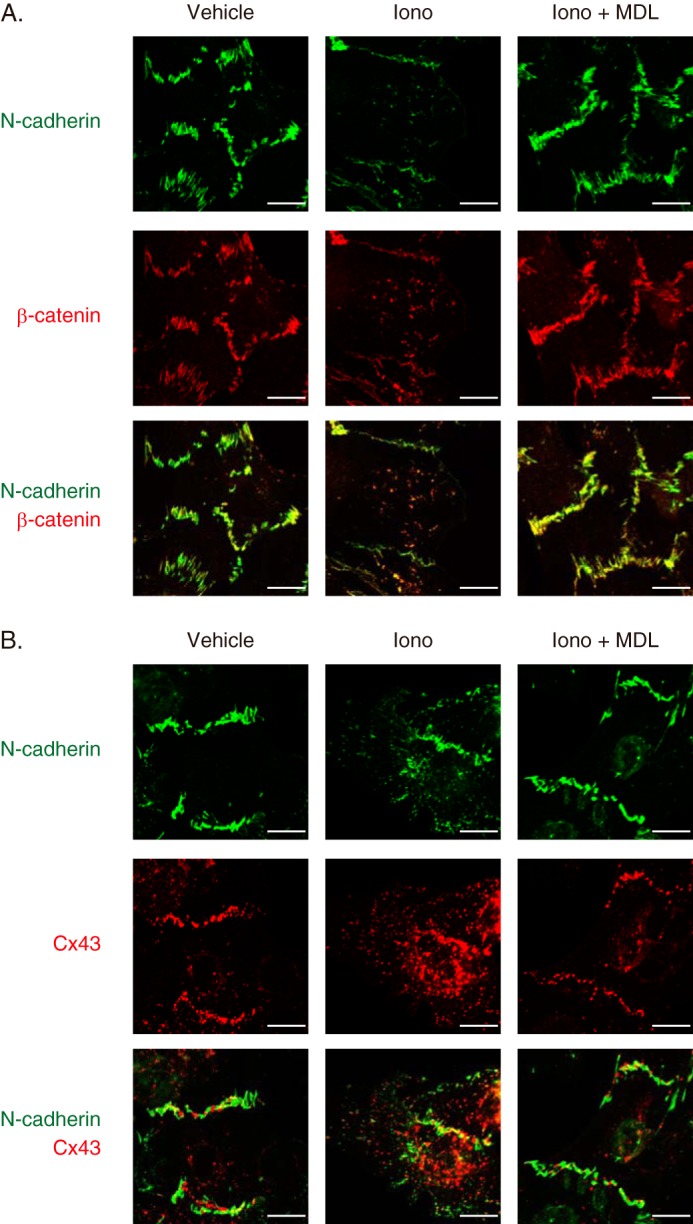
Calpain-mediated disassembly of intercalated disc proteins in rat neonatal cardiomyocytes. A, immunofluorescence of N-cadherin and β-catenin in rat neonatal cardiomyocytes. Cells were pretreated with MDL28170 (MDL, 10 μm) and stimulated with ionomycin (Iono, 10 μm) for 10 min. N-cadherin and β-catenin are represented in green and red, respectively. Experiments were repeated three times in triplicate, and the representative images are shown. Scale bars, 10 μm. B, immunofluorescence of N-cadherin and Cx43 in rat neonatal cardiomyocytes. Cells were pretreated with MDL28170 (10 μm) and stimulated with ionomycin (10 μm) for 10 min. N-cadherin and Cx43 are represented in green and red, respectively. Experiments were repeated three times in triplicate, and the representative images are shown. Scale bars, 10 μm.
DISCUSSION
In the present study we provided experimental evidence that calpains impaired the cell-cell interactions through degradation of cadherin-associated protein complex and thereby promoted LV remodeling after MI. Calpain-mediated proteolysis was increased in the chronic phase (7 days and later), not in the acute phase (before 24 h), after MI, and profound activation of calpains exacerbated LV remodeling without affecting myocardial cell death in Cast−/− mice. In the border zone of MI hearts, Cast−/− mice showed a decrease in N-cadherin expression concomitant with an increase in calpain activation and prominent myocardial remodeling. In cultured cardiomyocytes calpain activation caused degradation of N-cadherin and disorganization of cadherin-based cell adhesions.
The pathogenic role of calpains in MI hearts has remained unclear despite the efforts of many laboratories. One of the major obstacles for the study of calpains has been the lack of reliable methods to measure calpain activity accurately in vivo. Previous reports demonstrated that expression levels or enzymatic activities of calpains were increased in the hearts after MI (17–21), but these results must be interpreted cautiously. Obviously, the protein content of calpains does not necessary correlate with their proteolytic activity, and the in vitro enzymatic assays using homogenized tissue samples after the addition of Ca2+ merely indicate the proteolytic capacity of calpains, not the calpain activity in situ. However, in our study the use of a specific antibody against calpain-cleaved fragment of αII-spectrin has allowed for direct observation of the proteolytic activity of calpains in in vivo hearts in temporal and spatial terms. Another obstacle has been the lack of specific and effective calpain inhibitors (1). The best approach for identifying the role of calpains in cellular function is to introduce calpastatin, the specific and endogenous inhibitor of calpains. However, according to a recent study, cardiac overexpression of calpastatin inhibited basal calpain activity and resulted in spontaneous and progressive heart failure (29). Thus, we have analyzed Cast−/− and Cast+/+ mice to examine the consequences of exaggerated calpain activation in the hearts after MI.
In a variety of mammalian cells, activated calpains mediate cell death by multiple mechanisms. For example, calpains activate p53 (32), and pro-apoptotic BH3-only protein Bax (33) and Bid (34) inactivate anti-apoptotic Bcl-xL (35), facilitate the release of apoptosis-inducing factor (AIF) from mitochondria (36, 37), induce caspase-12 activation (35) and release of lysosomal cathepsins (38), cleave autophagy regulating protein Atg5 (39), degrade cytoskeletal proteins (40), disrupt ion homeostasis (9, 40, 41), and increase plasma membrane permeability (40). In vivo transgenic overexpression of calpain 1 in the heart has induced heart failure, which is associated with cardiomyocyte necrosis and mononuclear cell infiltration (29). In addition, profound calpain activation in Cast−/− mice has exacerbated excitotoxicity by kainite, leading to neuronal DNA fragmentation in the brain (25). However, the prevalence of cardiomyocyte apoptosis and the infarct size did not differ between Cast−/− and Cast+/+ mice throughout the observation period after MI (Figs. 2 and 3). These results suggest that calpastatin deficiency has little effect on myocardial cell death in MI hearts. Previous reports demonstrated that pharmacological inhibition of calpains reduced the infarct size (22, 23). In addition, recent reports showed that overexpression of Capn1 increased infarct size and enhanced myocardial cell death after MI (10), whereas genetic disruption of Capn1 or Capns1 provided the reciprocal results (10, 42). However, cardiac-specific Capns1 knock-out mice were susceptible for hemodynamic stress-induced myocardial injury because of defective membrane repair, indicating that calpains play a beneficial role as well (43). Because calpain activity in sham-operated hearts was indistinguishable between Cast−/− and Cast+/+ hearts (Fig. 1B), our study presented experimental evidence that inhibition of calpain activation without affecting the basal activity had little impact on myocardial cell death after MI.
Temporal and spatial analyses of calpain-cleaved αII-spectrin revealed that calpain activation was induced at the border zone of MI hearts in the chronic phase (Fig. 4C). After MI, the border zone expands in response to increased wall stress, promoting further LV dilatation and contractile dysfunction (44). Because calpain activation was followed by progressive contractile dysfunction and LV dilatation (Figs. 1 and 2), we speculate that profound activation of calpains at the border zone may contribute to exacerbation of LV remodeling in Cast−/− mice. According to in vitro assays, activated calpains cleave a large number of proteins including cytoskeletal proteins, membrane-associated proteins, signaling mediators, and transcription factors (1). Recent reports showed that calpain activation induced IκB degradation and NF-κB activation (45) and that disruption of Capns1 inhibited NF-κB signaling and inflammation, leading to improvement of LV remodeling after MI (42). In an attempt to explore potential calpain substrates in MI hearts, we found that calpain-cleaved αII-spectrin was co-localized with N-cadherin at the intercalated discs (Fig. 4, A–C). Importantly, increased calpain proteolysis was associated with a decrease in N-cadherin expression in cardiomyocytes at the border zone of Cast−/− hearts (Fig. 4C). In cultured cardiomyocytes, N-cadherin was cleaved in the presence of ionomycin, resulting in the generation of at least two fragments, an N-terminal 106-kDa and a C-terminal 37-kDa fragment. A calpain inhibitor MDL28170 significantly but not completely suppressed ionomycin-stimulated activation of calpains (Fig. 6A) and thereby prevented a significant degree of N-cadherin cleavage (Fig. 6B). In contrast to the degradation of full-length N-cadherin, the generation of cleaved fragments was not repressed by pretreatment with the calpain inhibitor (Fig. 6B). We speculate that the cleaved fragments may become unstable and susceptible to further degradation by undefined proteases. Previous reports demonstrated that calpains cleaved at least four regions of the intracellular domain of N-cadherin in neural cells (46, 47) and that calpain-mediated N-cadherin cleavage suppressed cell-cell adhesion in myogenic C2C12 cells (47). In cultured cardiomyocytes, calpain activation by ionomycin disassembled cadherin-based cell-cell adhesion consisting of intercalated disc proteins such as β-catenin and Cx43 (Fig. 7). It was reported that targeted disruption of N-cadherin in the hearts caused disassembly of intercalated discs, leading to LV dilatation and dysfunction (48). In addition, abnormal mechanical coupling through intercalated discs has been observed in the hearts of animal model and human patients with heart failure (49–51). Therefore, we assume that calpain activation-associated down-regulation of N-cadherin at the border zone is profoundly involved in the progression of LV remodeling after MI. It has been reported that a large number of proteins are cleaved by calpains in in vitro assays, including cytoskeletal proteins, membrane-associated proteins, kinases and phosphatases, and transcription factors (1). To prove the pathogenic significance of calpain-mediated cleavage of N-cadherin, our observation must be further investigated in future studies using knock-in mice of N-cadherin that is resistant to calpain cleavage for testing if these mice would rescue the exacerbated LV remodeling after MI in Cast−/− mice.
It remains an open question of what triggers calpain activation at the border zone cardiomyocytes in the chronic phase after MI. One of the candidates upstream of calpain activation is the renin-angiotensin system. It is well established that the local renin-angiotensin system is activated during the remodeling process after MI and that pharmacological or genetic blockade of renin-angiotensin system prevented LV remodeling after MI in many animal models and human patients (52). In cultured vascular smooth muscle cells, stimulation with angiotensin II (Ang II) has increased the calpain activity through transactivation of epidermal growth factor receptor and systemic overexpression of calpastatin blunted cardiac hypertrophy and perivascular inflammation in Ang II-infused mice (53). A recent study showed that calpain activation mediated Ang II-induced endothelial dysfunction in rodents (54). It is intriguing that activation of Ang II receptor signaling by locally produced Ang II and mechanical stress (28) may lead to calpain activation and thereby promote LV remodeling after MI. Even so, we have to assume that additional undefined factors play a regulatory role in activating calpains in a temporally and spatially restricted manner in MI hearts.
In conclusion, calpains are activated in the chronic phase after MI, and profound activation of calpains may exacerbate LV remodeling possibly through the alterations of intercalated disc organization in cardiomyocytes at the border zone. Therefore, pharmacological intervention of the calpain-calpastatin system may emerge as a promising strategy in the treatment of LV remodeling after MI unless it hampers the basal calpain activity.
Acknowledgments
We thank A. Furuyama, M. Ikeda, Y. Ohtsuki, I. Sakamoto, M. Shimizu, K. Kawaguchi, N. Miyagawa, H. Taniwaki, and Y. Ueda for excellent technical assistance.
This work was supported in part by grants from Japan Society for the Promotion of Science (KAKENHI 21229010, 23390213, 24659390, and 25-40014) and Health and Labor Sciences Research Grants (to I. K. and H. A.) and Japan Foundation for Applied Enzymology (to H. A.).
- MI
- myocardial infarction
- Ang II
- angiotensin II
- Cx
- connexin
- LV
- left ventricular.
REFERENCES
- 1. Goll D. E., Thompson V. F., Li H., Wei W., Cong J. (2003) The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 2. Hanna R. A., Campbell R. L., Davies P. L. (2008) Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456, 409–412 [DOI] [PubMed] [Google Scholar]
- 3. Zatz M., Starling A. (2005) Calpains and disease. N. Engl. J. Med. 352, 2413–2423 [DOI] [PubMed] [Google Scholar]
- 4. Murphy E., Steenbergen C. (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88, 581–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsumura Y., Saeki E., Inoue M., Hori M., Kamada T., Kusuoka H. (1996) Inhomogeneous disappearance of myofilament-related cytoskeletal proteins in stunned myocardium of guinea pig. Circ. Res. 79, 447–454 [DOI] [PubMed] [Google Scholar]
- 6. Gao W. D., Atar D., Liu Y., Perez N. G., Murphy A. M., Marban E. (1997) Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ. Res. 80, 393–399 [PubMed] [Google Scholar]
- 7. Papp Z., van der Velden J., Stienen G. J. (2000) Calpain-I induced alterations in the cytoskeletal structure and impaired mechanical properties of single myocytes of rat heart. Cardiovasc. Res. 45, 981–993 [DOI] [PubMed] [Google Scholar]
- 8. Tsuji T., Ohga Y., Yoshikawa Y., Sakata S., Abe T., Tabayashi N., Kobayashi S., Kohzuki H., Yoshida K. I., Suga H., Kitamura S., Taniguchi S., Takaki M. (2001) Rat cardiac contractile dysfunction induced by Ca2+ overload: possible link to the proteolysis of α-fodrin. Am. J. Physiol. Heart Circ. Physiol. 281, H1286–H1294 [DOI] [PubMed] [Google Scholar]
- 9. Inserte J., Garcia-Dorado D., Hernando V., Soler-Soler J. (2005) Calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ. Res. 97, 465–473 [DOI] [PubMed] [Google Scholar]
- 10. Kang M. Y., Zhang Y., Matkovich S. J., Diwan A., Chishti A. H., Dorn G. W., 2nd (2010) Receptor-independent cardiac protein kinase Cα activation by calpain-mediated truncation of regulatory domains. Circ. Res. 107, 903–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neuhof C., Fabiunke V., Deibele K., Speth M., Möller A., Lubisch W., Fritz H., Tillmanns H., Neuhof H. (2004) Reduction of myocardial infarction by calpain inhibitors A-705239 and A-705253 in isolated perfused rabbit hearts. Biol. Chem. 385, 1077–1082 [DOI] [PubMed] [Google Scholar]
- 12. Khalil P. N., Neuhof C., Huss R., Pollhammer M., Khalil M. N., Neuhof H., Fritz H., Siebeck M. (2005) Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. Eur. J. Pharmacol. 528, 124–131 [DOI] [PubMed] [Google Scholar]
- 13. Yoshikawa Y., Hagihara H., Ohga Y., Nakajima-Takenaka C., Murata K. Y., Taniguchi S., Takaki M. (2005) Calpain inhibitor-1 protects the rat heart from ischemia-reperfusion injury: analysis by mechanical work and energetics. Am. J. Physiol. Heart Circ. Physiol. 288, H1690–H1698 [DOI] [PubMed] [Google Scholar]
- 14. Hernando V., Inserte J., Sartório C. L., Parra V. M., Poncelas-Nozal M., Garcia-Dorado D. (2010) Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell Cardiol. 49, 271–279 [DOI] [PubMed] [Google Scholar]
- 15. Steenbergen C., Murphy E., Levy L., London R. E. (1987) Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart. Circ. Res. 60, 700–707 [DOI] [PubMed] [Google Scholar]
- 16. Imahashi K., Pott C., Goldhaber J. I., Steenbergen C., Philipson K. D., Murphy E. (2005) Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ. Res. 97, 916–921 [DOI] [PubMed] [Google Scholar]
- 17. Sandmann S., Yu M., Unger T. (2001) Transcriptional and translational regulation of calpain in the rat heart after myocardial infarction: effects of AT(1) and AT(2) receptor antagonists and ACE inhibitor. Br. J. Pharmacol. 132, 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandmann S., Prenzel F., Shaw L., Schauer R., Unger T. (2002) Activity profile of calpains I and II in chronically infarcted rat myocardium: influence of the calpain inhibitor CAL 9961. Br. J. Pharmacol. 135, 1951–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshida H., Takahashi M., Koshimizu M., Tanonaka K., Oikawa R., Toyo-oka T., Takeo S. (2003) Decrease in sarcoglycans and dystrophin in failing heart following acute myocardial infarction. Cardiovasc. Res. 59, 419–427 [DOI] [PubMed] [Google Scholar]
- 20. Takahashi M., Tanonaka K., Yoshida H., Oikawa R., Koshimizu M., Daicho T., Toyo-Oka T., Takeo S. (2005) Effects of ACE inhibitor and AT1 blocker on dystrophin-related proteins and calpain in failing heart. Cardiovasc. Res. 65, 356–365 [DOI] [PubMed] [Google Scholar]
- 21. Takahashi M., Tanonaka K., Yoshida H., Koshimizu M., Daicho T., Oikawa R., Takeo S. (2006) Possible involvement of calpain activation in pathogenesis of chronic heart failure after acute myocardial infarction. J. Cardiovasc. Pharmacol. 47, 413–421 [DOI] [PubMed] [Google Scholar]
- 22. Saitoh T., Nakajima T., Takahashi T., Kawahara K. (2006) Changes in cardiovascular function on treatment of inhibitors of apoptotic signal transduction pathways in left ventricular remodeling after myocardial infarction. Cardiovasc. Pathol. 15, 130–138 [DOI] [PubMed] [Google Scholar]
- 23. Mani S. K., Balasubramanian S., Zavadzkas J. A., Jeffords L. B., Rivers W. T., Zile M. R., Mukherjee R., Spinale F. G., Kuppuswamy D. (2009) Calpain inhibition preserves myocardial structure and function following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 297, H1744–H1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carragher N. O. (2006) Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr. Pharm. Des. 12, 615–638 [DOI] [PubMed] [Google Scholar]
- 25. Takano J., Tomioka M., Tsubuki S., Higuchi M., Iwata N., Itohara S., Maki M., Saido T. C. (2005) Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J. Biol. Chem. 280, 16175–16184 [DOI] [PubMed] [Google Scholar]
- 26. Kawai S., Takagi Y., Kaneko S., Kurosawa T. (2011) Effect of three types of mixed anesthetic agents alternate to ketamine in mice. Exp. Anim. 60, 481–487 [DOI] [PubMed] [Google Scholar]
- 27. Takagawa J., Zhang Y., Wong M. L., Sievers R. E., Kapasi N. K., Wang Y., Yeghiazarians Y., Lee R. J., Grossman W., Springer M. L. (2007) Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J. Appl. Physiol. 102, 2104–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zou Y., Akazawa H., Qin Y., Sano M., Takano H., Minamino T., Makita N., Iwanaga K., Zhu W., Kudoh S., Toko H., Tamura K., Kihara M., Nagai T., Fukamizu A., Umemura S., Iiri T., Fujita T., Komuro I. (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 6, 499–506 [DOI] [PubMed] [Google Scholar]
- 29. Galvez A. S., Diwan A., Odley A. M., Hahn H. S., Osinska H., Melendez J. G., Robbins J., Lynch R. A., Marreez Y., Dorn G. W., 2nd (2007) Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ. Res. 100, 1071–1078 [DOI] [PubMed] [Google Scholar]
- 30. Harris T. J., Tepass U. (2010) Adherens junctions: from molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 11, 502–514 [DOI] [PubMed] [Google Scholar]
- 31. Luo Y., Radice G. L. (2003) Cadherin-mediated adhesion is essential for myofibril continuity across the plasma membrane but not for assembly of the contractile apparatus. J. Cell Sci. 116, 1471–1479 [DOI] [PubMed] [Google Scholar]
- 32. Sedarous M., Keramaris E., O'Hare M., Melloni E., Slack R. S., Elce J. S., Greer P. A., Park D. S. (2003) Calpains mediate p53 activation and neuronal death evoked by DNA damage. J. Biol. Chem. 278, 26031–26038 [DOI] [PubMed] [Google Scholar]
- 33. Moubarak R. S., Yuste V. J., Artus C., Bouharrour A., Greer P. A., Menissier-de Murcia J., Susin S. A. (2007) Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 27, 4844–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen M., He H., Zhan S., Krajewski S., Reed J. C., Gottlieb R. A. (2001) Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 276, 30724–30728 [DOI] [PubMed] [Google Scholar]
- 35. Nakagawa T., Yuan J. (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150, 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polster B. M., Basañez G., Etxebarria A., Hardwick J. M., Nicholls D. G. (2005) Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 280, 6447–6454 [DOI] [PubMed] [Google Scholar]
- 37. Chen Q., Paillard M., Gomez L., Ross T., Hu Y., Xu A., Lesnefsky E. J. (2011) Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem. Biophys. Res. Commun. 415, 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Artal-Sanz M., Tavernarakis N. (2005) Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS Lett. 579, 3287–3296 [DOI] [PubMed] [Google Scholar]
- 39. Yousefi S., Perozzo R., Schmid I., Ziemiecki A., Schaffner T., Scapozza L., Brunner T., Simon H. U. (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8, 1124–1132 [DOI] [PubMed] [Google Scholar]
- 40. Liu X., Van Vleet T., Schnellmann R. G. (2004) The role of calpain in oncotic cell death. Annu. Rev. Pharmacol. Toxicol. 44, 349–370 [DOI] [PubMed] [Google Scholar]
- 41. Bano D., Young K. W., Guerin C. J., Lefeuvre R., Rothwell N. J., Naldini L., Rizzuto R., Carafoli E., Nicotera P. (2005) Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120, 275–285 [DOI] [PubMed] [Google Scholar]
- 42. Ma J., Wei M., Wang Q., Li J., Wang H., Liu W., Lacefield J. C., Greer P. A., Karmazyn M., Fan G. C., Peng T. (2012) Deficiency of Capn4 gene inhibits nuclear factor-κB (NF-κB) protein signaling/inflammation and reduces remodeling after myocardial infarction. J. Biol. Chem. 287, 27480–27489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taneike M., Mizote I., Morita T., Watanabe T., Hikoso S., Yamaguchi O., Takeda T., Oka T., Tamai T., Oyabu J., Murakawa T., Nakayama H., Nishida K., Takeda J., Mochizuki N., Komuro I., Otsu K. (2011) Calpain protects the heart from hemodynamic stress. J. Biol. Chem. 286, 32170–32177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jackson B. M., Gorman J. H., Moainie S. L., Guy T. S., Narula N., Narula J., John-Sutton M. G., Edmunds L. H., Jr., Gorman R. C. (2002) Extension of borderzone myocardium in postinfarction dilated cardiomyopathy. J. Am. Coll. Cardiol. 40, 1160–1167; discussion 1168–1171 [DOI] [PubMed] [Google Scholar]
- 45. Shumway S. D., Maki M., Miyamoto S. (1999) The PEST domain of IκBα is necessary and sufficient for in vitro degradation by μ-calpain. J. Biol. Chem. 274, 30874–30881 [DOI] [PubMed] [Google Scholar]
- 46. Sato N., Fujio Y., Yamada-Honda F., Funai H., Wada A., Kawashima S., Awata N., Shibata N. (1995) Elevated calcium level induces calcium-dependent proteolysis of A-CAM (N-cadherin) in heart: analysis by detergent-treated model. Biochem. Biophys. Res. Commun. 217, 649–653 [DOI] [PubMed] [Google Scholar]
- 47. Jang Y. N., Jung Y. S., Lee S. H., Moon C. H., Kim C. H., Baik E. J. (2009) Calpain-mediated N-cadherin proteolytic processing in brain injury. J. Neurosci. 29, 5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kostetskii I., Li J., Xiong Y., Zhou R., Ferrari V. A., Patel V. V., Molkentin J. D., Radice G. L. (2005) Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 96, 346–354 [DOI] [PubMed] [Google Scholar]
- 49. Matsushita T., Oyamada M., Fujimoto K., Yasuda Y., Masuda S., Wada Y., Oka T., Takamatsu T. (1999) Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ. Res. 85, 1046–1055 [DOI] [PubMed] [Google Scholar]
- 50. Wang X., Gerdes A. M. (1999) Chronic pressure overload cardiac hypertrophy and failure in guinea pigs. III. Intercalated disc remodeling. J. Mol. Cell Cardiol. 31, 333–343 [DOI] [PubMed] [Google Scholar]
- 51. Schaper J., Froede R., Hein S., Buck A., Hashizume H., Speiser B., Friedl A., Bleese N. (1991) Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 83, 504–514 [DOI] [PubMed] [Google Scholar]
- 52. Dorn G. W., 2nd (2009) Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat. Rev. Cardiol. 6, 283–291 [DOI] [PubMed] [Google Scholar]
- 53. Letavernier E., Perez J., Bellocq A., Mesnard L., de Castro Keller A., Haymann J. P., Baud L. (2008) Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ. Res. 102, 720–728 [DOI] [PubMed] [Google Scholar]
- 54. Scalia R., Gong Y., Berzins B., Freund B., Feather D., Landesberg G., Mishra G. (2011) A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling. Circ. Res. 108, 1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]