

Background: Ca2+-independent autonomous CaMKII activity and nitric oxide (NO) signaling regulate neuronal function and death.
Results: NO generated autonomous CaMKII activity by Ca2+/CaM-dependent S-nitrosylation, and CaMKII inhibition protected from NO-induced neuronal cell death.
Conclusion: NO-mediated regulation of CaMKII contributes to its pathological functions.
Significance: S-Nitrosylation is a novel path to CaMKII autonomy that connects Ca2+- and NO signaling.
Keywords: Ca2+/Calmodulin-dependent Protein Kinase II (CaMKII), Calmodulin (CaM), Excitotoxicity, Nitric Oxide, S-Nitrosylation, Autonomy, Kinase Regulation
Abstract
Both signaling by nitric oxide (NO) and by the Ca2+/calmodulin (CaM)-dependent protein kinase II α isoform (CaMKIIα) are implicated in two opposing forms of synaptic plasticity underlying learning and memory, as well as in excitotoxic/ischemic neuronal cell death. For CaMKIIα, these functions specifically involve also Ca2+-independent autonomous activity, traditionally generated by Thr-286 autophosphorylation. Here, we demonstrate that NO-induced S-nitrosylation of CaMKIIα also directly generated autonomous activity, and that CaMKII inhibition protected from NO-induced neuronal cell death. NO induced S-nitrosylation at Cys-280/289, and mutation of either site abolished autonomy, indicating that simultaneous nitrosylation at both sites was required. Additionally, autonomy was generated only when Ca2+/CaM was present during NO exposure. Thus, generation of this form of CaMKIIα autonomy requires simultaneous signaling by NO and Ca2+. Nitrosylation also significantly reduced subsequent CaMKIIα autophosphorylation specifically at Thr-286, but not at Thr-305. A previously described reduction of CaMKII activity by S-nitrosylation at Cys-6 was also observed here, but only after prolonged (>5 min) exposure to NO donors. These results demonstrate a novel regulation of CaMKII by another second messenger system and indicate its involvement in excitotoxic neuronal cell death.
Introduction
Ca2+/calmodulin (CaM)3-dependent protein kinase II α isoform (CaMKIIα) and its “autonomous” activity (Fig. 1) (1, 2) mediates both long-term potentiation (LTP) (3, 4) and depression (LTD) (5) of synaptic strength, as well as excitotoxic neuronal cell death during ischemia (6, 7). Several ways to generate CaMKII autonomy have been prominently described over three decades, and include several very recent additions: Thr-286 autophosphorylation (8, 9), T-site-mediated GluN2B binding (10), Met-281/282 oxidation (11), and Ser-279 glycosylation (12) (Fig. 1), the latter two for the CaMKIIó isoform that is dominant in the heart (13). In all cases, an initial Ca2+/CaM-stimulation is required to induce the CaMKII autonomy, which then persists when the Ca2+ stimulus has subsided and Ca2+/CaM has dissociated. However, autonomous CaMKII is by no means fully active, and is instead significantly further stimulated by Ca2+/CaM, at least for normal CaMKII substrates (8, 14–16). The highest level of CaMKII autonomy (the ratio of autonomous over maximally stimulated activity) is likely generated by Thr-286 autophosphorylation (1), but with an autonomy of ∼20%, even Thr-286-phosphorylated CaMKII is still ∼5-fold further stimulated by Ca2+/CaM (8, 14–16). This mechanism allows for a molecular memory of past Ca2+-signals by the autonomous activity, but may also prevent complete uncoupling from subsequent cellular Ca2+ signaling. However, physiological functions for such further stimulation of autonomous CaMKII have been described only very recently, specifically in determining whether autonomous CaMKII promotes potentiation or depression of synaptic strength (5).
FIGURE 1.
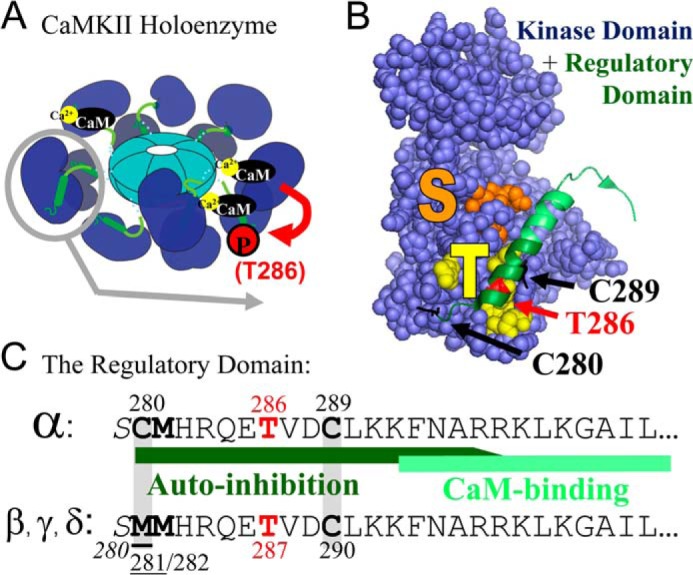
Model of CaMKII structure and regulation. A, CaMKII forms 12meric holoenzymes via C-terminal association domains, with the kinase domains radiating outward (1, 2). Each kinase subunit is activated separately by direct binding of Ca2+/CaM, which can also induce intra-holoenzyme inter-subunit Thr-286 autophosphorylation (9). B, structure of the CaMKII kinase (space-fill) and regulatory (ribbon) domains (2). In the basal inactive state, the substrate binding S-site (orange) is blocked by the regulatory domain, which is held in place in part by interactions with the neighboring T-site (yellow). Thr-286 (red) autophosphorylation (8, 9, 14–16) or T-site-mediated binding to GluN2B (10) generates “autonomous” CaMKII activity, likely by preventing complete re-association of the regulatory domain with the kinase domain (1). The Cys-280 and Cys-289 S-nitrosylation sites identified here are marked in black. C, sequence of the CaMKIIα autoregulatory domain with specific residues and regions marked as in panel B. The autoregulatory domain of the CaMKIIβ, γ, and ó isoforms is highly homologous, with only one amino acid exchanged (underlined). The Met-281/282 oxidation (11) and Ser-280 glycosylation (12) sites that also generate CaMKII autonomy are indicated. Note that all pathways to CaMKII autonomy require an initial Ca2+/CaM-stimulus, and that even autonomous activity is further stimulated by Ca2+/CaM (1).
In CaMKIIα, the major isoform in the brain (1), the residues homologous to Met-281/282 in the δ isoforms are Cys-280/Met-281 (Fig. 1), and the substitution of one methionine for a cysteine raised the possibility of physiological regulation by nitric oxide (NO)-mediated S-nitrosylation (17–19), in addition to pathological regulation by oxidation. Such potential CaMKII regulation was intriguing, as nitric oxide (NO) and NO synthase (NOS) are also implicated in hippocampal LTP, LTD, and excitotoxic neuronal cell death (19–23). The results of this study show that specific S-nitrosylation generated CaMKII autonomy and that CaMKII inhibition protected from NO-induced neuronal cell death.
EXPERIMENTAL PROCEDURES
Proteins
CaMKIIα wt and CaM were purified after baculovirus/sf9 cell expression or bacterial expression as previously described (16, 24). For comparisons of CaMKII wt and mutants, GFP-CaMKII fusion proteins were used (25), which were purified after expression in HEK cells. Briefly, HEK cells were homogenized with a motorized pellet pestle (Kontes) for 10 s in 0.4 ml of ice-cold 50 mm PIPES pH 7.2, 10% glycerol, 1 mm EDTA, 1 mm DTT, and complete protease inhibitor (Roche), then centrifuged at 16,000 × g for 20 min. The CaMKII concentration from the resulting supernatant was determined by their GFP-fluorescence measured in a spectrophotometer (Fluoromax 3; Horiba Jobin Yvon). For the comparison of CaMKII wt to T286A, these extracts were adjusted for equal CaMKII and total HEK cell protein amounts (26, 27). All other mutants were further purified as previously described (16, 24). Protein phosphatase 2A C subunit was purchased from Cayman Chemical. The substrate peptide Syntide-2 was from Genescript.
NO Release
NO release from the NO donor Diethylamine NONOate (DEA-NONOate) was measured spectrophotometrically in reaction buffer or deionized water by the disappearance of absorption at 250 nm due to loss of parent compound (ϵ = 6500 m−1cm−1).
CaMKII Autophosphorylation, Nitrosylation, and Oxidation
CaMKII was made autonomous by reacting 200–300 nm kinase for 5 min with Ca2+/CaM (1 mm/1 μm) and 50 mm PIPES pH 7.1 in the presence of either ATP/Mg2+ (0.1/10 mm) on ice (for Thr-286 autophosphorylation) or at room temperature with DEA-NONOate (3 mm; for nitrosylation; Cayman Chemical) or H202 (3 mm; for oxidation). The reaction was diluted and Ca2+ chelated with EDTA (5 mm). Where indicated, kinase was reacted with peroxynitrite (ONOO− Cayman Chemical) instead of NO, and/or in the absence or presence of the ONOO− scavengers tryptophan (Sigma) or MnTMPyP (A.G. Scientific).
For experiments where kinase was both phosphorylated and nitrosylated, CaMKII was reacted with DEA-NONOate at room temperature followed by phosphorylation of Thr-286 by addition of ATP/Mg2+ on ice, or this was reversed and kinase was first phosphorylated at Thr-286 on ice, phosphorylation was stopped by addition of EDTA, then the kinase was reacted with NO at room temperature. Thr-305/306 phosphorylation was induced by chelating Ca2+ in the presence of ATP (after Thr-286 phosphorylation or nitrosylation).
CaMKII Activity Assays
Kinase activity was assessed by 32P incorporation into the peptide substrate syntide 2, either in the presence of Ca2+/CaM (1.2 mm/1 μm) or EGTA (1.5 mm), as previously described (16, 24). To ensure that reactions are within the linear range, 1 min reaction times (at 30 °C) were used for stimulated activity and Thr-286-induced autonomy (16); reaction times of up to 5 min were used only for conditions with lower activity.
Western Analysis
Samples were boiled in SDS-PAGE loading buffer (2% SDS, 50 mm DTT, 67.5 mm Tris pH 6.8, 10% glycerol, 0.16 mg/ml bromphenol blue), separated on 10% acrylamide gels then transferred to PVDF membranes. Blots were blocked with 5% nonfat dry milk (or 5% BSA for the phospho-T305 detection) for 1 h at room temperature, then incubated overnight at 4 °C with either antibodies against either CaMKIIα (1:2000; CBα2, produced in-house), phospho-T286 (1:3000; Phosphosolutions), or phospho-T305 (1:1000; AssayBiotech). Blots were imaged with a chemiimager (Alpha Innotech) after exposure to Supersignal West Femto ECL reagent (Pierce).
Detection of S-Nitrosylation
Detection of S-nitrosylation was assessed using the biotin-switch method (28). CaMKII WT or mutant was nitrosylated in the presence of Ca2+/CaM in HEPES pH 7.1 buffer as described above, then diluted in HEN buffer (HEPES pH 7.7, 1 mm EDTA, and 0.1 mm neocuprione). Proteins were then precipitated at −20 °C with 4 volumes of cold acetone for 20 min and centrifuged at 15,000 × g for 10 min. The pellet was rinsed with ice cold acetone, resuspended in HEN buffer, then non-nitrosylated cysteines were blocked with 25 mm MMTS in 2.5% SDS for 30 min at 50 °C. Proteins were again acetone precipitated to remove unreacted MMTS and resuspended in HEN buffer with 1% SDS. The nitro-cysteine was then reduced with 1 mm ascorbate and the reduced cysteine labeled using the non-cleavable biotin-BMCC (250 μm) for 1 h at room temperature. Unreacted biotin was removed by acetone precipitating the proteins. Proteins were then run on Western blots, and biotinylation was detected using the avidin-based vectastain kit (Vector Laboratories).
Neuronal Cell Death Assays
Neuronal cell death was measured by release of LDH into the media using a cytotoxicity detection kit (Roche) as previously described (6). Briefly, medium density primary disassociated hippocampal cultures were prepared from newborn Sprague-Dawley rats and plated onto poly-d-lysine-coated 24-well plates. Cultures were maintained in Neurobasal A media with B-27 supplement, 50 units/ml penicillin/streptomycin and 2 mm Glutamax at 37 °C in 5% CO2. After 14 DIV, cell death was induced by addition of 100 μm glutamate or 300 μm DEA-NONOate to the media for 5 min. Wells with inhibitor had 100 μm APV or 5 μm tatCN21 or scrambled control peptide included during the treatment as well a 20-min pretreatment.
RESULTS
NO Generates Autonomous CaMKII Activity
Incubation of purified CaMKIIα (in the presence of Ca2+/CaM) either with an oxidizer (H2O2) or with an NO donor (DEA-NONOate) both generated autonomous activity (measured after chelation of Ca2+), with higher autonomy generated by the NO donor (Fig. 2A). NO release by the NO donor is well controlled, with no release under acidic conditions (as in deionized water) and with near maximal release within 5 min after addition to buffer at pH 7.2 (Fig. 2B). As expected, NO-induced autonomy was abolished by C280/M281V mutation (analogous to CaMKIIδ oxidation and M281/282V mutation; Ref. 11). However, surprisingly, autonomy was also abolished by C289V mutation (Fig. 2A), indicating that simultaneous S-nitrosylation of both Cys-280 and Cys-289 was required to induce autonomy, and that individual nitrosylation of either residue alone was not sufficient.
FIGURE 2.

NO induces Ca2+-independent autonomous CaMKII activity by a process that requires both Cys-280 and Cys-289. A, incubation of CaMKII (in the presence of Ca2+/CaM) either with the oxidizer H2O2 or the NO donor DEA-NONOate for 5 min generated autonomous CaMKII activity (measured by phosphorylation of syntide 2 after chelating Ca2+ with EGTA). NO-induced autonomy was abolished by mutation of either Cys-280 or Cys-289. Bar graphs indicate absolute kinase activity as mean ± S.E. The numbers inserted for each bar indicate the level of autonomy (in percent of maximal Ca2+/CaM-stimulated activity). B, NO release from the NO donor was readily triggered in buffer at pH 7.2, but not in acidic deionized water. C, NO-induced CaMKII autonomy (measured as in panel A) was not reduced by T286A mutation or by the Ca2+/CaM competitive inhibitor KN93 (10 μm), but effectively blocked by the peptide inhibitor tatCN21 (5 μm). These experiments were done with CaMKII from crude HEK cell extracts, and non-transfected mock extracts were thus used as additional control. Bar graphs indicate mean ± S.E.
NO-induced CaMKII Autonomy Is Independent of Thr-286 and Blocked by tatCN21 but Not KN93
To test the possibility that generation of autonomous CaMKII activity by the NO donor was dependent on promoting Thr-286 autophosphorylation, a CaMKII T286A mutant was used. The NO donor induced autonomy also for the CaMKII T286A mutant (Fig. 2C), indicating that the effect was independent of Thr-286 phosphorylation. If any, NO-induced autonomy of the T286A mutant was even higher compared with CaMKII wild type (Fig. 2C), an effect that could be due to reduced auto-inhibitory interactions within the T286A mutant. Additionally, the effects of two mechanistically distinct CaMKII inhibitors were tested: KN93, which is Ca2+/CaM-competitive (29), and tatCN21, which does not act through competition with Ca2+/CaM (27). As expected based on these different modes of inhibition, tatCN21 completely blocked NO-induced autonomous CaMKII activity, while KN93 had no significant effect (Fig. 2C). The same differential effect of these two inhibitors has been observed previously for Thr-286-induced CaMKII autonomy (6).
Induction of NO-mediated Autonomy Requires an Initial Ca2+/CaM Stimulus
While a 5-min incubation with the NO donor and Ca2+/CaM generated autonomous CaMKII activity (Figs. 2A and 3A), it did not affect maximally Ca2+/CaM-stimulated activity (Fig. 3A), as measured by subsequent activity assays with or without chelation of Ca2+ after the initial incubation. Presence of Ca2+/CaM was required to induce CaMKII autonomy by the NO donor, as no autonomous activity was induced when Ca2+/CaM was omitted from the initial incubation (Fig. 3A). Thus, generation of this novel form of CaMKII autonomy requires coinciding NO and Ca2+ signals.
FIGURE 3.

NO requires Ca2+/CaM to induce CaMKII autonomy and inhibits stimulated activity after prolonged exposure. A, 5 min incubation with NO donor induced autonomous CaMKII activity only in the presence but not in the absence of Ca2+/CaM. Maximal Ca2+/CaM-stimulated CaMKII activity was not affected. Bar graphs indicate absolute kinase activity as mean ± S.E. The numbers inserted for each bar of the right panel indicate the level of autonomy (in percent of maximal Ca2+/CaM-stimulated activity). B, NO donors inhibits Ca2+/CaM-stimulated and NO-induced autonomous CaMKII activity, but only after prolonged exposure (>5 min). Stimulated and autonomous CaMKII activities were normalized to their respective maximums. The quantifications indicate mean ± S.E.
Prolonged Exposure to Nitric Oxide Reduces CaMKII Activity
A previous study showed that NO inhibits CaMKII activity through S-nitrosylation at C6 (30). To address this apparently conflicting finding, CaMKII was exposed to NO for varying periods of time (Fig. 3B). Indeed, while Ca2+/CaM-stimulated CaMKII activity was unaffected by a 5 min exposure to NO, it was significantly reduced after prolonged exposure times (Fig. 3B). A similar time-dependent reduction was also observed for the NO-induced autonomous activity (Fig. 3B), indicating that the observed reduction of CaMKII activity was not mediated by interference with Ca2+/CaM-stimulation. Thus, while NO can indeed inhibit both stimulated and autonomous CaMKII activity, this opposing effect requires prolonged NO signaling, while shorter NO signals are sufficient to generate autonomous activity.
NO Reduces Ca2+/CaM-induced CaMKII Thr-286 Autophosphorylation
For other forms of Thr-286-independent forms of CaMKII autonomy, a positive cross regulation with Thr-286-induced autonomy has been suggested (10–12). By contrast, previous exposure to NO in presence of Ca2+/CaM dramatically reduced subsequent CaMKII autophosphorylation at Thr-286, which was induced by addition of ATP (Fig. 4A). While both Thr-286 phosphorylation and NO generate autonomous CaMKII activity, the level of Thr-286-induced autonomy is higher: While autonomy by Thr-286 phosphorylation is ∼20% of maximally stimulated activity (8, 16), autonomy by NO was only ∼10% (see Fig. 2A). Indeed, addition of NO donors prior to a Thr-286 autophosphorylation reaction reduced not only Thr-286 phosphorylation (Fig. 4A), but also the resulting autonomous activity (Fig. 4B). A similar effect was caused by the oxidizer H2O2 (Fig. 4B) that generated a lower level of autonomy than NO (see Fig. 2A).
FIGURE 4.

NO exposure inhibits subsequent CaMKII Thr-286 autophosphorylation. A, pre-incubation with NO donor inhibited subsequent Ca2+/CaM-induced CaMKII autophosphorylation at Thr-286. Western-analysis with a phospho-specific antibody is shown. B, pre-incubation with NO donor or H2O2 significantly reduced the level of CaMKII autonomy induced by a 5 min Thr-286 autophosphorylation reaction. Bar graphs indicate mean ± S.E.
NO Has No Significant Effects on CaMKII Thr-305 Autophosphorylation
CaMKII Thr-305/306 autophosphorylation inhibits subsequent Ca2+/CaM binding (31, 32). Vice versa, Ca2+/CaM binding also suppresses Thr-305/306 phosphorylation. Thus, Thr-305/306 is most efficiently autophosphorylated after dissociation of CaM from Thr-286-phosphorylated autonomous CaMKII. Additional NO exposure of previously Thr-286-phosphorylated CaMKII did not reduce the Thr-305 autophosphorylation that was induced by chelating Ca2+ to dissociate CaM (Fig. 5). Thus, the NO-mediated inhibition of CaMKII autophosphorylation was specific to Thr-286 (see Fig. 4) and did not extend to Thr-305.
FIGURE 5.

NO exposure does not affect subsequent CaMKII T305 autophosphorylation. After NO exposure and/or Thr-286 autophosphorylation (as indicated), CaMKII autophosphorylation at Thr-305 was induced by chelation of Ca2+ and continued for 5 min at 30 °C. Thr-305 phosphorylation was detected by Western analysis (lower panel) and quantified by normalized immunodetection values (IDVs; upper panel). Basal background phospho-T305 detection prior to the reactions is indicated by the dotted line. While previous Thr-286 phosphorylation dramatically enhanced subsequent Thr-305 phosphorylation, previous NO exposure had no significant effects. Bar graphs indicate mean ± S.E.
This finding raised the possibility that generation of autonomous CaMKII activity by NO exposure may even substitute for Thr-286 phosphorylation in enabling efficient Thr-305/306 phosphorylation. However, NO exposure did not cause any significant increase in Thr-305 autophosphorylation for CaMKII that was not previously Thr-286 phosphorylated (Fig. 5).
NO-induced CaMKII Autonomy Is Mediated by Specific S-Nitrosylation
In addition to S-nitrosylation of cysteine residues, NO can induce oxidation of cysteine and methionine residues, the latter through formation of peroxynitrite (ONOO−). However, four independent lines of evidence showed that the NO-induced CaMKII autonomy was indeed caused by S-nitrosylation of Cys-280/Cys-289 and not by ONOO−-mediated oxidation:
The effect of NO donors was larger compared with oxidation by H2O2 (see Fig. 2A), consistent with a bulkier modification by nitrosylation causing more efficient relief from autoinhibition.
NO donors indeed caused S-nitrosylation of CaMKII (Fig. 6A), as detected by the biotin-switch method (28). This specifically included nitrosylation of Cys-280/Cys-289, as mutation of these residues clearly reduced the detected nitrosylation (Fig. 6B).
The ONOO− scavengers tryptophan (Trp) or MnTMPyP (MnTP) did not reduce the effect of NO donors, but instead even further enhanced it (Fig. 7A), consistent with suppression of Cys-280/289 oxidation allowing more nitrosylation of these residues. Comparing scavenger addition before and after the nitrosylation reaction (both before the kinase activity assays) confirmed that the scavengers affected the nitrosylation reaction and did not have any other direct effects on kinase activity (Fig. 7A).
The effect of directly adding ONOO− was much smaller compared with adding NO donors. Importantly, the effect of ONOO− was abolished by the ONOO− scavenger Trp, but dramatically enhanced by MnTP, an ONOO− scavenger that releases NO during ONOO− consumption (Fig. 7B).
FIGURE 6.
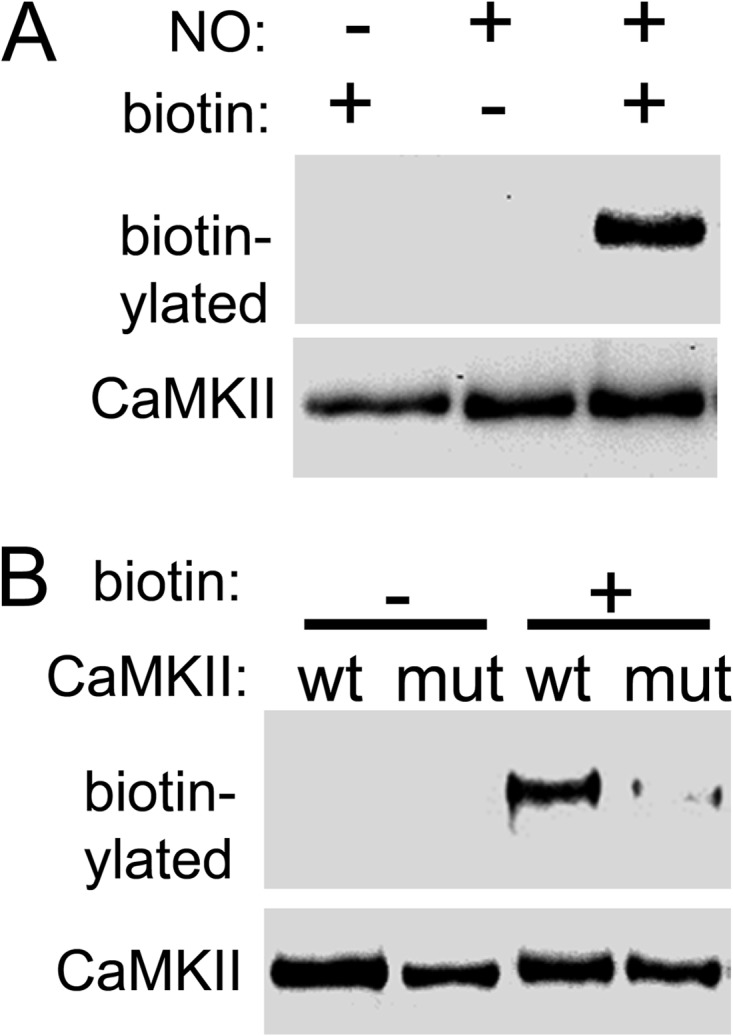
NO induces CaMKII S-nitrosylation at Cys-280/289. A, NO donor induced S-nitrosylation of CaMKII, as detected by the biotin-switch method. B, nitrosylation included Cys-280/289, as the detected nitrosylation was reduced by mutation of these residues (mut).
FIGURE 7.

NO induces CaMKII autonomy via S-nitrosylation. A, ONOO− scavengers tryptophan (Trp) and MnTMPyP (MnTP) did not block but enhance the NO donor effect on CaMKII autonomy. The scavengers had no effect on CaMKII activity when instead added only immediately before the kinase assays. B, direct ONOO− addition resulted only in a small increase in CaMKII autonomy, which was blocked by Trp but enhanced by MnTP, an ONOO− scavenger that releases NO during ONOO− consumption. The quantifications indicate mean ± S.E.
NO-induced Cell Death Is Reduced by CaMKII Inhibition
In dissociated hippocampal neuron cultures, similar levels of cell death were triggered by excitotoxic glutamate and by direct application of NO donors (Fig. 8). Importantly, CaMKII inhibition with tatCN21 significantly protected from cell death not only when induced by glutamate (6, 7), but also when induced by NO donors (Fig. 8). Notably, both glutamate- and NO-induced cell death were also reduced by inhibiting the NMDA-type glutamate receptor with APV (Fig. 8), indicating that NO-induced cell death also required Ca2+ influx through this receptor (as induced by the high spontaneous neuronal activity within the cultures). The observed Ca2+ requirement for NO-induced cell death is also consistent with the regulation of NO-induced CaMKII autonomy, which also required a Ca2+ stimulus (see Fig. 3A).
FIGURE 8.

CaMKII inhibition protects from neuronal cell death induced by NO donors. Glutamate (100 μm) or NO donor (300 μm) triggered similar levels of death in hippocampal cultures, as measured 24 h later by lactate dehydrogenase (LDH) release. The NMDA receptor antagonist APV (100 μm) significantly reduced cell death in either case. As described for glutamate insults, the CaMKII inhibitor tatCN21 (5 μm) also protected from NO-induced cell death. Bar graphs indicate mean ± S.E.
DISCUSSION
The results of this study demonstrate that NO-induced CaMKII autonomy is a novel regulatory mechanism for an enzyme critically involved in mediating synaptic plasticity (1, 3–5) and ischemic/excitotoxic neuronal cell death (6, 7). Indeed, CaMKII inhibition protected also from neuronal cell death directly induced by NO donors. NO can cause S-nitrosylation or oxidation of proteins (the latter via formation of ONOO−); however, the results of this study showed that CaMKII autonomy was induced by specific S-nitrosylation that occurs at residues Cys-280/289. Mutation of either residue abolished NO-induced autonomy, indicating requirement of their simultaneous nitrosylation.
Generation of CaMKII autonomy by autophosphorylation at Thr-286 has been considered to be a hallmark feature of CaMKII regulation for almost 30 years, as it enables the kinase to “remember” past Ca2+ stimuli (8, 14–16) (for review see Ref. 1). Several additional ways to generate autonomous CaMKII activity have been prominently described more recently: Binding to GluN2B (10), Met-280/281 oxidation (11), and Ser-279 O-linked glucosylation (12). The novel form of autonomy that is generated by NO-induced S-nitrosylation shares several features with these previously described autonomy mechanisms. All known forms of CaMKII autonomy require an initial Ca2+/CaM-stimulus for induction (8–12). Thus, they all provide a molecular memory of past Ca2+ stimuli. However, in case of autonomy induced by oxidation, glucosylation, or S-nitrosylation, this molecular memory is only formed in presence of a coinciding second type of signal. For oxidation and glucosylation, the second signal appears to be typically pathological (such as oxidative stress or elevated blood sugar in diabetes; Refs. 11, 12). By contrast, NO signals that are required for S-nitrosylation could additionally be generated by physiological signaling. All forms of autonomous CaMKII are also significantly further stimulated by Ca2+/CaM (for review see Ref. 1). This can retain Ca2+ sensitivity even of “autonomous” CaMKII and may prevent complete uncoupling from subsequent Ca2+ stimuli. Indeed, recent evidence suggests that autonomous CaMKII activity can have opposing effects on synaptic strength, depending on presence or absence of additional Ca2+/CaM stimulation (5).
With ∼20% of maximal Ca2+/CaM-stimulated activity, Thr-286 phosphorylation likely generates the highest level of CaMKII. While S-nitrosylation generated lower levels of autonomy (∼10–15%), this was significantly higher than autonomy induced by oxidation (∼5%), an effect that may be correlated to the bulk of the respective modification, as well as with the position of the modified residues within the auto-inhibitory region of the regulatory domain (see Fig. 1). Autonomy induced by NO is likely also higher compared with autonomy induced by GluN2B binding or glycosylation, although this is more difficult to compare. Assessment of GluN2B induced autonomy (10, 25) was done using a substrate peptide that enhances autonomy through a special mechanism not shared with normal substrates (16). Consequently, the actual level of autonomy is likely lower than originally reported (10, 25) and ∼5–10% autonomy was put forward as a more likely estimate (1). The level of autonomy induced by glucosylation (specifically O-linked N-acetylglucosamine modification of Ser-279) is unclear (12). Using the Camui FRET-sensor of CaMKII activity (33), conditions that induced CaMKII glucosylation caused the same extent of loss in FRET as full stimulation with Ca2+/CaM (12). However, these conditions increased actual CaMKII activity only 1.7-fold over baseline, and activity in response to full Ca2+/CaM stimulation was not compared (12). Based on the ∼1000-fold stimulation of CaMKII by Ca2+/CaM (1), these results could indicate as little as 0.17% autonomy (or up to 7% autonomy, in case that the baseline was largely due to background and contribution of basal CaMKII activity was only 1%). Furthermore, while the FRET change was clearly sensitive to mutation of the glucosylated residue, this remains to be tested for the modest increase in actual CaMKII activity (12). Also, while specific CaMKII glucosylation clearly had important functional consequences, it is still unclear if these function were mediated by autonomous CaMKII activity, by stimulated activity, or by activity-independent CaMKII effects (12), especially since examples of the latter have been described for some neuronal CaMKII functions (34, 35). Obviously, the FRET sensor reports structural changes rather than actual kinase activity, and the sensor is known to over-report Thr-286-dependent autonomous activity (12, 33). Notably, this over-reporting can be an advantage for the detection of autonomous CaMKII compared with the significantly larger activity of stimulated CaMKII (36). However, it should also be noted that there is a novel FRET sensor of CaMKII activity (with different placement of the fluorescent proteins) that more accurately reports the levels of autonomous activity relative to stimulated levels, at least for the Thr-286-dependent autonomy (37).
The secondary effects of the different forms of CaMKII autonomy on levels of Ca2+/CaM-stimulated activity also differ. Thr-286 phosphorylation mildly enhances stimulated activity by 10–20% (26), but also promotes additional Thr-305/306 phosphorylation that prevents subsequent Ca2+/CaM binding (26, 31, 32, 38). By contrast, GluN2B binding actually reduces stimulated activity (10, 39), but inhibits Thr-305/306 phosphorylation (10) (for review see Ref. 1). By contrast S-nitrosylation did not directly affect Ca2+/CaM-stimulated activity, but inhibited subsequent Thr-286 phosphorylation (with no effects on Thr-305 phosphorylation). This prevented generation of the higher level of Thr-286-dependent autonomy, an effect observed here also after CaMKII oxidation. Thus, the different forms of CaMKII autonomy can have intricate cross-regulatory effects with each other as well as with stimulated CaMKII activity. NO-induced CaMKII autonomy is a novel aspect also in the cross-talk between CaMKII and different NOS isoforms, as these NO-producing enzymes are in turn regulated by CaMKII activity (40–45).
Both CaMKII and neuronal NOS (nNOS) are localized at synapses via association with the NMDA-receptor complex, are involved in excitotoxic/ischemic cell death, and inhibition of either is neuroprotective (for review see Refs. 7, 23). The CaMKII-mediated regulation of nNOS cannot explain this effect, as CaMKII inhibits rather than activates nNOS (40, 43, 44). By contrast, the NO-induced CaMKII autonomy indicates that nNOS is instead upstream of CaMKII in excitotoxic death signaling. Indeed, CaMKII inhibition protected not only from excitotoxic/ischemic death, but also from neuronal cell death directly induced by an NO donor. Intriguingly, even disruption of nNOS from the NMDA-receptor complex provides neuroprotection (22), and it will be interesting to see if this disruption acts by preventing efficient S-nitrosylation of the co-localized CaMKII.
Two recent studies indicated NO-mediated regulation of CaMKII also in the heart (46, 47), where CaMKII is known to have crucial pathological functions (13). However, in this case, CaMKII activation by NO did not appear to be through direct S-nitrosylation, as CaMKII activation (and the downstream functions) was instead dependent on PKG (47), which is activated by cGMP after NO-induced guanylyl cyclase activation (17–19). Notably, however, one of these studies showed that NO donors did also cause a direct 2.5-fold increase of CaMKIIó activity over baseline (46). However, this rather small effect may have been caused by ONOO−-mediated oxidation (an effect that was ruled out in our experiments on the CaMKIIα isoform). Indeed, in this case, the NO effect was no larger than the effect of oxidation (46), and the CaMKIIó isoform does not have a Cys residue at the position homologous to Cys-280 in CaMKIIα (and the nitrosylation effect on CaMKIIα autonomy demonstrated here was sensitive not only to mutation of the Cys-289 that is conserved among the isoforms, but also to mutation of the unique Cys-280).
It should be noted that the absolute activity of CaMKII made autonomous by oxidation reported here (∼1 μmol/min/mg) is higher than previously reported, while the level of relative autonomy (∼5% of maximal stimulated activity) is lower than reported previously (∼25 nmol/min/mg and ∼40%, respectively) (11). The previously reported relative autonomy may be an overestimate due to kinase reactions that were beyond the linear range, for instance due to long reaction times (1). This is consistent with the low apparent activity reported after maximal stimulation and with the high apparent autonomy also after Thr-286 phosphorylation (70%) (11), even though the assays used a regular substrate described to support only lower levels of ∼20% autonomy (8, 16). However, as previously discussed (1), we wish to point out that such overestimates of the relative autonomy levels after oxidation (11) or GluN2B binding (10) do not diminish the importance of these studies that elucidated new principle pathways toward CaMKII autonomy. The NO mechanisms described here add another path to CaMKII autonomy, and it will be interesting to investigate the functions in physiological CaMKII signaling, in addition to the pathological functions in neuronal cell death described here.
Acknowledgment
We thank Jacqueline Kulbe for help with the neuronal cell culture.
This research was supported, in whole or in part, by National Institutes of Health Grants NS081248 and NS080851 (to K. U. B.). The University of Colorado is currently seeking patent protection for a CaMKII inhibitor used in this study.
- CaM
- calmodulin
- LTP
- long-term potentiation
- CaMKIIα
- CaM-dependent protein kinase II α isoform
REFERENCES
- 1. Coultrap S. J., Bayer K. U. (2012) CaMKII regulation in information processing and storage. Trends Neurosci. 35, 607–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chao L. H., Stratton M. M., Lee I. H., Rosenberg O. S., Levitz J., Mandell D. J., Kortemme T., Groves J. T., Schulman H., Kuriyan J. (2011) A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell 146, 732–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giese K. P., Fedorov N. B., Filipkowski R. K., Silva A. J. (1998) Autophosphorylation at Thr286 of the α-calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873 [DOI] [PubMed] [Google Scholar]
- 4. Buard I., Coultrap S. J., Freund R. K., Lee Y. S., Dell'Acqua M. L., Silva A. J., Bayer K. U. (2010) CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J. Neurosci. 30, 8214–8220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coultrap S. J., Freund R. K., O'Leary H., Sanderson J. L., Roche K. W., Dell'Acqua M. L., Bayer K. U. (2014) Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Reports 6, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vest R. S., O'Leary H., Coultrap S. J., Kindy M. S., Bayer K. U. (2010) Effective post-insult neuroprotection by a novel Ca(2+)/ calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem. 285, 20675–20682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coultrap S. J., Vest R. S., Ashpole N. M., Hudmon A., Bayer K. U. (2011) CaMKII in cerebral ischemia. Acta. Pharmacol. Sin. 32, 861–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller S. G., Kennedy M. B. (1986) Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44, 861–870 [DOI] [PubMed] [Google Scholar]
- 9. Hanson P. I., Meyer T., Stryer L., Schulman H. (1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12, 943–956 [DOI] [PubMed] [Google Scholar]
- 10. Bayer K. U., De Koninck P., Leonard A. S., Hell J. W., Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805 [DOI] [PubMed] [Google Scholar]
- 11. Erickson J. R., Joiner M. L., Guan X., Kutschke W., Yang J., Oddis C. V., Bartlett R. K., Lowe J. S., O'Donnell S. E., Aykin-Burns N., Zimmerman M. C., Zimmerman K., Ham A. J., Weiss R. M., Spitz D. R., Shea M. A., Colbran R. J., Mohler P. J., Anderson M. E. (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erickson J. R., Pereira L., Wang L., Han G., Ferguson A., Dao K., Copeland R. J., Despa F., Hart G. W., Ripplinger C. M., Bers D. M. (2013) Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anderson M. E., Brown J. H., Bers D. M. (2011) CaMKII in myocardial hypertrophy and heart failure. J Mol. Cell Cardiol. 51, 468–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lou L. L., Lloyd S. J., Schulman H. (1986) Activation of the multifunctional Ca2+/calmodulin-dependent protein kinase by autophosphorylation: ATP modulates production of an autonomous enzyme. Proc. Natl. Acad. Sci. U.S.A. 83, 9497–9501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schworer C. M., Colbran R. J., Soderling T. R. (1986) Reversible generation of a Ca2+-independent form of Ca2+(calmodulin)-dependent protein kinase II by an autophosphorylation mechanism. J. Biol. Chem. 261, 8581–8584 [PubMed] [Google Scholar]
- 16. Coultrap S. J., Buard I., Kulbe J. R., Dell'Acqua M. L., Bayer K. U. (2010) CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J. Biol. Chem. 285, 17930–17937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura T., Tu S., Akhtar M. W., Sunico C. R., Okamoto S., Lipton S. A. (2013) Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78, 596–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hess D. T., Stamler J. S. (2012) Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 287, 4411–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sen N., Snyder S. H. (2010) Protein modifications involved in neurotransmitter and gasotransmitter signaling. Trends Neurosci. 33, 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schuman E. M., Madison D. V. (1991) A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 21. Stanton P. K., Winterer J., Bailey C. P., Kyrozis A., Raginov I., Laube G., Veh R. W., Nguyen C. Q., Müller W. (2003) Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J. Neurosci. 23, 5936–5944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aarts M., Liu Y., Liu L., Besshoh S., Arundine M., Gurd J. W., Wang Y. T., Salter M. W., Tymianski M. (2002) Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science 298, 846–850 [DOI] [PubMed] [Google Scholar]
- 23. Aarts M. M., Tymianski M. (2004) Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr. Mol. Med. 4, 137–147 [DOI] [PubMed] [Google Scholar]
- 24. Coultrap S. J., Bayer K. U. (2012) Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII). In Neuromethods: Protein Kinase Technologies (Mukai H., ed) pp. 49–72, Springer [Google Scholar]
- 25. Bayer K. U., LeBel E., McDonald G. L., O'Leary H., Schulman H., De Koninck P. (2006) Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coultrap S. J., Barcomb K., Bayer K. U. (2012) A significant but rather mild contribution of T286 autophosphorylation to Ca2+/CaM-stimulated CaMKII activity. PLoS One 7, e37176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vest R. S., Davies K. D., O'Leary H., Port J. D., Bayer K. U. (2007) Dual Mechanism of a Natural CaMKII Inhibitor. Mol. Biol. Cell 18, 5024–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaffrey S. R., Snyder S. H. (2001) The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001, pl1. [DOI] [PubMed] [Google Scholar]
- 29. Sumi M., Kiuchi K., Ishikawa T., Ishii A., Hagiwara M., Nagatsu T., Hidaka H. (1991) The newly synthesized selective Ca2+/calmodulin-dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 181, 968–975 [DOI] [PubMed] [Google Scholar]
- 30. Song T., Hatano N., Kambe T., Miyamoto Y., Ihara H., Yamamoto H., Sugimoto K., Kume K., Yamaguchi F., Tokuda M., Watanabe Y. (2008) Nitric oxide-mediated modulation of calcium/calmodulin-dependent protein kinase II. Biochem. J. 412, 223–231 [DOI] [PubMed] [Google Scholar]
- 31. Hanson P. I., Schulman H. (1992) Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J. Biol. Chem. 267, 17216–17224 [PubMed] [Google Scholar]
- 32. Colbran R. J. (1993) Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J. Biol. Chem. 268, 7163–7170 [PubMed] [Google Scholar]
- 33. Takao K., Okamoto K., Nakagawa T., Neve R. L., Nagai T., Miyawaki A., Hashikawa T., Kobayashi S., Hayashi Y. (2005) Visualization of synaptic Ca2+ /calmodulin-dependent protein kinase II activity in living neurons. J. Neurosci. 25, 3107–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hojjati M. R., van Woerden G. M., Tyler W. J., Giese K. P., Silva A. J., Pozzo-Miller L., Elgersma Y. (2007) Kinase activity is not required for alphaCaMKII-dependent presynaptic plasticity at CA3-CA1 synapses. Nat. Neurosci. 10, 1125–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borgesius N. Z., van Woerden G. M., Buitendijk G. H., Keijzer N., Jaarsma D., Hoogenraad C. C., Elgersma Y. (2011) betaCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting αCaMKII to synapses. J. Neurosci. 31, 10141–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S. J., Escobedo-Lozoya Y., Szatmari E. M., Yasuda R. (2009) Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujii H., Inoue M., Okuno H., Sano Y., Takemoto-Kimura S., Kitamura K., Kano M., Bito H. (2013) Nonlinear decoding and asymmetric representation of neuronal input information by CaMKIIα and calcineurin. Cell Rep. 3, 978–987 [DOI] [PubMed] [Google Scholar]
- 38. Hashimoto Y., Schworer C. M., Colbran R. J., Soderling T. R. (1987) Autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. Effects on total and Ca2+-independent activities and kinetic parameters. J. Biol. Chem. 262, 8051–8055 [PubMed] [Google Scholar]
- 39. Robison A. J., Bartlett R. K., Bass M. A., Colbran R. J. (2005) Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-D-aspartate receptor NR2B subunits and α-actinin. J. Biol. Chem. 280, 39316–39323 [DOI] [PubMed] [Google Scholar]
- 40. Hayashi Y., Nishio M., Naito Y., Yokokura H., Nimura Y., Hidaka H., Watanabe Y. (1999) Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J. Biol. Chem. 274, 20597–20602 [DOI] [PubMed] [Google Scholar]
- 41. Watanabe Y., Song T., Sugimoto K., Horii M., Araki N., Tokumitsu H., Tezuka T., Yamamoto T., Tokuda M. (2003) Post-synaptic density-95 promotes calcium/calmodulin-dependent protein kinase II-mediated Ser847 phosphorylation of neuronal nitric oxide synthase. Biochem. J. 372, 465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fleming I., Fisslthaler B., Dimmeler S., Kemp B. E., Busse R. (2001) Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 88, E68–E75 [DOI] [PubMed] [Google Scholar]
- 43. Rameau G. A., Tukey D. S., Garcin-Hosfield E. D., Titcombe R. F., Misra C., Khatri L., Getzoff E. D., Ziff E. B. (2007) Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 27, 3445–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rameau G. A., Chiu L. Y., Ziff E. B. (2004) Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-D-aspartate receptor. J. Biol. Chem. 279, 14307–14314 [DOI] [PubMed] [Google Scholar]
- 45. Jones R. J., Jourd'heuil D., Salerno J. C., Smith S. M., Singer H. A. (2007) iNOS regulation by calcium/calmodulin-dependent protein kinase II in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 292, H2634–H2642 [DOI] [PubMed] [Google Scholar]
- 46. Gutierrez D. A., Fernandez-Tenorio M., Ogrodnik J., Niggli E. (2013) NO-dependent CaMKII activation during β-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 100, 392–401 [DOI] [PubMed] [Google Scholar]
- 47. Zhang D. M., Chai Y., Erickson J. R., Brown J. H., Bers D. M., Lin Y. F. (2014) Intracellular signalling mechanism responsible for modulation of sarcolemmal ATP-sensitive potassium channels by nitric oxide in ventricular cardiomyocytes. J. Physiol. 592, 971–990 [DOI] [PMC free article] [PubMed] [Google Scholar]