

Background: Plasma membrane proteins can be degraded by a number of mechanisms.
Results: Turnover rates are determined by endocytosis signals and trafficking to lysosomes, ubiquitination, and ectodomain cleavage.
Conclusion: Membrane trafficking to lysosomes and proteolysis at the cell surface determine protein half-life.
Significance: Learning how plasma membrane proteins are turned over is critical for understanding protein homeostasis.
Keywords: Endocytosis, Lysosome, Plasma Membrane, Shedding, Trafficking
Abstract
Proteins targeted to the plasma membrane (PM) of cells are degraded at different rates. Sorting motifs contained within the cytoplasmic domains of transmembrane proteins, post-translational modifications (e.g. ubiquitination), and assembly into multiprotein or protein-lipid complexes all may affect the efficiency of endocytosis and recycling and influence the delivery to degradative compartments. Using the SNAP-tag labeling system, we examined the turnover of a model PM protein, the α chain of the interleukin-2 receptor (Tac). The surface lifetimes of SNAP-Tac fusions were influenced by their mode of entry into cells (clathrin-dependent versus clathrin-independent), their orientation in the PM (transmembrane versus glycosylphosphatidylinositol-anchored), and ubiquitination in their cytosolic domains. In addition, shedding of SNAP-Tac into the medium was greatly influenced by its O-linked glycosylation status. For a number of PM proteins, delivery to lysosomes and ectodomain shedding represent distinct parallel mechanisms to determine protein half-life.
Introduction
Cells regulate the synthesis and degradation of cellular proteins to maintain physiological homeostasis. For integral membrane proteins at the plasma membrane (PM),2 changes in the abundance, trafficking, and stability of these proteins, including nutrient transporters, ion channels, and cell signaling receptors, are important factors in many human diseases (1, 2). Endocytosis appears to play a major role in the stability of PM proteins. Proteins that reside for the most part at the PM are generally more slowly degraded than proteins that undergo endocytosis (3). In addition, recycling mechanisms may also limit the time a protein is exposed to the sorting machinery that traffics endocytosed proteins to lysosomes for degradation (4).
This relationship between endocytosis and degradation is often regulated by post-translational modifications such as ubiquitination. This regulation involves endocytosis through clathrin-mediated (CME) or clathrin-independent (CIE) pathways, selective recognition of cargo for ubiquitination, sorting of ubiquitinated cargo within endosomes, packaging of ubiquitinated cargo into vesicles that bud into the lumen of the endosome (forming multivesicular bodies), and fusion of these endosomes with lysosomes, resulting in cargo degradation (2, 4–7).
CME has been extensively studied and involves the selection and rapid internalization of PM proteins that contain distinct cytoplasmic sorting sequences recognized by adaptor proteins that are part of the clathrin coat (8). CME is the primary mechanism for endocytosis of transferrin receptor, low density lipoprotein (LDL) receptor, and signaling receptors after ligand stimulation (9). CIE, by contrast, is a slower non-selective internalization pathway that appears to lack a distinctive cytoplasmic coat and may function as a bulk form of endocytosis (10). There has been increased interest in CIE because it is the mode of entry for a number of bacterial toxins and cell surface proteins involved in immune function, ion and nutrient transport, and cell adhesion (11).
In addition to lysosomal degradation, a number of PM proteins undergo proteolytic cleavage of the luminal domain into the extracellular space, also known as ectodomain shedding. Although generally overlooked as a mechanism for protein turnover, ectodomain shedding is an essential post-translational mechanism that plays a critical role in development, cellular homeostasis, and immune function and during pathological conditions in vivo (12–14). Whether turnover of specific PM proteins represents a balance between lysosomal targeting and ectodomain shedding and how this may be regulated in different cell types are unclear. We are interested in factors that determine PM protein turnover, especially proteins that enter cells by CIE, and in developing a systematic method to study this in cells.
To establish a method to analyze PM protein turnover, we used a chemical labeling approach in which the self-labeling SNAP-tag was appended to the N terminus of the α chain of the interleukin-2 receptor, also known as Tac, as well as to a number of Tac variants that differ only in their mode of internalization (CIE versus CME), anchorage in the PM (transmembrane versus lipid-anchored), ability to be ubiquitinated, and presence or absence of juxtamembrane O-linked sugars. Pulse-chase studies with PM labeled SNAP-Tac chimeras demonstrated the usefulness of a model protein that undergoes a variety of trafficking and cell surface modifications.
EXPERIMENTAL PROCEDURES
Cells, Reagents, and Antibodies
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Lonza) with 10% fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin and grown in a 5% CO2 atmosphere at 37 °C. BG-S-S-488, BG-488, BG-800, and BG-PEG12-biotin were synthesized as described previously (15, 16). Rabbit anti-early endosomal antigen 1 (EEA1) and rabbit anti-Lamp1 were purchased from BD Biosciences and Abcam (Cambridge, MA), respectively. All secondary antibodies were purchased from Invitrogen Molecular Probes (Eugene, OR). Batimastat (BB-94) and the O-linked glycosylation inhibitor benzyl 2-acetamido-2-deoxy-α-d-galactopyranoside were from Sigma.
Plasmid Construction
For all SNAP-Tac chimeras, the signal peptide of hen egg lysozyme (MRSLLILVLCFLPLAALG) was introduced just before the second amino acid of the SNAP-tag (DKDCEMKR …). Following the SNAP-tag, a (GGGGS)2 linker was introduced followed by restriction sites (XbaI and ApaI) to insert the indicated extracellular, transmembrane, and cytoplasmic tail domains of Tac. The native signal peptide of Tac was omitted, and the beginning of the extracellular domain (1ELCDDD …) followed the linker. For SNAP-Tac-LI, the sequence ERAPLIRT from the rat lysosomal membrane protein LimpII (NCBI Reference Sequence NM_054001) was appended to the cytoplasmic tail of Tac. This contains the dileucine signal for clathrin adaptor-protein interactions and internalization via CME (17) but lacks a second acidic residue at position −5 from LI necessary for binding to the AP-3 adaptor (18), thus preventing the protein from being directly targeted from the Golgi complex to lysosomes. For SNAP-Tac-KR, the single lysine residue in the cytoplasmic tail of Tac was mutated to an arginine (K246R). For SNAP-Tac-GPI, the transmembrane domain and cytoplasmic tail of Tac was replaced by the glycosylphosphatidylinositol (GPI) anchor sequence from the low affinity immunoglobulin γ Fc region receptor III-B (CD16b; NCBI Reference Sequence NM_001271037) (19). For the SNAP-Tac(T→A) glycosylation mutant, threonine residues corresponding to amino acids 197, 203, 208, and 216 in the mature Tac protein (20) were replaced by alanine residues. For truncated SNAP-Tac chimeras containing control and mutated O-linked glycosylation sites, a 27-amino acid stalk region from the membrane proximal side of the extracellular domain of Tac (193LVTTTDFQIQTEMAATMETSIFTTEYQ219 …) was introduced together with the Tac transmembrane domain and cytoplasmic tail (amino acids 220–251) into the SNAP vector described above. The putative cleavage site that generates the soluble form of Tac resides immediately prior to the stalk domain between residues Cys192 and Leu193 (see “Discussion”). The threonine residues (in bold) were changed to alanine residues in the glycosylation mutants. All constructs were cloned into the mammalian expression vector pcDNA3.1 using standard PCR and cloning techniques and sequence-verified.
Transfections
HeLa cells were grown on glass coverslips (for immunofluorescence) or 10-cm plates (for degradation and pulldown assays) and transfected using X-tremeGENE 9 (Roche Applied Science) according to the manufacturer's specifications. Experiments were performed 15–18 h after transfection.
Immunofluorescence and Localization of SNAP Proteins
Cells expressing the chimeric proteins were labeled with BG-S-S-488 (1–2 μm) at 4 °C for 30 min in DMEM with 20 mm HEPES, pH 7.4. The cells were then rinsed two times in Hanks' buffered saline solution (HBSS) containing calcium and magnesium, transferred to fresh medium, and incubated at 37 °C. At the indicated time points, cells were incubated with 10 mm tris(2-carboxyethyl)phosphine (TCEP) for 2 min at 37 °C to remove excess surface label. After two washes in PBS, cells were fixed for 10 min in 3% formaldehyde in PBS; rinsed with PBS, 10% fetal bovine serum (FBS); and incubated with rabbit anti-EEA1 monoclonal antibody in PBS, 10% FBS containing 0.2% saponin for 1 h followed by Alexa Fluor 594 goat anti-rabbit secondary antibody (Invitrogen Molecular Probes). Cells were mounted with Fluoromount G (Southern Biotechnologies, Birmingham, AL).
To monitor trafficking of cargo to lysosomal compartments, cells were incubated with BG-S-S-488 for 15 min at 37 °C, rinsed in HBSS, then transferred to fresh medium containing 20 mm NH4Cl and 20 μm BG-NH2 to block subsequent SNAP-tag labeling, and incubated for 6–22 h at 37 °C. The cells were rinsed in PBS, treated or not with TCEP, fixed, stained with rabbit anti-Lamp1 monoclonal antibody in PBS, 10% FBS containing 0.2% saponin for 1 h, and processed as described above. All images were obtained using either a 510 or 780 LSM confocal microscope (Zeiss, Thornwood, NY) with a 63× Plan Apo objective and processed using Adobe Photoshop. The MetaMorph colocalization application (Molecular Devices) was used to quantify colocalization between SNAP-tag cargo and EEA1 or Lamp-1 acquired as maximum intensity projections. After separating the confocal images in the two different colors, individual cells were outlined using the MetaMorph trace region tool. After thresholding, quantification of the amount of overlapping fluorescence pixels per cell was obtained after applying the colocalization function. Data analysis was done using Excel and Prism software.
Degradation and Extracellular Shedding Assay
HeLa or COS cells were transfected with plasmids encoding SNAP constructs in 10-cm dishes. After 18 h, cells were removed from the dishes with 10 mm EDTA and replated into 12-well plates to assure similar levels of expression in each well. The next day, cells were labeled with BG-800 at 4 °C for 30 min, rinsed, and then incubated in medium at 37 °C for the indicated time points in the presence of 20 μm BG-NH2 to block further SNAP labeling. At each time point, medium was collected, and cells were solubilized in 0.25 ml of lysis buffer I (50 mm Tris-Cl, pH 7.4, 0.15 m NaCl, 1.0% Triton X-100) with protease inhibitors (Roche Applied Science Complete tablets) and 20 μm BG-NH2. For SNAP-Tac-GPI, cells were solubilized in lysis buffer I containing 1.0% octyl β-d-glucopyranoside instead of Triton X-10. Cell media and lysates (in duplicates or triplicates) were run on 12% SDS-polyacrylamide gels and directly scanned and quantified by the Odyssey infrared scanner (LI-COR Biosciences, Lincoln, NE). To determine protein concentration, gels were stained with Coomassie stain as described (21) and rescanned. Protein detection was in the 700 nm channel. In brief, after electrophoresis, gels were stained for 20 min with 0.25% Coomassie Blue R-250 (Sigma B-7920) in 50% methanol and 10% acetic acid. They were then rinsed with 40% methanol and 7% acetic acid followed by destaining for 30 min to overnight in the same solution. Bicinchoninic acid (BCA) or other protein quantification assays can be used; however, we found that the values given by Coomassie staining generated more accurate results as it directly accounts for protein loading. To measure protein loading, the intensity for single or multiple protein bands was determined within each lane. The fluorescence signal for the protein of interest, as detected by BG-800, was then normalized to the Coomassie signal. To calculate the fraction shed into the medium, the values of the media fractions accounted for volume were divided by the total signal represented by shed plus cell-associated values. Prism and Excel software were used to analyze data, generate degradation plots, and calculate half-lives.
Pulldown of Ubiquitinated SNAP-Tac Proteins
This protocol is essentially from Eyster et al. (15) with minor modifications and corrections. HeLa cells (10-cm dishes) were transfected with the indicated SNAP-Tac construct (4 μg/dish) with HA-ubiquitin (1 μg/dish) and with or without MARCH8-FLAG (1 μg/dish). After 18 h, cells were labeled with BG-PEG4-biotin (1–2 μm) for 1 h at 37 °C. Cells were rinsed twice with PBS, lifted, and pelleted at 300 × g. Cell pellets were incubated with 100 μm BG-NH2 in 0.05 ml of PBS for 15 min at 4 °C and then solubilized by adding 0.2 ml of lysis buffer II (50 mm Tris-Cl, pH 7.4, 0.15 m NaCl, 0.1% Triton X-100) with 10 mm N-ethylmaleimide plus Complete protease inhibitor tablets. The cell extract was denatured by adding 20% SDS to obtain a 1% final concentration and boiled for 10 min. The SDS was quenched by adding 0.02 ml of 20% Triton X-100 and 1.8 ml of lysis buffer I, and the cell extract was placed on ice for 30 min. The lysate was centrifuged at 13,000 × g for 5 min, and 0.05 ml of the supernatant was saved for SDS-PAGE. 0.05 ml of 1:1 slurry of NeutrAvidin-agarose resin (Thermo Scientific) was added to the supernatant and rocked at 4 °C for 1 h. The beads were washed three times with lysis buffer I and once with water. 20 μl of 3× SDS sample buffer was added, and the beads were boiled for 10 min before protein separation by SDS-PAGE (6% Tris, glycine for analysis of ubiquitination; 4–20% for analysis of MARCH8-FLAG expression; Novex, Invitrogen), transfer to nitrocellulose, and immunoblotting. HA-ubiquitin was probed with monoclonal HA.11 (Covance), MARCH8-FLAG was probed with mouse anti-FLAG (M2 from Sigma), and SNAP-Tac proteins were probed with rabbit polyclonal anti-SNAP (New England Biolabs). Species-specific infrared secondary antibodies were used for subsequent detection. Biotinylated SNAP-Tac was detected with DyLight 800-conjugated NeutrAvidin (Thermo Scientific). Membranes were incubated with primary and secondary antibodies (each for 1 h at room temperature), then washed three times with 0.1% Tween 20 in PBS, and visualized by scanning with an Odyssey infrared scanner.
Inhibition of Extracellular Shedding
HeLa cells were transfected with SNAP constructs and replated into 12-well plates as described above. The next day, cells were labeled with BG-800 at 4 °C for 30 min in the presence or absence of 500 nm batimastat (BB-94). Cells were then incubated at 37 °C for 30 min in the presence or absence of BB-94. Media were collected, and cells were solubilized in 0.25 ml of lysis buffer I with protease inhibitors and 20 μm BG-NH2. Cell media and lysates (in triplicates or duplicates) were run on SDS-polyacrylamide gels and directly scanned and quantified by the Odyssey infrared scanner. To determine protein concentration, gels were stained with Coomassie stain as described above and rescanned. Quantification was described above.
Inhibition of O-Linked Glycosylation
For studies with inhibitors of O-linked glycosylation, HeLa cells were transiently transfected for 16 h with SNAP-Tac in 12-well dishes. Where indicted, GalNAc-O-Bn (Sigma) was added during the overnight transfection (2 mm final concentration). Cells were labeled with BG-800 for 15 min at 37 °C, washed with HBSS, and then incubated with Opti-MEM (0.5 ml) containing 40 μm BG-NH2. Cells were maintained for 10 min at room temperature and then shifted to 37 °C. The medium was collected, and cell lysates were made at 0 and 8 h of chase. Where indicated, the “sheddase” inhibitor BB-94 was added during the chase (1 μm final concentration). To qualitatively assess the amount of SNAP-Tac shed into the medium during the overnight incubation with GalNAc-O-Bn, the medium from control versus GalNAc-O-Bn-treated cells was incubated with BG-800 for 4 h at 4 °C. SDS sample buffer was added, and samples were separated by SDS-PAGE.
Shedding of SNAP-Tac and Endogenous Proteins in HeLa and COS Cells
10-cm dishes of subconfluent HeLa or COS cells were treated with 1 mm EZ-Link NHS-PEG4-biotin (Thermo Scientific) in HBSS containing 50 mm HEPES, pH 7.4 for 30 min at room temperature. Cells were rinsed once with HBSS and then incubated with HBSS containing 50 mm Tris, pH 7.4 for 15 min at room temperature. Cells were then chased for 5 h at 37 °C in serum-free DMEM. Media were collected, and the cells were rinsed twice with PBS and then solubilized in lysis buffer I plus protease inhibitors. SDS sample buffer was added, and samples were heated to 95 °C for 3 min before SDS-PAGE. Equal amounts of protein were loaded from cell lysates as determined by BCA assay (Thermo Scientific). Biotinylated proteins were labeled with DyLight 800-conjugated NeutrAvidin and then detected on an Odyssey scanner.
RESULTS
The α chain of the interleukin-2 receptor, also known as CD25 or Tac, is a type I membrane protein with a short, 13-amino acid cytoplasmic tail that lacks known targeting sequences. It has been used extensively as a reporter protein onto which cytoplasmic sorting sequences can be appended to demonstrate the trafficking function of these sequences (22–26). Tac normally enters cells via CIE (27, 28).
The SNAP-tag is a ∼20-kDa protein derived from the DNA repair enzyme alkylguanine-DNA alkyltransferase. The SNAP-tag has an active site cysteine residue that covalently binds O6-benzylguanine (BG) and its synthetic derivatives (29). BG can be conjugated to a variety of fluorophores and other labels (30), making it possible to tag and track the protein of interest directly by a variety of methods such as fluorescence microscopy, gel electrophoresis, and in vivo imaging (31–33). The SNAP-tag has been demonstrated not to affect the function of a large number of fusion proteins (34, 35) and is an optimal approach for pulse-chase labeling experiments (34, 36). The covalent bond between BG and the SNAP-tag, however, makes fluorescence studies of endocytosis difficult because the probe cannot be removed from labeled proteins on the cell surface, concealing the intracellular endocytosed pool. Hence, we introduced a modification into this system that allows for the removal of the surface label (16). Briefly, a cleavable disulfide bond is introduced between the BG moiety and Alexa Fluor 488, creating BG-S-S-488, which allows us to remove surface fluorescence after a brief (1–2-min) treatment with the cell-impermeable reducing agent TCEP.
To examine how different cytoplasmic sorting motifs affect the trafficking and turnover of cell surface proteins, different cytoplasmic tails were appended onto a reporter protein, SNAP-Tac. These modifications include an ERAPLIRT extension to generate a clathrin internalization signal (SNAP-Tac-LI); replacement of the transmembrane domain of Tac with a GPI anchor (SNAP-Tac-GPI); and mutation of the single lysine residue in the cytoplasmic tail of Tac (SNAP-Tac-K246R), a mutant expected to lack cytoplasmic sequences for ubiquitination (Fig. 1A) (see “Discussion”).
FIGURE 1.

Endocytosis of SNAP-Tac chimeras. A, schematic representation of various SNAP-Tac constructs. TM, transmembrane domain. B, HeLa cells expressing SNAP-Tac fusion proteins were labeled with BG-S-S-488 at 4 °C for 30 min and then incubated for 10 min at 37 °C. Cells were not treated (left panels) or treated (right panels) with 10 mm TCEP for 2 min at 37 °C to remove the surface label and then fixed. After fixation, cells were immunolabeled with antibodies to EEA1 followed by species-specific secondary antibodies to detect EEA1-positive endosomes. The insets illustrate colocalization between internalized SNAP-Tac proteins and EEA1. C, colocalization analysis between SNAP-Tac chimeras and EEA1. One-way analysis of variance followed by Tukey's post hoc multiple comparison test was used to compare colocalization values. Error bars represent mean ± S.E. **, p < 0.05; ***, p < 0.001. D, HeLa cells expressing SNAP-Tac fusions were labeled with BG-S-S-488 at 37 °C for 1 h and then transferred to media containing 25 mm NH4Cl to block lysosomal degradation. After 6 h, cells were treated or not with 10 mm TCEP and fixed as above. After fixation, cells were immunolabeled with antibodies to Lamp-1 followed by species-specific secondary antibodies to detect Lamp1-positive lysosomes. The insets illustrate colocalization between internalized SNAP-Tac proteins and Lamp1. E, one-way analysis of variance was used to compare colocalization values between SNAP-Tac chimeras and Lamp-1 as above. No significant colocalization differences were observed between various constructs. Bars, 10 μm.
Endocytosis of SNAP-Tac Fusion Proteins
We first examined the trafficking itinerary of SNAP-Tac proteins in cells using the cleavable BG-S-S-488 ligand. HeLa cells expressing SNAP-Tac, SNAP-Tac-LI, SNAP-Tac-KR, or SNAP-Tac-GPI were labeled at 4 °C with BG-S-S-488 for 30 min, then placed in fresh medium, and incubated at 37 °C for 10 min to allow endocytosis to occur. After 10 min, SNAP-Tac was still primarily at the cell surface, but some internal punctate structures could also be observed (Fig. 1B, top row). The internal pool of SNAP-Tac was revealed after the addition of TCEP, and it was found that these endosomes colocalized with EEA1, a marker for sorting endosomes (Fig. 1B, top row, right panels). In contrast to SNAP-Tac, after 10 min, most of the total labeled SNAP-Tac-LI was observed in endosomes that labeled with EEA1 (Fig. 1B, second row), leaving a little surface label behind that could be removed with TCEP. This rapid and efficient entry of SNAP-Tac-LI is consistent with its ability to enter cells via CME and was observed previously for Tac-LI (27). SNAP-Tac-KR and SNAP-Tac-GPI were internalized similarly to SNAP-Tac for which internal endosomes, which colocalized with EEA1, were only revealed after removal of the surface label with TCEP (Fig. 1B, third and fourth rows, respectively). Colocalization of internalized SNAP-Tac cargo variants with EEA1 after 10 min of internalization is shown in Fig. 1C. Delivery of SNAP-Tac-GPI to EEA1-positive compartments was significantly reduced when compared with SNAP-Tac and SNAP-Tac-LI. Whether this reflects reduced endocytosis of SNAP-Tac-GPI or reduced sorting to EEA1-positive endosomes is unclear. Non-ionic detergents extracted SNAP-Tac, -LI, and -KR, whereas SNAP-Tac-GPI resided in detergent-resistant lipid rafts similar to its parent protein, Tac-GPI (data not shown) (27).
Next, we wished to follow these cargo proteins for longer periods of time to monitor their trafficking to late endosomal and lysosomal compartments marked by the presence of Lamp1. To do this, ammonium chloride was added to the medium to inhibit lysosomal degradation. After 6 h, SNAP-Tac was observed in compartments that colocalized with Lamp1, a marker for lysosomes, whereas some was still observed at the cell surface (Fig. 1D, top row). The lysosomal pool of SNAP-Tac was more readily observed after treatment with TCEP to remove the surface pool (Fig. 1D, top row, right panels). In contrast, after 6 h, nearly all of the SNAP-Tac-LI colocalized with Lamp1 structures (Fig. 1D, second row), and little if any surface SNAP-Tac-LI remained. Thus, uptake via CME results in the efficient delivery to lysosomes compared with SNAP-Tac cargo that enters cells by CIE. At 6 h of uptake, localization of SNAP-Tac-KR (Fig. 1D, third row) and SNAP-Tac-GPI (Fig. 1D, fourth row) appeared similar to that of SNAP-Tac. Quantification of SNAP-Tac cargo colocalized with Lamp-1 after 6 h of uptake is shown in Fig. 1E and showed no significant differences in SNAP-Tac cargo delivery to lysosomes. However, after longer than 10 h of internalization, most of the SNAP-Tac had cleared the surface (observable without TCEP) and accumulated in Lamp1-positive compartments, whereas a significant amount of SNAP-Tac-KR and SNAP-Tac-GPI still remained on the surface (data not shown). This suggests that SNAP-Tac fusion proteins incapable of being ubiquitinated resided longer at the cell surface.
Pulse-Chase Analysis of SNAP-Tac Cargo Proteins
To analyze the turnover of labeled SNAP-Tac fusion proteins, we designed a degradation assay taking advantage of the covalent labeling of the SNAP-tagged protein by BG (34, 37). We labeled cells with BG-800, which allowed us to run total cellular lysates on gels and quantify protein bands directly on an infrared scanner with built-in software to quantify protein bands. We used a Coomassie staining protocol to normalize for total protein content (see “Experimental Procedures”).
We found that the surface pool of SNAP-Tac was degraded with a half-life of 5.5 h, which was extended to 11 h in the presence of lysosomal inhibitors (Fig. 2, A, B, and C). SNAP-Tac-LI, in contrast, was degraded more rapidly with a half-life of 3.2 h, consistent with its rapid entry by CME and routing to late endosomes (Fig. 2, A, B, and C). Degradation of SNAP-Tac-LI was also impaired in the presence of ammonium chloride (t½ of 8 h), indicating that this degradation also occurred in lysosomes. These results are consistent with previous findings of turnover for Tac and Tac-LI using surface biotinylation assays (27), confirming that addition of the SNAP-tag to the extracellular portion of these proteins does not alter their trafficking or degradation.
FIGURE 2.

Pulse-chase analyses of SNAP-Tac proteins. HeLa cells were pulse-labeled with BG-800, blocked with cold BG-NH2, and then chased for various time points before cells were harvested. A, examples of turnover of BG-800-labeled SNAP-Tac (left) and SNAP-Tac-LI (right) from a representative experiment. Top panels show the BG-800 signals, and the bottom panels show the Coomassie signals from stained gels. Duplicate or triplicate independent samples are shown in the different lanes. The migration of prestained molecular mass markers (in kDa) is shown. B, quantification of fluorescent signals from gels of SNAP-Tac (n = 13), SNAP-Tac-LI (n = 8), SNAP-Tac-KR (n = 6), and SNAP-Tac-GPI (n = 4) where n is the number of independent experiments, each done in triplicate. Graphs show the BG-800 signal remaining at different time points as a percentage of the signal at time 0. The means ± S.D. (error bars) are indicated for untreated samples (circles) and in the presence of lysosomal inhibitors (lys inh) (triangles). For time points without error bars, the means of two independent experiments are shown. Data are fitted to a single exponential decay equation. C, one-way analysis of variance analysis was used to compare t½ values for SNAP-Tac, SNAP-Tac-LI, SNAP-Tac-KR, and SNAP-Tac-GPI. Error bars represent average t½ ±S.D. *, p < 0.05; ***, p < 0.001.
The half-lives of surface-labeled SNAP-Tac-KR (t½ of 7.9 h) and SNAP-Tac-GPI (t½ of 12.1 h) were significantly longer than that of SNAP-Tac (Fig. 2, B and C). Although lysosomal inhibitors significantly extended the half-life of SNAP-Tac-KR (t½ of 12 h), the half-life of SNAP-Tac-GPI was extended only for an hour. The reason for this is unclear. SNAP-Tac-LI that also contains the K246R mutation is degraded with a similar half-life as SNAP-Tac-LI (data not shown). This may be due to the dominant lysosomal targeting of SNAP-Tac-LI. These findings demonstrate that the turnover rate of our SNAP-Tac proteins is determined by sequences in their cytoplasmic tails that affect the mode of endocytosis (CIE versus CME) and the orientation in the membrane (transmembrane versus lipid-linked) coupled to the likely presence of a lysine, which is subject to ubiquitination.
SNAP-Tac and SNAP-Tac-LI, but Not SNAP-Tac-KR or SNAP-Tac-GPI, Are Polyubiquitinated by MARCH8
We have assumed that mutating the single lysine residue in the cytoplasmic tail of Tac would prevent its ubiquitination, hence explaining its longer half-life. To formally test this, we transfected our SNAP-Tac constructs with HA-ubiquitin and MARCH8, a ubiquitin ligase known to ubiquitinate a number of CIE cargo molecules (15). SNAP-tagged proteins were surface-biotinylated with BG-PEG12-biotin and recovered under denaturing conditions (see “Experimental Procedures”). In the presence of MARCH8, but not in its absence, a slower migrating ladder of HA-ubiquitin can be observed above SNAP-Tac and SNAP-Tac-LI but not with SNAP-Tac-KR or SNAP-Tac-GPI (Fig. 3). Four ubiquitin bands could be seen for both SNAP-Tac and SNAP-Tac-LI, which also labeled with antibodies to the SNAP protein, thus ruling out nonspecific binding of ubiquitinated proteins. Thus, SNAP-Tac is polyubiquitinated by MARCH8 on its one lysine residue. The proportion of ubiquitinated SNAP-Tac and SNAP-Tac-LI in the presence of MARCH 8 is similar (data not shown), demonstrating that overexpressed MARCH8 does not discriminate CIE from CME cargo. In cells not overexpressing MARCH8, the faster rate of degradation of SNAP-Tac-LI is likely due to its faster inherent rate of internalization (see Fig. 1, B and C) and rapid delivery to lysosomes via its LI motif. No ubiquitination of SNAP-Tac-GPI was observed as expected (Fig. 3). Thus, the slower turnover of surface-labeled SNAP-Tac-KR and SNAP-Tac-GPI is likely due to their lack of ubiquitin modifications.
FIGURE 3.
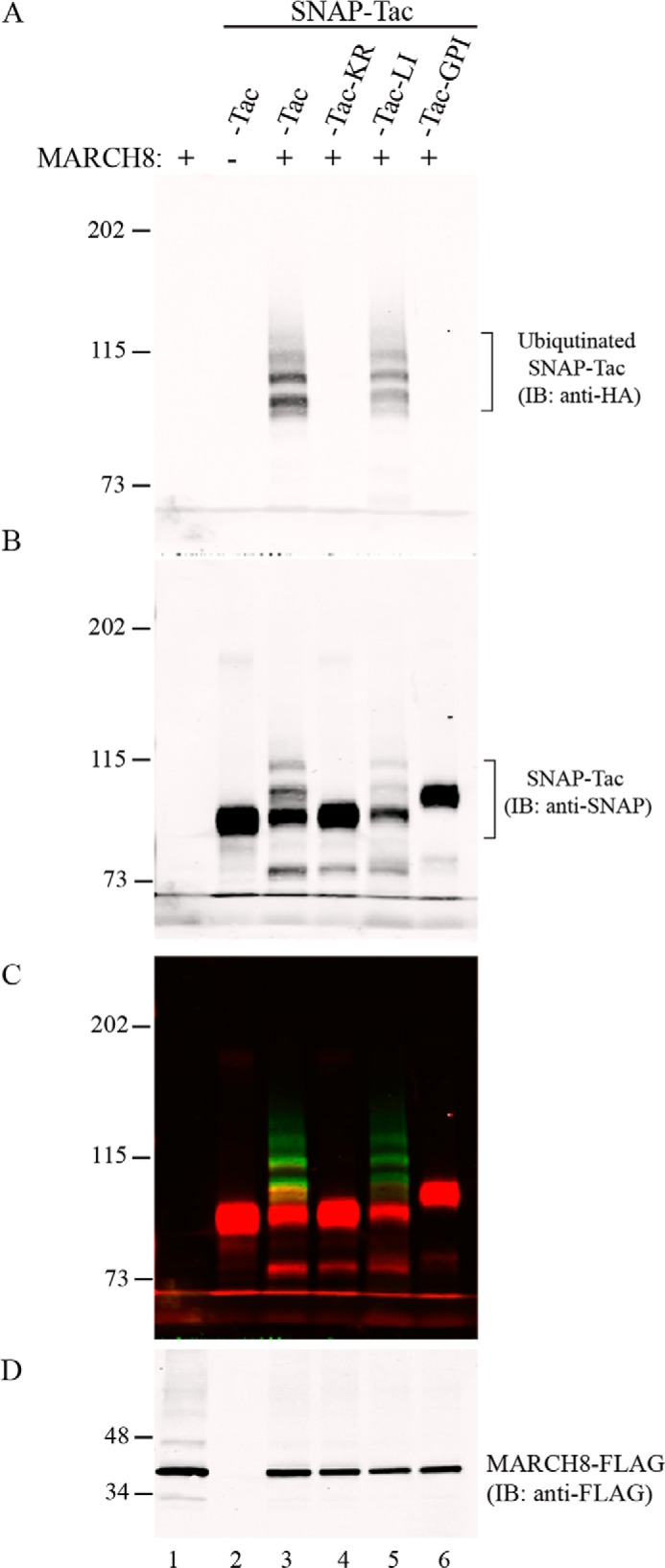
Ubiquitination of SNAP-Tac proteins. HeLa cells were transfected with the indicated SNAP-Tac construct together with HA-ubiquitin and MARCH8-FLAG where indicated. Cells were labeled with BG-PEG12-biotin for 1 h at 37 °C and then harvested for denaturing pulldown with NeutrAvidin-agarose and subsequent immunolabeling. A, immunoblot from pulldown with anti-HA and detection with DyLight 800 goat anti-mouse secondary antibody. B, immunoblot (IB) pulldown with anti-SNAP and detection with Alexa Fluor 680 goat anti-rabbit secondary antibody. C, merged image. D, immunoblot of MARCH8 expression from the cell lysate ( of the total volume) with anti-FLAG. The migration of prestained molecular mass markers (in kDa) is shown to the left of each panel.
Extracellular Domain O-Linked Glycosylation Protects Tac from Proteolytic Shedding
In addition to cytosolic determinants, multisubunit interactions and post-translational modifications on the extracellular domain may also affect protein half-life. Tac is a monomeric protein with two asparagine-linked (Asn70 and Asn89) glycosylation sites and a number of O-linked glycosylation sites in the stalk region near the transmembrane domain (20, 38) (Fig. 4A). To investigate whether these sites for carbohydrate modification could affect the itinerary and turnover of SNAP-Tac, we mutated four threonine residues (at amino acids 197, 203, 208, and 216 of the mature Tac protein) proposed to be O-glycosylated (20) to alanine residues (SNAP-Tac(T→A)) (Fig. 4A). We also created a truncated version of SNAP-Tac in which the bulk of the luminal domain of Tac was deleted, but its 27-amino acid juxtamembrane stalk region as well as its transmembrane domain and cytoplasmic tail (SNAP-Tac trunc) was included. A threonine mutant variant of this construct was also generated (SNAP-Tac trunc(T→A)) (Fig. 4A). A SNAP-Tac construct with two asparagine-linked (Asn70 and Asn89) glycosylation sites mutated to serine residues had no effect on protein delivery to the cell surface or its turnover and will not be described further (data not shown).
FIGURE 4.

Mutation of O-linked glycosylation sites results in rapid loss of SNAP-Tac and SNAP-Tac trunc from the cell surface. A, schematic of SNAP-Tac and SNAP-Tac trunc together with their Thr → Ala mutants. The numbering system reflects the amino acids from the mature Tac protein. B–E, HeLa cells were labeled for 30 min at 4 °C with BG-488 and either fixed directly or chased for 30 min at 37 °C before fixation. Representative images for SNAP-Tac (B), SNAP-Tac(T→A) (C), SNAP-Tac trunc (D), and SNAP-Tac trunc(T→A) (E) are shown. DAPI staining shows cell nuclei. Bars, 10 μm.
We examined the trafficking of these proteins by fluorescence microscopy. After labeling with non-cleavable BG-488 on ice, surface staining was observed for all of the constructs (Fig. 4, B–E, top panels). After 30-min incubation to allow endocytosis, surface labeling was still observed for SNAP-Tac and SNAP-Tac trunc (Fig. 4, B and D); however, surface staining was gone from cells that were transfected with the threonine mutants SNAP-Tac(T→A) and SNAP-Tac trunc(T→A) (Fig. 4, C and E). Not only was the surface label lost, but also there was only a small amount of label, possibly in endosomes, associated with these cells.
To examine this rapid loss biochemically, we performed the degradation assay on these proteins using BG-800. We found that the turnover of SNAP-Tac and SNAP-Tac trunc was similar with half-lives of 4–5 h (Fig. 5, A, C, and E). Thus, the turnover of SNAP-Tac was not influenced by the absence of the bulk of its extracellular domain. By contrast, within 30 min, the cell-associated protein levels for both threonine to alanine mutants, SNAP-Tac(T→A) and SNAP-Tac trunc(T→A), were down to 10% of initial surface label (Fig. 5, B, D, and E). This rapid loss from the cell seems incompatible with lysosomal degradation, and given that Tac can be shed from the surface of some cells (39), we examined whether these proteins were shed into the media. Whereas only about 20% of SNAP-Tac and truncated SNAP-Tac were detected in the media by 4 h (Fig. 5, A, C, and F), over 80% of the glycosylation mutants were found in the media by 30 min of incubation (Fig. 5, B, D, and F). Shedding was confirmed by a shift in molecular weight seen between the bands in the cell-associated and media fractions. Lysosomal inhibitors did not prevent shedding (data not shown). A SNAP-Tac-GPI(T→A) construct was also rapidly shed into the medium, demonstrating that shedding does not require the Tac transmembrane or cytoplasmic tail domains (data not shown). Thus, the lack of O-linked glycans on the juxtamembrane stalk of SNAP-Tac sensitizes the protein to proteolytic cleavage on the PM.
FIGURE 5.

Mutation of O-linked glycosylation results in shedding of SNAP-Tac into the medium. HeLa cells were transfected with SNAP-Tac, SNAP-Tac(T→A), SNAP-Tac trunc, or SNAP-Tac trunc(T→A) and labeled with BG-800 for 30 min on ice in serum-free DMEM. Cells were chased for 30 min, 2 h, and 4 h in serum-free DMEM containing excess BG-NH2. Media and lysates were collected and run on SDS-polyacrylamide gels. Duplicate or triplicate independent lysate samples were loaded in individual lanes. For the media samples only, one sample is loaded per time point. Coomassie staining was used to determine total protein loaded. A–D, representative gels from the indicated full-length or truncated constructs. The full-length proteins and shed products are indicated (in A and B, m represents medium). In B, the fourth lane is empty. Quantification of cell-associated (normalized to time 0) (E) and shed (the fraction of the total signal shed into the medium; see “Experimental Procedures”) (F) SNAP-Tac proteins is shown. Bar graphs represent data averaged from three independent experiments ±S.D. (error bars). The migration of prestained molecular mass markers (in kDa) is shown to the left of each panel.
Matrix metalloprotease-9 (MMP-9) has been implicated in the shedding of Tac from the cell surface (40). We used batimastat (BB-94), a broad spectrum MMP inhibitor, to see whether we could inhibit the release of SNAP-Tac and SNAP-Tac(T→A) into the media. We found that, after 30 min, shedding of both SNAP-Tac (Fig. 6A, lanes 5 and 6) and SNAP-Tac(T→A) (Fig. 6B, lanes 5 and 6) was completely prevented by batimastat. In addition, the loss of cell-associated SNAP-Tac(T→A) was also reversed by batimastat (Fig. 6B, lanes 7 and 8). These results were quantified for both cell-associated and shed proteins (Fig. 6, C and D). These observations suggest that the extracellular juxtamembrane stalk of Tac provides a potential recognition site for cleavage by a batimastat-sensitive protease and that O-linked sugars in this region protect Tac from this cleavage. Thus, glycosylated Tac should be resistant to shedding, whereas Tac lacking O-linked sugars might be more sensitive. To test this, SNAP-Tac-expressing cells were treated overnight with an O-linked glycosylation inhibitor, GalNAc-O-Bn, a competitive inhibitor of N-acetylgalactosamine (41). SNAP-Tac showed an increased mobility in cells treated overnight with GalNAc-O-Bn compared with untreated cells (see Fig. 6E, compare lanes 1 and 2 with lanes 3 and 4), suggesting that O-glycans are not added. When shedding was examined in GalNAc-O-Bn-treated cells, an increased proportion of SNAP-Tac was cleaved from the cell surface and released into the medium (Fig. 6 E, compare lanes 15 and 16 with lanes 17 and 18). This shedding was BB-94-sensitive (Fig. 5E, lanes 19 and 20). In addition, enhanced labeling of shed SNAP-Tac with BG-800 was observed from the conditioned medium derived from cells treated overnight with GalNAc-O-Bn (Fig. 6F). These data confirm that the presence or absence of O-linked sugars added to the membrane-proximal domain of Tac protects or sensitizes Tac from shedding activities to prolong or shorten its cellular half-life.
FIGURE 6.

O-Linked glycosylation protects SNAP-Tac from ectodomain cleavage. HeLa cells expressing SNAP-Tac or SNAP-Tac(T→A) were labeled on ice with BG-800 for 30 min in the presence or absence of 500 nm BB-94, a broad spectrum MMP inhibitor. All experiments were performed in serum-free media. Media (med) and lysates (lys) for the 0-min time point were collected, and the rest of the cells were incubated at 37 °C for 30 min in the presence or absence of 500 nm BB-94. A, gel from SNAP-Tac-expressing cells. B, gel from SNAP-Tac(T→A)-expressing cells. Note that BB-94 inhibits the shedding of both proteins at 30 min (lanes 5 and 6) and prevents the loss of rapidly shed SNAP-Tac(T→A) from the lysate fraction (lanes 7 and 8) (64). Quantification of cell-associated proteins (normalized to time 0) (C) and shed (the fraction of the total signal shed into the medium) (D) SNAP-Tac proteins is shown. Bar graphs represent data averaged from three independent experiments ±S.D. (error bars). E, SNAP-Tac-expressing cells were pulse-labeled with BG-800 on ice for 30 min and then chased for 0 or 8 h in the presence of excess BG-NH2. Where indicated, the MMP inhibitor BB-94 was added during the labeling period and the chase. Also, where indicated, an O-linked glycosylation inhibitor was included in the medium during the overnight transfection as well as the pulse and chase periods. Note that treatment with this inhibitor produces a faster migrating, presumably O-linked sugar-depleted, SNAP-Tac protein (compare lanes 1 and 2 with lanes 3 and 4). During the chase, an increase in shed SNAP-Tac is also observed (compare lanes 15 and 16 with lanes 17 and 18) that is prevented with BB-94 (lanes 19 and 20). For lanes 9–20, the sensitivity of the image was enhanced 2-fold so that the minor signal from lanes 19 and 20 could be seen. F, conditioned medium from SNAP-Tac-transfected cells was labeled with BG-800. Note the increase in shed SNAP-Tac when cells were treated with the O-glycosylation inhibitor. The migration of prestained molecular mass markers (in kDa) is shown to the left of each panel.
A higher rate of shedding of SNAP-Tac was observed in COS cells as compared with HeLa cells both by fluorescence labeling and biochemically (Fig. 7A). This suggests that COS cells may have increased surface levels or activities of proteases responsible for shedding. To examine this, HeLa and COS cells were surface-biotinylated, and the release of biotinylated proteins into the medium was monitored during a chase. Both cells were extensively labeled by the biotinylation procedure, but COS cells released considerably more proteins into the medium compared with HeLa cells (Fig. 7B). Thus, for particular PM proteins, turnover is likely determined by a combination of shedding from the surface as well as delivery to lysosomes, the proportion of which may differ in different cell types.
FIGURE 7.
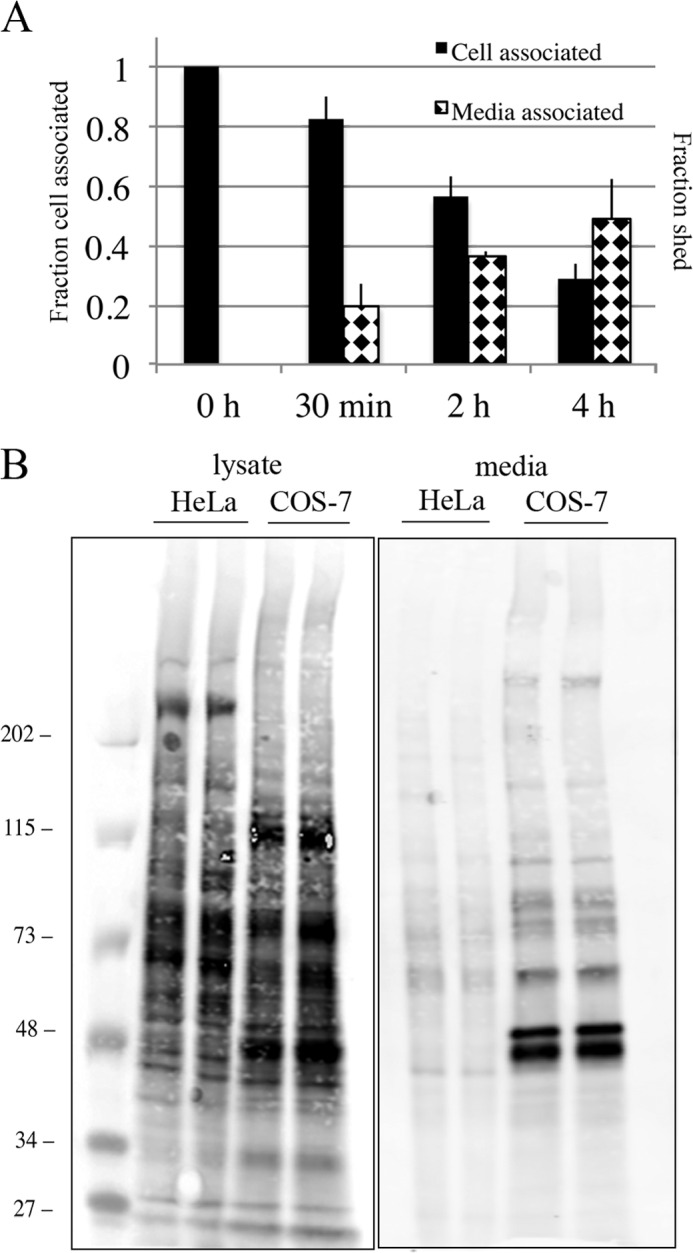
Elevated level of shedding of SNAP-Tac in COS cells. A, COS cells transfected with SNAP-Tac were labeled on ice with BG-800 for 30 min. At the indicated time points after shift to 37 °C, lysates and media were collected and processed as described under “Experimental Procedures.” The black bars indicate the fraction of cell-associated SNAP-Tac remaining after each time point (left axis), and the diamond-hatched bars indicate the fraction shed into the medium (right axis). Shedding of SNAP-Tac in COS cells is significantly enhanced when compared with HeLa cells (see Fig. 5, E and F). The bar graph represents normalized data averaged from three independent experiments ±S.D. (error bars). B, COS or HeLa cells were plated in 10-cm dishes, surface-labeled with NHS-PEG4-biotin for 30 min at room temperature, and then chased for 5 h in serum-free medium. After collecting media and lysing cells, samples were run on an SDS-polyacrylamide gel, then transferred onto nitrocellulose, and incubated with DyLight 800-conjugated NeutrAvidin to detect biotinylated proteins. Equivalent protein levels and media volumes were loaded for each cell type. The scanned sensitivity of the lysate samples was reduced 6-fold to visualize individual protein bands. The migration of prestained molecular mass markers (in kDa) is shown.
DISCUSSION
Here we developed a model system to identify features of cell surface proteins that determine cellular half-life. We used SNAP-Tac, a simple type I transmembrane protein and CIE cargo molecule, as a reporter protein. We modified selected regions of the protein to analyze protein turnover via entry by CME, replacing the cytoplasmic and transmembrane sequences with a GPI anchor, preventing ubiquitination via removal of the sole cytoplasmic lysine in Tac, and eliminating O-linked glycosylation. Protein half-life was affected by each of these modifications.
Cytoplasmic sequences affected protein turnover by regulating the mode of entry and intracellular itinerary. Dileucine motifs allow entry by CME and rapid targeting to lysosomes (8, 27). Consistent with this, the rapid turnover of SNAP-Tac-LI (t½ of 3.2 h) contrasts with the degradation of SNAP-Tac, which enters by CIE and has a longer cell surface lifetime (t½ of 5.5 h). Tac that lacks its single cytoplasmic lysine, a modification that prevents ubiquitination, still enters by CIE but now has a reduced turnover rate (t½ of 7.9 h). SNAP-Tac-LI that also contains the K246R mutation is degraded with a similar half-life as SNAP-Tac-LI (data not shown). This may be due to the dominant lysosomal targeting of SNAP-Tac-LI. The longest cell surface lifetime (t½ of 12.1 h) is achieved when Tac has a GPI anchor. SNAP-Tac-GPI resides in detergent-resistant lipid raft domains similar to its parent molecule, Tac-GPI, and enters cells via CIE (19). The extended cellular half-life of SNAP-Tac-GPI may be due to the absence of cytoplasmic domain for ubiquitination and/or may be due to its residence in a particular membrane lipid environment. Using a biotinylation/recovery assay, Hare and Taylor (3) demonstrated that proteins that were endocytosed had, in general, a shorter stability than proteins restricted to the cell surface. Quantitation of internalization and recycling rates of the various SNAP-Tac proteins will better define this relationship between internalization and degradation (16). Another issue regarding the longer half-lives of SNAP-Tac-KR and SNAP-Tac-GPI is whether, in the absence of ubiquitination, these proteins fail to be packaged into intraluminal vesicles of multivesicular bodies, which may reduce their susceptibility to lysosomal hydrolysis, thus extending their apparent lifetimes (2).
Recently, Trono and co-workers (34) used the SNAP-tag technology to measure the half-life of fusion proteins in cultured Jurkat T cells and Jurkat tumors in living mice. The half-life of surface-labeled unstimulated SNAP-CD4 was 5.2 h, which is similar to that of wild-type SNAP-Tac observed here (5 h). Surprisingly, in Jurkat cells, SNAP-GPI had a half-life of 3.8 h, which is considerably shorter than what we observed for SNAP-Tac-GPI (t½ of 12 h). One possibility is that the different structures of the two proteins affect their turnover rate. SNAP-Tac-GPI contains the O-glycosylated stalk domain from Tac, which may prolong its lifetime by reducing its trafficking to lysosomes or its susceptibility to lysosomal hydrolysis. The sequence of SNAP-GPI from Bojkowska et al. (34) was not provided but likely contains the extracellular SNAP-tag fused directly to a GPI anchor targeting sequence. Alternatively, Jurkat cells may have a higher endogenous sheddase activity than HeLa cells similar to what we have observed in COS cells. The medium was not analyzed in Bojkowska et al. (34). GPI-anchored proteins usually have long half-lives due to their inclusion in lipid rafts and entry by CIE (11, 42); however, different GPI anchors within the same cell type can dramatically affect the amounts of shedding of these proteins (43).
Tac was chosen as a model reporter construct for a number of reasons. First, Tac is targeted to the plasma membrane by default and has been extensively used to delineate cytoplasmic sorting signals (22, 25, 26, 44). Tac itself is a model CIE protein whose internalization and trafficking kinetics mimic those of endogenous CIE cargo such as major histocompatibility complex class I (MHCI) (27). Second, except in immune cells where Tac is associated with β and γ chains of the interleukin 2 receptor (38), it is not stably associated with other endogenous molecules that may affect its trafficking and turnover kinetics. In contrast, when we measured the turnover rates of another CIE cargo, SNAP-CD98 (15), which associates covalently at the PM with the large neutral amino acid transporter 1 (LAT1), the heterodimeric form of the molecule (SNAP-CD98/LAT1) was stabilized compared with monomeric SNAP-CD98.3 Thus, assembly into multisubunit complexes can influence PM protein turnover.
It is generally thought that ubiquitin tagging of cytoplasmic domains either at the cell surface or on endosomes targets them to late endosomes/lysosomes for degradation. Deubiquitinating activities may permit them to avoid ultimate destruction and recycle back to the cell surface (4, 45). For proteins that lack cytoplasmic sequences (e.g. GPI-anchored proteins) or have mutations that block ubiquitination, degradation still occurs, albeit with reduced kinetics, at least for the proteins we have examined here. Whether this is via a bulk flow mechanism in which a fractional proportion of endocytosed membrane reaches lysosomes by default or these proteins are subject to an undefined surveillance mechanism is not known.
The extracellular domains of a number of transmembrane proteins are released from the cell surface as soluble proteins through a regulated proteolytic mechanism called ectodomain shedding (46, 47). Only about 2% of cell surface proteins are released from the surface by ectodomain shedding, indicating that cells selectively shed their protein ectodomains (46). Cleavage often occurs in a juxtamembrane stalk region adjacent to the membrane anchor (47–49) and may require an unstructured or extended stalk of minimal length (50). In addition to a compactly folded stalk that may protect membrane protein from proteolysis, proteins such as the LDL receptor (51, 52), transferrin receptor (53, 54), and decay accelerating factor (55) have relatively short juxtamembrane stalk sequences that are O-glycosylated, and when these sites are either mutated or the proteins are expressed in a mutant Chinese hamster ovary (CHO) cell line defective in mucin-type O-glycan synthesis (ldlD), they are proteolytically shed. Conversely, stalk O-glycosylation of angiotensin-converting enzyme increases susceptibility to proteolysis (47).
Normally, little Tac is expressed on unstimulated lymphoid cells. Rubin et al. (39) demonstrated that activated normal peripheral blood mononuclear cells and certain lines of T and B cell origin release a soluble form of Tac into the culture medium. Significant increases in both Tac and the soluble form of Tac are seen in autoimmune diseases, organ transplant rejection, and in many T cell and B cell neoplasms (56, 57). Although Tac has been known to be O-glycosylated for years (20, 38), the relationship between O-glycosylation and shedding has not been addressed. For the SNAP-Tac protein studied here, removal of four (of a possible five) (20) O-glycosylation sites resulted in shedding soon after SNAP-Tac reached the cell surface. This effect was mimicked with wild-type SNAP-Tac in the presence of the O-glycosylation inhibitor benzyl 2-acetamido-2-deoxy-α-d-galactopyranoside and was inhibited by the general MMP inhibitor batimastat (BB-94). The loss of cell-associated Tac observed in ldlD CHO cells (58) may, therefore, be due to proteolytic shedding. Structural analysis revealed that cleavage of Tac occurs in the extracellular domain between Cys192 and Leu193 in the mature protein (59). Although this region contains no consensus site for proteolytic cleavage, studies have indicated that MMP-9, a member of the MMP family, has the ability to cleave Tac (Refs. 40 and 60, but also see Ref. 61). MMPs are important proteolytic enzymes involved in cancer metastasis, hydrolysis of the extracellular matrix, invasion, and immune cell development (62, 63). Interestingly, we observed that COS cells had an elevated shedding activity compared with HeLa cells both with SNAP-Tac and when total surface proteins were biotinylated. Whether this is a result of differences in the structure of O-glycans synthesized in the Golgi complex or increased surface MMP activity in COS cells is not known.
We have used a model PM protein to discover that parallel processing pathways exist to regulate the turnover of PM proteins, endocytosis and trafficking to lysosomes, and proteolytic shedding. The extent of each of these pathways was protein- and cell type-specific and dependent upon a number of factors, including whether the proteins were endocytosed by clathrin-dependent or -independent pathways, whether they were ubiquitinated, and whether they contained structural motifs that could be recognized by PM proteases. The labeling methods used here can be extended to PM proteins found in multisubunit complexes and those that are involved in ligand-dependent cell signaling pathways.
Acknowledgments
We thank members of the Donaldson laboratory for comments on this work. We also acknowledge the Light Microscopy Core Facility of the National Heart, Lung, and Blood Institute.
This work was supported, in whole or in part, by National Institutes of Health Grant HL006060 from the Intramural Research Program in the NHLBI.
D. Karabasheva, N. B. Cole, and J. G. Donaldson, unpublished observations.
- PM
- plasma membrane
- GPI
- glycosylphosphatidylinositol
- CME
- endocytosis through clathrin-mediated pathways
- CIE
- endocytosis through clathrin-independent pathways
- HBSS
- Hanks' buffered saline solution
- TCEP
- tris(2-carboxyethyl)phosphine
- BG
- O6-benzylguanine
- GalNAc-O-Bn
- benzyl 2-acetamido-2-deoxy-α-d-galactopyranoside
- NHS
- N-hydroxysuccinimide
- EEA1
- early endosomal antigen 1
- trunc
- truncated
- MMP
- matrix metalloproteinase
- 488
- Alexa Fluor 488.
REFERENCES
- 1. Okiyoneda T., Apaja P. M., Lukacs G. L. (2011) Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 23, 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. MacGurn J. A., Hsu P. C., Emr S. D. (2012) Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81, 231–259 [DOI] [PubMed] [Google Scholar]
- 3. Hare J. F., Taylor K. (1991) Mechanisms of plasma membrane protein degradation: recycling proteins are degraded more rapidly than those confined to the cell surface. Proc. Natl. Acad. Sci. U.S.A. 88, 5902–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clague M. J., Liu H., Urbé S. (2012) Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 23, 457–467 [DOI] [PubMed] [Google Scholar]
- 5. Henne W. M., Buchkovich N. J., Emr S. D. (2011) The ESCRT pathway. Dev. Cell 21, 77–91 [DOI] [PubMed] [Google Scholar]
- 6. Tanno H., Komada M. (2013) The ubiquitin code and its decoding machinery in the endocytic pathway. J. Biochem. 153, 497–504 [DOI] [PubMed] [Google Scholar]
- 7. Raiborg C., Stenmark H. (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452 [DOI] [PubMed] [Google Scholar]
- 8. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 9. Godlee C., Kaksonen M. (2013) Review series: from uncertain beginnings: initiation mechanisms of clathrin-mediated endocytosis. J. Cell Biol. 203, 717–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howes M. T., Kirkham M., Riches J., Cortese K., Walser P. J., Simpson F., Hill M. M., Jones A., Lundmark R., Lindsay M. R., Hernandez-Deviez D. J., Hadzic G., McCluskey A., Bashir R., Liu L., Pilch P., McMahon H., Robinson P. J., Hancock J. F., Mayor S., Parton R. G. (2010) Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J. Cell Biol. 190, 675–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maldonado-Báez L., Williamson C., Donaldson J. G. (2013) Clathrin-independent endocytosis: a cargo-centric view. Exp. Cell Res. 319, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horiuchi K. (2013) A brief history of tumor necrosis factor α-converting enzyme: an overview of ectodomain shedding. Keio J. Med. 62, 29–36 [DOI] [PubMed] [Google Scholar]
- 13. Scheller J., Chalaris A., Garbers C., Rose-John S. (2011) ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32, 380–387 [DOI] [PubMed] [Google Scholar]
- 14. Blobel C. P. (2005) ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43 [DOI] [PubMed] [Google Scholar]
- 15. Eyster C. A., Cole N. B., Petersen S., Viswanathan K., Früh K., Donaldson J. G. (2011) MARCH ubiquitin ligases alter the itinerary of clathrin-independent cargo from recycling to degradation. Mol. Biol. Cell 22, 3218–3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cole N. B., Donaldson J. G. (2012) Releasable SNAP-tag probes for studying endocytosis and recycling. ACS Chem. Biol. 7, 464–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pond L., Kuhn L. A., Teyton L., Schutze M. P., Tainer J. A., Jackson M. R., Peterson P. A. (1995) A role for acidic residues in di-leucine motif-based targeting to the endocytic pathway. J. Biol. Chem. 270, 19989–19997 [DOI] [PubMed] [Google Scholar]
- 18. Höning S., Sandoval I. V., von Figura K. (1998) A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 17, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naslavsky N., Weigert R., Donaldson J. G. (2004) Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol. Biol. Cell 15, 3542–3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miedel M. C., Hulmes J. D., Pan Y. C. (1989) Limited proteolysis of recombinant human soluble interleukin-2 receptor. Identification of an interleukin-2 binding core. J. Biol. Chem. 264, 21097–21105 [PubMed] [Google Scholar]
- 21. Luo S., Wehr N. B., Levine R. L. (2006) Quantitation of protein on gels and blots by infrared fluorescence of Coomassie Blue and Fast Green. Anal. Biochem. 350, 233–238 [DOI] [PubMed] [Google Scholar]
- 22. Kinlough C. L., Poland P. A., Gendler S. J., Mattila P. E., Mo D., Weisz O. A., Hughey R. P. (2011) Core-glycosylated mucin-like repeats from MUC1 are an apical targeting signal. J. Biol. Chem. 286, 39072–39081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schapiro F. B., Soe T. T., Mallet W. G., Maxfield F. R. (2004) Role of cytoplasmic domain serines in intracellular trafficking of furin. Mol. Biol. Cell 15, 2884–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Letourneur F., Klausner R. D. (1992) A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell 69, 1143–1157 [DOI] [PubMed] [Google Scholar]
- 25. Humphrey J. S., Peters P. J., Yuan L. C., Bonifacino J. S. (1993) Localization of TGN38 to the trans-Golgi network: involvement of a cytoplasmic tyrosine-containing sequence. J. Cell Biol. 120, 1123–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marks M. S., Roche P. A., van Donselaar E., Woodruff L., Peters P. J., Bonifacino J. S. (1995) A lysosomal targeting signal in the cytoplasmic tail of the beta chain directs HLA-DM to MHC class II compartments. J. Cell Biol. 131, 351–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naslavsky N., Weigert R., Donaldson J. G. (2003) Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14, 417–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radhakrishna H., Donaldson J. G. (1997) ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 139, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89 [DOI] [PubMed] [Google Scholar]
- 30. Keppler A., Arrivoli C., Sironi L., Ellenberg J. (2006) Fluorophores for live cell imaging of AGT fusion proteins across the visible spectrum. BioTechniques 41, 167–175 [DOI] [PubMed] [Google Scholar]
- 31. Gautier A., Nakata E., Lukinavicius G., Tan K. T., Johnsson K. (2009) Selective cross-linking of interacting proteins using self-labeling tags. J. Am. Chem. Soc. 131, 17954–17962 [DOI] [PubMed] [Google Scholar]
- 32. Emami-Nemini A., Roux T., Leblay M., Bourrier E., Lamarque L., Trinquet E., Lohse M. J. (2013) Time-resolved fluorescence ligand binding for G protein-coupled receptors. Nat. Protoc. 8, 1307–1320 [DOI] [PubMed] [Google Scholar]
- 33. Cole N. B. (2013) Site-specific protein labeling with SNAP-tags. Curr. Protoc. Protein Sci. 73, Unit 30.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bojkowska K., Santoni de Sio F., Barde I., Offner S., Verp S., Heinis C., Johnsson K., Trono D. (2011) Measuring in vivo protein half-life. Chem. Biol. 18, 805–815 [DOI] [PubMed] [Google Scholar]
- 35. Foraker A. B., Camus S. M., Evans T. M., Majeed S. R., Chen C. Y., Taner S. B., Corrêa I. R., Jr., Doxsey S. J., Brodsky F. M. (2012) Clathrin promotes centrosome integrity in early mitosis through stabilization of centrosomal ch-TOG. J. Cell Biol. 198, 591–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bodor D. L., Rodríguez M. G., Moreno N., Jansen L. E. (2012) Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. Curr. Protoc. Cell Biol. Chapter 8, Unit 8.8 [DOI] [PubMed] [Google Scholar]
- 37. Harder J. L., Whiteman E. L., Pieczynski J. N., Liu C. J., Margolis B. (2012) Snail destabilizes cell surface Crumbs3a. Traffic 13, 1170–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Greene W. C., Leonard W. J. (1986) The human interleukin-2 receptor. Annu. Rev. Immunol. 4, 69–95 [DOI] [PubMed] [Google Scholar]
- 39. Rubin L. A., Kurman C. C., Fritz M. E., Biddison W. E., Boutin B., Yarchoan R., Nelson D. L. (1985) Soluble interleukin 2 receptors are released from activated human lymphoid cells in vitro. J. Immunol. 135, 3172–3177 [PubMed] [Google Scholar]
- 40. De Paiva C. S., Yoon K. C., Pangelinan S. B., Pham S., Puthenparambil L. M., Chuang E. Y., Farley W. J., Stern M. E., Li D. Q., Pflugfelder S. C. (2009) Cleavage of functional IL-2 receptor α chain (CD25) from murine corneal and conjunctival epithelia by MMP-9. J. Inflamm. 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuan S. F., Byrd J. C., Basbaum C., Kim Y. S. (1989) Inhibition of mucin glycosylation by aryl-N-acetyl-α-galactosaminides in human colon cancer cells. J. Biol. Chem. 264, 19271–19277 [PubMed] [Google Scholar]
- 42. Lemansky P., Fatemi S. H., Gorican B., Meyale S., Rossero R., Tartakoff A. M. (1990) Dynamics and longevity of the glycolipid-anchored membrane protein, Thy-1. J. Cell Biol. 110, 1525–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li R., Liu T., Yoshihiro F., Tary-Lehmann M., Obrenovich M., Kuekrek H., Kang S. C., Pan T., Wong B. S., Medof M. E., Sy M. S. (2003) On the same cell type GPI-anchored normal cellular prion and DAF protein exhibit different biological properties. Biochem. Biophys. Res. Commun. 303, 446–451 [DOI] [PubMed] [Google Scholar]
- 44. Haft C. R., Klausner R. D., Taylor S. I. (1994) Involvement of dileucine motifs in the internalization and degradation of the insulin receptor. J. Biol. Chem. 269, 26286–26294 [PubMed] [Google Scholar]
- 45. Wright M. H., Berlin I., Nash P. D. (2011) Regulation of endocytic sorting by ESCRT-DUB-mediated deubiquitination. Cell Biochem. Biophys. 60, 39–46 [DOI] [PubMed] [Google Scholar]
- 46. Hayashida K., Bartlett A. H., Chen Y., Park P. W. (2010) Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. 293, 925–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schwager S. L., Chubb A. J., Scholle R. R., Brandt W. F., Mentele R., Riordan J. F., Sturrock E. D., Ehlers M. R. (1999) Modulation of juxtamembrane cleavage (“shedding”) of angiotensin-converting enzyme by stalk glycosylation: evidence for an alternative shedding protease. Biochemistry 38, 10388–10397 [DOI] [PubMed] [Google Scholar]
- 48. Hinkle C. L., Sunnarborg S. W., Loiselle D., Parker C. E., Stevenson M., Russell W. E., Lee D. C. (2004) Selective roles for tumor necrosis factor α-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J. Biol. Chem. 279, 24179–24188 [DOI] [PubMed] [Google Scholar]
- 49. Weskamp G., Schlöndorff J., Lum L., Becherer J. D., Kim T. W., Saftig P., Hartmann D., Murphy G., Blobel C. P. (2004) Evidence for a critical role of the tumor necrosis factor α convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J. Biol. Chem. 279, 4241–4249 [DOI] [PubMed] [Google Scholar]
- 50. Baran P., Nitz R., Grötzinger J., Scheller J., Garbers C. (2013) Minimal interleukin 6 (IL-6) receptor stalk composition for IL-6 receptor shedding and IL-6 classic signaling. J. Biol. Chem. 288, 14756–14768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamamoto T., Davis C. G., Brown M. S., Schneider W. J., Casey M. L., Goldstein J. L., Russell D. W. (1984) The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell 39, 27–38 [DOI] [PubMed] [Google Scholar]
- 52. Kozarsky K., Kingsley D., Krieger M. (1988) Use of a mutant cell line to study the kinetics and function of O-linked glycosylation of low density lipoprotein receptors. Proc. Natl. Acad. Sci. U.S.A. 85, 4335–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rutledge E. A., Root B. J., Lucas J. J., Enns C. A. (1994) Elimination of the O-linked glycosylation site at Thr 104 results in the generation of a soluble human-transferrin receptor. Blood 83, 580–586 [PubMed] [Google Scholar]
- 54. Rutledge E. A., Enns C. A. (1996) Cleavage of the transferrin receptor is influenced by the composition of the O-linked carbohydrate at position 104. J. Cell. Physiol. 168, 284–293 [DOI] [PubMed] [Google Scholar]
- 55. Reddy P., Caras I., Krieger M. (1989) Effects of O-linked glycosylation on the cell surface expression and stability of decay-accelerating factor, a glycophospholipid-anchored membrane protein. J. Biol. Chem. 264, 17329–17336 [PubMed] [Google Scholar]
- 56. Morris J. C., Waldmann T. A. (2000) Advances in interleukin 2 receptor targeted treatment. Ann. Rheum. Dis. 59, Suppl. 1, i109–i114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rubin L. A., Nelson D. L. (1990) The soluble interleukin-2 receptor: biology, function, and clinical application. Ann. Intern. Med. 113, 619–627 [DOI] [PubMed] [Google Scholar]
- 58. Kozarsky K. F., Call S. M., Dower S. K., Krieger M. (1988) Abnormal intracellular sorting of O-linked carbohydrate-deficient interleukin-2 receptors. Mol. Cell. Biol. 8, 3357–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robb R. J., Kutny R. M. (1987) Structure-function relationships for the IL 2-receptor system. IV. Analysis of the sequence and ligand-binding properties of soluble Tac protein. J. Immunol. 139, 855–862 [PubMed] [Google Scholar]
- 60. Sheu B. C., Hsu S. M., Ho H. N., Lien H. C., Huang S. C., Lin R. H. (2001) A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 61, 237–242 [PubMed] [Google Scholar]
- 61. El Houda Agueznay N., Badoual C., Hans S., Gey A., Vingert B., Peyrard S., Quintin-Colonna F., Ravel P., Bruneval P., Roncelin S., Lelongt B., Bertoglio J., Fridman W. H., Brasnu D., Tartour E. (2007) Soluble interleukin-2 receptor and metalloproteinase-9 expression in head and neck cancer: prognostic value and analysis of their relationships. Clin. Exp. Immunol. 150, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khokha R., Murthy A., Weiss A. (2013) Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 13, 649–665 [DOI] [PubMed] [Google Scholar]
- 63. Visse R., Nagase H. (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827–839 [DOI] [PubMed] [Google Scholar]
- 64. Haglund K., Ivankovic-Dikic I., Shimokawa N., Kruh G. D., Dikic I. (2004) Recruitment of Pyk2 and Cbl to lipid rafts mediates signals important for actin reorganization in growing neurites. J. Cell Sci. 117, 2557–2568 [DOI] [PubMed] [Google Scholar]