

Background: GPIHBP1 binds lipoprotein lipase (LPL) and transports it to the capillary lumen.
Results: A GPIHBP1 missense mutation (S107C) leads to the formation of GPIHBP1 multimers that cannot bind LPL.
Conclusion: An extra cysteine leads to GPIHBP1 multimerization, defective LPL binding, and hypertriglyceridemia.
Significance: This study identifies a novel mechanism by which GPIHBP1 mutations interfere with LPL binding and cause hypertriglyceridemia.
Keywords: Disulfide, Genetics, Lipolysis, Protein Folding, Protein-Protein Interaction, Hypertriglyceridemia, Lipoprotein Lipase, Mutation
Abstract
GPIHBP1, a glycosylphosphatidylinositol-anchored glycoprotein of microvascular endothelial cells, binds lipoprotein lipase (LPL) within the interstitial spaces and transports it across endothelial cells to the capillary lumen. The ability of GPIHBP1 to bind LPL depends on the Ly6 domain, a three-fingered structure containing 10 cysteines and a conserved pattern of disulfide bond formation. Here, we report a patient with severe hypertriglyceridemia who was homozygous for a GPIHBP1 point mutation that converted a serine in the GPIHBP1 Ly6 domain (Ser-107) to a cysteine. Two hypertriglyceridemic siblings were homozygous for the same mutation. All three homozygotes had very low levels of LPL in the preheparin plasma. We suspected that the extra cysteine in GPIHBP1-S107C might prevent the trafficking of the protein to the cell surface, but this was not the case. However, nearly all of the GPIHBP1-S107C on the cell surface was in the form of disulfide-linked dimers and multimers, whereas wild-type GPIHBP1 was predominantly monomeric. An insect cell GPIHBP1 expression system confirmed the propensity of GPIHBP1-S107C to form disulfide-linked dimers and to form multimers. Functional studies showed that only GPIHBP1 monomers bind LPL. In keeping with that finding, there was no binding of LPL to GPIHBP1-S107C in either cell-based or cell-free binding assays. We conclude that an extra cysteine in the GPIHBP1 Ly6 motif results in multimerization of GPIHBP1, defective LPL binding, and severe hypertriglyceridemia.
Introduction
Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1)3 is an endothelial cell protein that binds lipoprotein lipase (LPL) within the interstitial spaces and transports it to the capillary lumen (1). In the absence of GPIHBP1, LPL remains in the interstitial spaces (1), resulting in defective processing of triglyceride-rich lipoproteins, severe hypertriglyceridemia (chylomicronemia), and impaired delivery of lipid nutrients to parenchymal cells (2, 3).
GPIHBP1 belongs to the lymphocyte antigen 6 (Ly6) family of proteins. This family, which is also referred to as the LU (Ly6/uPAR) family (4), includes the urokinase-type plasminogen activator receptor (uPAR) and CD59 (an inhibitor of autologous complement activation) (5). GPIHBP1 and most mammalian members of the Ly6 family of proteins are tethered to the plasma membrane by a glycosylphosphatidylinositol anchor. The hallmark of Ly6 proteins is a 70–80-amino acid motif containing 8 cysteines, which are arranged in a characteristic spacing pattern and are oxidized to yield an identical disulfide bonding. The formation of disulfide bonds is one of several structural features that lead these proteins to adopt a three-fingered structure (6). The consensus three-fingered folding topology of mammalian Ly6 proteins is identical to the canonical three-fingered structure in α-neurotoxins of snake venoms (7). Many mammalian Ly6 proteins, including GPIHBP1, have a total of 10 cysteines, leading to the formation of an extra disulfide bond that stabilizes the first loop in the three-fingered structural motif.
The Ly6 domain is crucial for ligand interactions. In the case of uPAR, three Ly6 domains cooperate to form the binding interface for the serine protease urokinase-type plasminogen activator (8) as well as a low affinity binding site for the extracellular matrix protein vitronectin (9); this cooperative arrangement facilitates allosteric control of ligand binding (10). In the case of GPIHBP1, the Ly6 domain is crucial for binding LPL. Six GPIHBP1 missense mutations, all involving conserved amino acids in the Ly6 domain, have already been linked to chylomicronemia in humans (11–18). In four of these cases, the mutant GPIHBP1 was tested and shown to lack the ability to bind LPL (12, 13, 15, 16). Subsequent studies uncovered LPL mutations that abolish the ability of LPL to bind to wild-type GPIHBP1 (19).
In the present study, we screened 92 patients with unexplained chylomicronemia for GPIHBP1 mutations. We uncovered a novel missense mutation that converted Ser-107 in the GPIHBP1 Ly6 domain to a cysteine. Our studies revealed the mechanism by which this GPIHBP1 mutation leads to chylomicronemia.
EXPERIMENTAL PROCEDURES
Subjects
Ninety-two patients with severe hypertriglyceridemia, defined as fasting plasma triglyceride levels >10 mmol/liter (>885 mg/dl) on at least two occasions, were identified at King Chulalongkorn Memorial Hospital. After excluding coding-sequence and splice-site mutations in LPL, APOC2, and APOA5, we screened for GPIHBP1 mutations. A homozygous missense mutation in GPIHBP1 (c.320C>G; p.S107C) was identified in a 46-year-old woman with chylomicronemia. Unrelated normolipidemic subjects (n = 111) were recruited as experimental controls. All subjects provided informed consent, and all studies were performed according to the Declaration of Helsinki for human studies.
Genomic DNA Analyses
Genomic DNA was isolated from whole blood. Each exon of GPIHBP1 and the exon-intron junctions was amplified from genomic DNA for sequencing. The primers used are shown in Table 1. A c.320C>G; p.S107C mutation was detected in a single patient and confirmed by additional DNA sequencing reactions. The functional significance of the variant was predicted in silico with the PolyPhen-2 and SNPs3D programs. Apolipoprotein E genotypes were determined by PCR and DNA sequencing.
TABLE 1.
PCR primers for amplifying the exons of GPIHBP1
| GPIHBP1 exon | Forward primer 5′ → 3′ | Reverse primer 5′ → 3′ |
|---|---|---|
| 1 | ACAATGACTTCTCCTTCCCCTCT | CTCATCCCCTCCTTCTTCCTAAG |
| 2 | TGAGTAGGGTGAGTAGGGTGTTC | TACTCTGGAAGGCAACTGAGTGT |
| 3 | CACAGCTTACAGGACCAAGTCA | AGTGGGGAACAAGTGCTGAG |
| 4 | TGCAGAGCCACCTCAGAGAC | CCAAGACACTCCAAATCCATTCT |
Biochemical Measurements
Total plasma cholesterol, triglycerides, and high density lipoprotein cholesterol (HDL-C) levels were measured with enzymatic methods (Roche Diagnostics). LPL mass levels in the preheparin plasma and in the plasma after an intravenous injection of heparin (50 IU/kg) were measured with a sandwich ELISA using monoclonal antibodies 5F9 and 5D2 (20).
GPIHBP1 Constructs
Mammalian expression vectors for untagged soluble mouse GPIHBP1 and S-protein-tagged human GPIHBP1 have been described previously (21–23). An expression vector for GPIHBP1-S107C was generated by site-directed mutagenesis with the QuikChange Lightning kit (Stratagene).
To express soluble versions of GPIHBP1 (i.e. GPIHBP1 lacking the GPI anchor), we used a Drosophila S2 cell expression system using the carboxyl-terminal Ly6 domain (domain III) of human uPAR as a tag (24, 25). DNA sequences encoding uPAR domain III, followed by sequences encoding human GPIHBP1 amino acids 21–136 and mouse GPIHBP1 amino acids 136–198 (which contain the epitope for monoclonal antibody 11A12) were ligated into pMT/V5-His (Invitrogen) with the In-Fusion HD cloning kit (Clontech). This vector contains a metallothionein promoter that allows metal-inducible expression of the protein. Mutant versions of this GPIHBP1 expression vector were generated with the QuikChange Lightning kit.
Cell Surface Expression Assay
To express GPIHBP1 in Chinese hamster ovary cells (CHO-K1 cells; American Type Culture Collection), we electroporated 5 × 106 cells with expression vectors (5 μg) encoding S-protein-tagged versions of GPIHBP1. After 24 h, we assessed the ability of GPIHBP1 to reach the cell surface. The GPIHBP1-transfected cells were first incubated with a rabbit polyclonal antibody against the S-protein tag (21). After the cells were washed, the amount of GPIHBP1 on the cell surface was assessed by performing Western blotting of cell extracts with an IRdye800-conjugated donkey anti-rabbit IgG (1:800; Li-Cor). The total amount of GPIHBP1 in cells was assessed by Western blotting with a goat polyclonal antibody against the S-protein tag (followed by an IRdye680-conjugated donkey anti-goat IgG; 1:5,000).
Releasing GPIHBP1 from the Surface of Cells with Phosphatidylinositol-specific Phospholipase C (PIPLC)
To determine whether GPIHBP1 on the cell surface was monomeric or was in disulfide-linked multimers, we released GPIHBP1 from the surface of cells with PIPLC (16 units/ml for 20 min at 37 °C). GPIHBP1 levels in the PIPLC-released material and in cell extracts were assessed by Western blotting with a goat polyclonal antibody against the S-protein tag (Abcam). In these studies, the PIPLC-released proteins were analyzed by SDS-PAGE under both nonreducing and reducing conditions (50 mm dithiothreitol).
Cell-based LPL-GPIHBP1 Binding Assay
CHO-K1 cells were electroporated with GPIHBP1 expression vectors (5 μg). 24 h after the electroporation, the GPIHBP1-expressing cells were incubated for 2 h at 4 °C with V5-tagged human LPL (26) in the absence or presence of heparin (250 units/ml) (21). At the end of the incubation, cells were washed six times with ice-cold PBS containing 1 mm MgCl2 and 1 mm CaCl2. Relative levels of GPIHBP1 and LPL in cell extracts were assessed by Western blotting with a goat polyclonal antibody against the S-protein tag (Abcam) and a mouse monoclonal against the V5 tag (Invitrogen).
Studies with Drosophila S2 Cells
Soluble versions of GPIHBP1 were expressed in Drosophila S2 cells (Invitrogen) as fusion proteins (described earlier). The cells were plated on 6-well plates (12 × 106 cells/well) and transfected with 19 μg of plasmid DNA with the Calcium Phosphate Transfection kit (Invitrogen). 24 h after the transfection, protein expression was induced with Schneider's medium (Sigma) containing 1% heat-inactivated fetal bovine serum (Invitrogen), 0.1% Pluronic F-68 (Sigma), and 500 μm CuSO4. For all experiments, the cells were grown in suspension culture for 3 days. To assess the electrophoretic migration of GPIHBP1, the conditioned medium and cell extracts were subjected to electrophoresis under reducing and nonreducing conditions. The GPIHBP1 protein was then detected with IRdye680-conjugated monoclonal antibody 11A12 (which recognizes mouse GPIHBP1 sequences) and IRdye800-conjugated monoclonal antibody R24 (which recognizes uPAR domain III) (27).
Cell-free LPL-GPIHBP1 Binding Assay
Soluble GPIHBP1 from CHO-K1 cells or Drosophila S2 cells was incubated for 1 h at 4 °C with V5-tagged human LPL (26) and agarose beads coated with monoclonal antibody 11A12 (23). After washing the beads, soluble GPIHBP1 and GPIHBP1-bound LPL were eluted from the antibody-coated beads with 0.1 m glycine, pH 2.7. The amounts of GPIHBP1 and LPL in the starting material, unbound fractions, wash fractions, and elution fractions were assessed by Western blotting with IRdye680-conjugated antibody 11A12 and an IRdye800-conjugated V5-antibody. To determine whether LPL binds preferentially to GPIHBP1 monomers, the same assay was used except that the agarose beads were coated with the LPL-specific monoclonal antibody 5D2. In those experiments, the LPL and any GPIHBP1-bound LPL captured by the antibody-coated beads were released by boiling in sample loading buffer. The different fractions were then separated by SDS-PAGE under reducing and nonreducing conditions, and Western blotting was performed with LPL- and GPIHBP1-specific antibodies.
Western Blotting
All samples were denatured in 1% lithium dodecyl sulfate for 10 min at 70 °C. Proteins were separated on 12% Bis-Tris SDS-polyacrylamide gels (Invitrogen) under reducing or nonreducing conditions and transferred to nitrocellulose membrane for Western blotting. The antibody dilutions were: 1:1,000 for a goat polyclonal against the S-protein tag (Abcam), 1:200 for a mouse monoclonal against the V5 tag (Invitrogen), 1:500 for a rabbit polyclonal against β-actin (Abcam), 1:1,000 for IRdye680-conjugated rat monoclonal antibody 11A12 (23), 1:1,000 for IRdye800-conjugated mouse monoclonal antibody R24 (24), 1:500 for an IRdye800-conjugated mouse monoclonal against the V5 tag, 1:5,000 for an IRdye680-conjugated donkey anti-goat IgG, 1:5,000 for an IRdye800-conjugated donkey anti-goat IgG, 1:2,000 for an IRdye680-conjugated donkey anti-rabbit IgG, and 1:2,000 for an IRdye800-conjugated donkey anti-mouse IgG (Li-Cor).
RESULTS
Identification of a GPIHBP1 Missense Mutation
Ninety-two patients with severe hypertriglyceridemia but lacking mutations in LPL, APOC2, or APOA5 were screened for GPIHBP1 mutations. A C-to-G transversion in exon 4 of GPIHBP1 (c.320C>G; p.S107C) was identified in a 46-year-old female with chylomicronemia. Her body mass index was normal. Her fasting plasma triglyceride level was 3,164 mg/dl; the fasting glucose and thyroid-stimulating hormone levels were normal.
The proband was first noted to have chylomicronemia at age 40 after presenting with epigastric discomfort and a plasma triglyceride level of 2,050 mg/dl. Subsequently, her fasting plasma triglyceride levels ranged between 1,247 and 6,448 mg/dl, although one value was as low as 505 mg/dl (when she adhered to a low fat diet and gemfibrozil treatment). The epigastric discomfort recurred episodically, but a diagnosis of pancreatitis was never established. She did not have eruptive xanthomas. She reported two uneventful pregnancies.
The family pedigree is shown in Fig. 1, and the plasma lipid levels for all available family members are recorded in Table 2. Two of the proband's brothers had a history of chylomicronemia but had lower plasma triglyceride levels than the proband. Both were homozygous for the p.S107C mutation (Fig. 1). Six family members were heterozygous for the p.S107C mutation, and all but subject II-2 had normal plasma triglyceride levels (Table 2). No GPIHBP1 mutations were encountered in 111 normolipemic control subjects.
FIGURE 1.

Family pedigree. The arrow indicates the proband. The p.S107C mutation is indicated by a half-filled circle or square. The slash mark denotes deceased; the question mark (?) denotes individuals who could not be tested. Insets show DNA sequencing chromatograms for a control subject (right) and the proband (left).
TABLE 2.
Clinical and molecular characteristics of the proband and her family
TG, triglyceride; TC, total cholesterol; HDL-C, high density lipoprotein cholesterol.
| Subject | Age | Sex | APOE genotype | GPIHBP1 genotype | TG | TC | HDL-C | Pre-heparin LPL mass | Fibrate therapy |
|---|---|---|---|---|---|---|---|---|---|
| Years | mg/dl | mg/dl | mg/dl | ng/ml | |||||
| I-2 | 86 | F | E3/E3 | wt/p.S107C | 108 | 206 | 43 | 368 | No |
| II-1 | 64 | M | E3/E3 | p.S107C/p.S107C | 842 | 158 | 16 | 32 | Yes |
| II-2 | 62 | F | E3/E3 | wt/p.S107C | 338 | 265 | 47 | 615 | No |
| II-6 | 52 | F | E3/E3 | wt/p.S107C | 152 | 248 | 46 | 535 | No |
| II-7 | 50 | F | E3/E3 | wt/p.S107C | 127 | 288 | 69 | 283 | No |
| II-8 | 46 | F | E3/E4 | p.S107C/p.S107C | 3,164 | 385 | 32 | 23 | Yes |
| II-9 | 49 | M | E3/E3 | wt/wt | 97 | 234 | 43 | 184 | No |
| II-10 | 43 | M | E3/E4 | p.S107C/p.S107C | 673 | 107 | 13 | 23 | No |
| III-1 | 24 | F | E3/E4 | wt/p.S107C | 87 | 188 | 44 | 295 | No |
| III-2 | 19 | F | E3/E3 | wt/p.S107C | 40 | 148 | 50 | 336 | No |
The preheparin plasma LPL levels in the three homozygotes were much lower than in the other family members (Table 2). The postheparin LPL plasma levels in the proband were extremely low (127 ng/ml), <5% of those in normolipidemic control subjects (3,240 ± 321 ng/ml, n = 9).
Testing the Effect of the S107C Mutation on the Trafficking of GPIHBP1 to the Cell Surface
Ser-107 in the GPIHBP1 Ly6 domain is conserved from the egg-laying platypus to humans, and both PolyPhen-2 and SNP3D predicted that the S107C substitution would be deleterious to protein function. Because the S107C mutation introduces a new cysteine and a free thiol group into the GPIHBP1 Ly6 domain, we suspected that the mutant protein might be misfolded and manifest impaired trafficking to the cell surface. To test this idea, we expressed wild-type GPIHBP1 and GPIHBP1-S107C in CHO cells and quantified the amount of GPIHBP1 on the cell surface. As an experimental control, we tested GPIHBP1-N78Q/N82Q, where N-linked glycosylation is absent and trafficking to the cell surface is known to be impaired (21). We also tested GPIHBP1-S107A. The S107C and S107A mutations had little effect on the amount of GPIHBP1 that reached the cell surface (95.9 ± 0.6 and 87.7 ± 2.1% of wild-type GPIHBP1, respectively) (Fig. 2). As we expected, reduced amounts of GPIHBP1-N78Q/N82Q reached the cell surface (39.7 ± 0.1% of wild-type GPIHBP1).
FIGURE 2.

Assessing the cell surface expression of GPIHBP1-S107C. A, Western blot to assess the amount of GPIHBP1 on the surface of CHO-K1 cells. Cells were electroporated with S-protein-tagged wild-type or mutant GPIHBP1 constructs. 24 h later, the cells were washed and incubated for 2 h at 4 °C with a rabbit polyclonal antibody against the S-protein tag (10 μg/ml). After washing the cells, Western blotting was performed on cell lysates with an antibody against rabbit IgG (green; to assess the amount of the rabbit anti-S-protein antibody bound to GPIHBP1 on the surface of cells) and a goat polyclonal against the S-protein tag (red; to assess total levels of GPIHBP1 expression in cells). B, bar graph of GPIHBP1 expression on the cell surface (mean ± S.E. (error bars) of four replicate measurements). The amount of GPIHBP1 on the cell surface was normalized to total GPIHBP1 expression in cells and plotted as a percentage of the ratio observed with wild-type GPIHBP1 (set at 100%). Statistical analysis was by one-way analysis of variance; ***, p < 0.05.
Testing Whether GPIHBP1-S107C Forms Multimers
We speculated that the extra, unpaired cysteine in the Ly6 domain of GPIHBP1-S107C might lead to the formation of disulfide bonds between two GPIHBP1 proteins, resulting in dimer formation. To test this idea, we transfected CHO cells with wild-type GPIHBP1, GPIHBP1-S107C, or GPIHBP1-S107A and then released GPIHBP1 from the surface of the cells with PIPLC. The PIPLC-released proteins were then analyzed by SDS-PAGE and Western blotting under both reducing and nonreducing conditions. In the setting of reducing conditions, Western blots revealed similar amounts of wild-type GPIHBP1, GPIHBP1-S107C, and GPIHBP1-S107A; all proteins migrated at ∼28 kDa (Fig. 3A, middle panel). However, in nonreduced samples, there were obvious differences. Virtually all of the GPIHBP1-S107C was in the form of dimers and multimers, and 28-kDa monomers were undetectable. With wild-type GPIHBP1, there were substantial amounts of monomers, and with GPIHBP1-S107A, there were intermediate levels of monomers (Fig. 3A, top panel).
FIGURE 3.

Assessing the electrophoretic migration of GPIHBP1-S107C under reducing and nonreducing conditions. A, electrophoretic migration of the GPIHBP1 released by PIPLC from the surface of GPIHBP1-transfected cells. CHO-K1 cells were electroporated with S-protein-tagged wild-type or mutant GPIHBP1 constructs. 24 h later, the cells were washed and incubated for 20 min at 37 °C with PIPLC (16 units/ml) to release GPI-anchored proteins. Western blotting was performed on cell lysates under reducing conditions (R) and on the PIPLC-released proteins under nonreducing (NR) and reducing conditions. All samples were denatured in 1% lithium dodecyl sulfate for 10 min at 70 °C. Actin (red) was used as a loading control. The location of GPIHBP1 monomers (∼28 kDa) is indicated with an arrowhead. B, electrophoretic migration of soluble human GPIHBP1 secreted by Drosophila S2 cells. Soluble GPIHBP1 proteins containing an amino-terminal uPAR tag (detectable with antibody R24) and a carboxyl-terminal mouse GPIHBP1 motif (detectable with antibody 11A12) were expressed in Drosophila S2 cells. Antibody R24 binds preferentially to properly folded monomers, whereas antibody 11A12 binds to both monomers and multimers. All samples were denatured in 1% lithium dodecyl sulfate for 10 min at 70 °C. Proteins were separated under nonreducing conditions (NR; conditioned media, top three panels) and reducing conditions (R; conditioned media and cell lysates, bottom two panels). GPIHBP1 proteins were detected with IRdye680-conjugated antibody 11A12 (red) and an IRdye800-conjugated antibody R24 (green). The GPIHBP1 monomer (∼38 kDa) is indicated with an arrowhead. C, quantification of the ratio of monomeric to total GPIHBP1 expressed as the percentage of the ratio observed with wild-type GPIHBP1 (set at 100%). The intensity of the GPIHBP1 monomers and the entire lane (total GPIHBP1) was quantified with a Li-Cor infrared scanner. Shown are mean ratios ± S.E. (error bars) of three independent experiments. Statistical analysis was by two-way analysis of variance (mutant versus wild-type); *, p < 0.001; **, p < 0.0001.
To test whether the formation of GPIHBP1-S107C multimers was a unique feature of the CHO expression system, we expressed soluble versions of human GPIHBP1 proteins in Drosophila S2 cells, a cell line that is commonly used to express proteins of the Ly6 family (24, 25, 28). The soluble GPIHBP1 contained an amino-terminal uPAR tag (detectable with antibody R24) and a carboxyl-terminal tag from mouse GPIHBP1 (detectable with antibody 11A12). High levels of wild-type GPIHBP1, GPIHBP1-S107C, and GPIHBP1-S107A were secreted from the Drosophila cells, as judged by Western blots of media samples run under reducing conditions (Fig. 3B, bottom two panels). When we examined samples that had been run under nonreducing conditions, most of the wild-type GPIHBP1 was monomeric although some was in the form of dimers and multimers (as judged by the antibody 11A12 Western blot) (Fig. 3B, top three panels). In contrast, nearly all of the GPIHBP1-S107C was in the form of dimers and multimers. GPIHBP1-S107A exhibited intermediate amounts of monomers (Fig. 3B, top three panels). The S107C mutation reduced the amount of monomers in the media by 85–95%, and the S107A mutation by 45–60%, as judged by quantitative analysis of the Western blots with a Li-Cor scanner (Fig. 3C). In the case of the antibody R24 Western blot, we detected almost exclusively monomeric protein, simply because antibody R24 has a strong preference for the properly folded uPAR Ly6 domain III. R24 does not bind to uPAR after it has been subjected to reducing agents.
To determine whether multimerization occurs inside the cell or only after reaching the cell surface, we examined the migration pattern under nonreducing conditions of cell lysates prepared from CHO-K1 cells that had been transfected with wild-type GPIHBP1 or GPIHBP1-S107C and then treated with PIPLC. Multimers could be detected in the cell lysates both with wild-type GPIHBP1 and GPIHBP1-S107C (Fig. 4), suggesting that multimerization begins before GPIHBP1 reaches the cell surface. Interestingly, 16% of intracellular GPIHBP1-S107C was monomeric (Fig. 4), whereas monomers represented only 1% of the PIPLC-released material (Fig. 3A), implying that multimerization continues at the cell surface.
FIGURE 4.
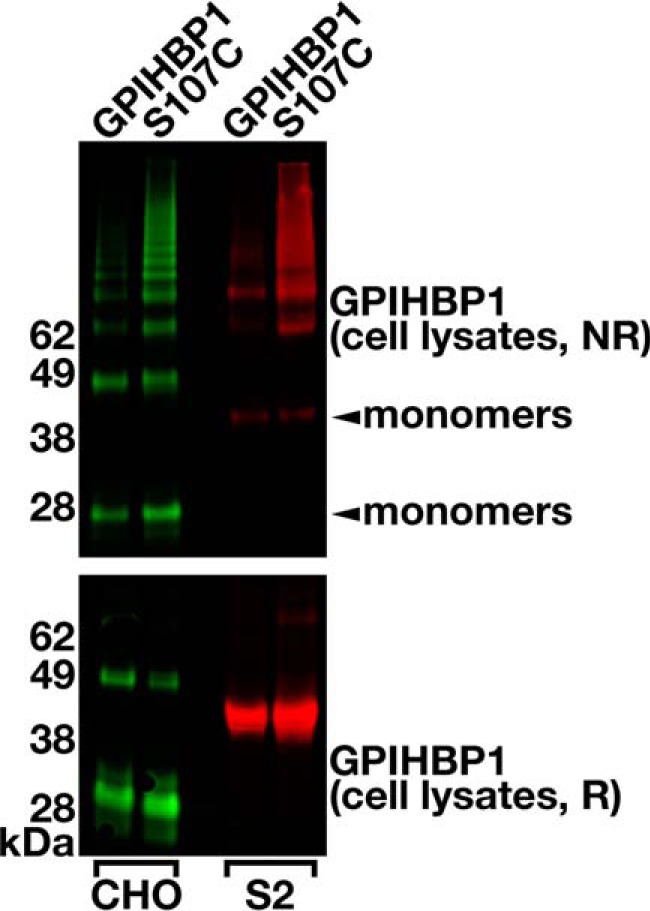
Assessing the electrophoretic migration of intracellular GPIHBP1-S107C under reducing and nonreducing conditions. CHO-K1 cells were electroporated with S-protein-tagged wild-type or GPIHBP1-S107C constructs. 24 h later, the cells were washed and incubated for 20 min at 37 °C with PIPLC (16 units/ml) to release GPI-anchored proteins. Soluble forms of GPIHBP1 proteins containing a carboxyl-terminal mouse GPIHBP1 motif (detectable with antibody 11A12) were expressed in Drosophila S2 cells. Western blotting was performed on cell lysates under nonreducing (NR, top panel) and reducing (R, bottom panel) conditions. All samples were denatured in 1% lithium dodecyl sulfate for 10 min at 70 °C. GPIHBP1 proteins were detected with a goat polyclonal antibody against the S-protein tag (CHO cells; green) or an IRdye680-conjugated antibody 11A12 (S2 cells; red). The location of GPIHBP1 monomers is indicated with an arrowhead (∼28 kDa for CHO-K1 cells; ∼38 kDa for Drosophila S2 cells).
Multimers were also detected in cells lysates of Drosophila S2 cells that had been transfected with wild-type GPIHBP1 and GPIHBP1-S107C (Fig. 4). For wild-type GPIHBP1, monomers represented 12% of intracellular GPIHBP1 (Fig. 4) whereas up to 50% of the secreted protein was monomeric (Fig. 3, B and C). This suggests that some misfolded proteins are targeted for degradation in Drosophila cells. For GPIHBP1-S107C, monomers only represented 4.4% of the intracellular protein (Fig. 4) and 3.5% of the secreted mutant protein (Fig. 3, B and C), suggesting that large amounts of misfolded proteins could overwhelm the quality control systems of the cell.
Testing the LPL-binding Properties of the Wild-type GPIHBP1 Produced by Insect Cells
Because the uPAR domain III-GPIHBP1 fusion protein contains two sequential Ly6 domains, we first determined whether the soluble wild-type GPIHBP1 secreted by Drosophila cells is capable of binding LPL. We mixed the medium from the GPIHBP1-expressing Drosophila S2 cells with the medium of CHO cells that expressed a V5-tagged human LPL and agarose beads coated with the LPL-specific monoclonal antibody 5D2 (20). After 1 h, the beads were washed, and the LPL (including any LPL-GPIHBP1 complexes) were eluted from the antibody-coated beads by heating in SDS-PAGE sample buffer. The starting material, unbound fractions, wash samples, and elution samples were then examined by SDS-PAGE under reducing conditions. Western blots revealed both the human GPIHBP1 protein and LPL in the elution material, indicating that the immobilized LPL captured the soluble uPAR-GPIHBP1 fusion protein (Fig. 5A, top panels) as efficiently as the soluble untagged GPIHBP1 produced in CHO-K1 cells (Fig. 5B, top panels).
FIGURE 5.

Testing whether GPIHBP1 dimers and multimers are capable of binding LPL. The conditioned medium of GPIHBP1-transfected CHO-K1 or S2 cells was incubated with V5-tagged human LPL and antibody 5D2-coated agarose beads for 1 h at 4 °C. After washing the beads, the LPL along with any LPL-bound GPIHBP1 was released from the beads by heating in sample loading buffer. Western blotting under reducing conditions (top panels) or nonreducing conditions (bottom panels) was performed on the starting material, the unbound fractions, the wash fractions, and elution fractions with an IRdye800-conjugated anti-V5 antibody (top panels, green), and an IRdye680-conjugated antibody 11A12 (top and bottom panels, red). A, 5D2 bead assay performed with conditioned medium from GPIHBP1-transfected Drosophila S2 cells. The intensity of the GPIHBP1 monomers and the entire lane (total GPIHBP1) was quantified in each fraction with a Li-Cor infrared scanner. The bar graph represents the ratio of monomeric to total GPIHBP1 in each fraction. B, 5D2 bead assay performed with conditioned medium from GPIHBP1-transfected CHO-K1 cells. The bar graph represents the ratio of monomeric to total GPIHBP1 detected in each fraction with a Li-Cor infrared scanner. Of note, monomeric GPIHBP1 eluted from the 5D2-coated beads even in the absence of added V5-tagged human LPL. This is likely due to the production of hamster LPL by CHO-K1 cells (37). Inset, Western blot showing the presence of endogenous hamster LPL (haLPL) in the conditioned medium from CHO-K1 cells (CHO) and CHO cells that had been stably transfected with V5-tagged human LPL (CHO-huLPL) (26). The haLPL was detected with a goat anti mouse LPL antibody followed by an IRdye680-conjugated donkey anti-goat IgG (red, panel A). V5-tagged human LPL (huLPL-V5) was detected with antibody 5D2 followed by an IRdye800-conjugated donkey anti-mouse IgG (green, panel A) or with an IRdye800-conjugated anti-V5 antibody (green, panel B). Antibody 5D2 cross-reacted with haLPL (panel A).
We hypothesized that only monomeric GPIHBP1 would bind LPL. To explore this idea, we ran the same samples under nonreducing conditions and performed Western blotting with monoclonal antibody 11A12. For the GPIHBP1 produced in Drosophila cells, substantial amounts of dimers and multimers were present in the starting material, unbound fractions, and wash fractions. Only 26% of GPIHBP1 was monomeric in the starting material, whereas 91% of the GPIHBP1 that co-eluted with LPL was monomeric (Fig. 5A, bar graph), demonstrating that LPL binds almost exclusively to monomeric GPIHBP1 (Fig. 5A, bottom panels). Similar results were obtained with untagged wild-type mouse GPIHBP1 secreted by CHO-K1 cells (Fig. 5B). Only 11% of GPIHBP1 was monomeric in the starting material, whereas 66% of the GPIHBP1 that co-eluted with LPL was monomeric (Fig. 5B).
Assessing the Ability of GPIHBP1-S107C to Bind LPL
Because multimers appeared to have little capacity to bind LPL and because GPIHBP1-S107C exists almost exclusively in the form of dimers and multimers, we suspected that GPIHBP1-S107C would not bind to LPL. To test this prediction, we used both cell-based and cell-free LPL-GPIHBP1 binding assays. In the cell-based assay, we incubated GPIHBP1-transfected CHO cells with V5-tagged human LPL in the presence or absence of heparin (which inhibits LPL binding to GPIHBP1) (29). After washing the cells, the binding of LPL to the GPIHBP1-expressing cells was assessed by performing Western blotting on cell extracts. Cells expressing wild-type GPIHBP1 bound LPL avidly, and the amount of binding was reduced by heparin (Fig. 6A). The amount of LPL binding to cells expressing GPIHBP1-S107C and GPIHBP1-C68G was negligible (0.2 and 1% of wild-type control, respectively) (Fig. 6A). The inability of GPIHBP1-C68G to bind to LPL is consistent with an earlier study by Olivecrona et al. (16). GPIHBP1-S107A, which displayed an intermediate ability to form monomers (Fig. 3), retained some capacity to bind LPL (14% of wild-type GPIHBP1) (Fig. 6A).
FIGURE 6.

Assessing the ability of GPIHBP1-S107C to bind LPL. A, testing the binding of LPL to GPIHBP1 on the surface of mammalian cells. CHO-K1 cells were electroporated with empty vector or S-protein-tagged versions of wild-type or mutant GPIHBP1 constructs. 24 h later, the cells were incubated for 2 h at 4 °C with V5-tagged human LPL in the absence or presence of heparin (250 units/ml). After washing the cells, LPL binding to cells was assessed by performing Western blotting of cell lysates with an anti-V5 antibody (green). In a separate Western blot, GPIHBP1 expression was assessed with an S-protein-specific antibody (green). Actin (red) was used as a loading control. B, a cell-free assay to test the ability of LPL to bind to soluble GPIHBP1. Drosophila S2 cells were transfected in triplicate wells with secreted versions of wild-type or mutant GPIHBP1 proteins containing an amino-terminal uPAR tag and carboxyl-terminal mouse GPIHBP1 sequences (detected by antibody 11A12). At the end of the 3-day induction, GPIHBP1-transfected cells were pooled and pelleted, and the conditioned medium was concentrated 6-fold with Amicon Ultra 10 MWCO filters. The conditioned medium from wild-type and mutant GPIHBP1-transfected cells was then incubated with V5-tagged human LPL and 11A12-coated beads for 1 h at 4 °C. After washing the beads, GPIHBP1 and any GPIHBP1-bound LPL were released from the beads with 0.1 m glycine, pH 2.7. Western blotting was performed on the starting material, unbound fractions, wash fractions, and elution fractions with an IRdye800-conjugated anti-V5 antibody (green) and an IRdye680-conjugated 11A12 antibody (red).
In the cell-free assay, we mixed the soluble GPIHBP1 from Drosophila S2 cells with V5-tagged human LPL and antibody 11A12-coated agarose beads. After washing the beads, GPIHBP1 and any GPIHBP1-bound LPL were eluted with 0.1 m glycine, pH 2.7. The amounts of GPIHBP1 and LPL present in the eluate were assessed with Western blotting. With wild-type GPIHBP1, both GPIHBP1 and LPL were present in the eluate, indicating that the immobilized GPIHBP1 binds LPL. With GPIHBP1-S107C and GPIHBP1-C65Y, LPL binding was only 0.3 and 2%, respectively, of that with wild-type GPIHBP1, indicating that these mutant proteins were incapable of binding LPL (Fig. 6B).
DISCUSSION
GPIHBP1 is responsible for picking up LPL within the interstitial spaces and shuttling it across endothelial cells to the capillary lumen (1). Earlier studies showed that missense mutations in the GPIHBP1 Ly6 domain interfere with LPL binding and transport and cause severe hypertriglyceridemia (12, 13, 15, 16, 21, 23). However, how these mutations alter GPIHBP1 structure and impair LPL binding has been unclear. Our lack of understanding of GPIHBP1 missense mutations stands in contrast to the situation with the LDL receptor, where multiple missense mutations have been classified into distinct categories based on molecular mechanisms (e.g. defective trafficking of the receptor to the cell surface, defective ligand binding, inability to localize to clathrin-coated pits) (30). In the present study, we identified a new GPIHBP1 missense mutation, S107C, in a family with hypertriglyceridemia and uncovered the mechanism of disease. This mutation, which introduces an extra cysteine into the Ly6 domain, resulted in multimerization of GPIHBP1. The propensity of GPIHBP1-S107C to form dimers and multimers was documented with the GPI-anchored form of the protein in mammalian cells and with a secreted version of the protein in Drosophila S2 cells. We went on to show, using cell-based and cell-free binding assays, that the GPIHBP1-S107C lacked the ability to bind LPL. The failure of the GPIHBP1-S107C to bind LPL made perfect sense because we showed, using an immunoprecipitation assay, that only GPIHBP1 monomers are capable of binding LPL. The most parsimonious explanation is that the formation of intermolecular disulfide bounds destabilizes GPIHBP1, results in the misfolding of the Ly6 domain and aggregation, disrupting the GPIHBP1-LPL binding site and/or sterically blocking the access of LPL to its binding site. Additional studies will be needed to determine whether the same mechanism applies to some or all of the previously identified GPIHBP1 mutations associated with chylomicronemia (11–18).
The S107C substitution had little effect on the trafficking of GPIHBP1 to the surface of CHO cells or on the secretion of soluble forms of GPIHBP1 in Drosophila S2 cells. This result is consistent with earlier studies showing that the substitution of conserved cysteines in the GPIHBP1 Ly6 domain with alanines had little effect on the trafficking of GPIHBP1 to the cell surface (23). The absence of significant effects of cysteine mutations on GPIHBP1 trafficking in mammalian cells (or on the secretion of soluble GPIHBP1 from insect cells) was somewhat surprising and may represent a property specific to GPIHBP1. In our own studies, we found that a cysteine mutation in SLURP1 (a secreted Ly6 protein) virtually eliminates the secretion of SLURP1 from CHO-K1 cells.4 Also, a cysteine mutation in CD59 (a GPI-anchored Ly6 protein) markedly reduces the amount of CD59 on the surface of blood mononuclear cells (31). Moreover, cysteine mutants in the ligand-binding domain of the LDL receptor do not traffic to the cell surface (30). On the other hand, cysteine mutations in the extracellular domain of Notch3 can cause vascular dementia without affecting the trafficking of Notch3 to the cell surface (32). Why GPIHBP1 cysteine mutations have little or no impact on protein trafficking is unclear but it is tempting to speculate that protein dimerization and multimerization might allow GPIHBP1 to escape the surveillance mechanisms that would ordinarily target misfolded proteins for degradation.
It was surprising that some disulfide-linked dimers and multimers also formed with wild-type GPIHBP1. This was the case in both the CHO cell and insect cell expression systems. The in vivo relevance of this observation is unclear. It is possible that multimerization of wild-type GPIHBP1 is a peculiarity of CHO cells or Drosophila S2 cells or that it is a consequence of protein overexpression. However, there is evidence that multimers of Ly6 proteins could occur normally. Fletcher et al. (33) released CD59 (a GPI-anchored Ly6 protein) from the surface of blood cells with PIPLC, separated the proteins by SDS-PAGE under nonreducing conditions, and then performed Western blotting with a CD59-specific monoclonal antibody that was capable of binding to improperly folded versions of CD59. In their Western blot, CD59 monomers were the predominant species in the PIPLC-released material, but there were also significant amounts of dimers and higher order multimers (33).
Our study was also informative from the clinical perspective. First, our studies indicate that GPIHBP1 mutations are uncommon in Thailand, even in a cohort of hypertriglyceridemic patients where mutations in LPL, APOC2, and APOA5 had been excluded. The low frequency of GPIHBP1 mutations in the Thai population is consistent with findings from similar patient populations in North America and Europe (11–15, 34). Second, our study revealed that GPIHBP1 mutations are associated with very low levels of LPL in the preheparin plasma. There had been suggestions that this might be the case (15, 16), but the data in the current study solidify this finding and also provide a good reason to believe that the levels of LPL in the preheparin plasma reflect levels of LPL along capillary lumen. Third, the proband had very low levels of LPL mass in the postheparin plasma, suggesting that most of the LPL released into the plasma in humans is from intravascular stores of LPL. In Gpihbp1 knock-out mice, the postheparin LPL levels increase to levels approaching those in wild-type mice, but the dose of heparin administered to mice (on a mg/kg basis) is much higher than the dose given to human subjects (35).
An intriguing finding in the present studies was that two homozygotes identified through the family investigation had only moderately increased plasma triglyceride levels. In contrast, the proband had severe hypertriglyceridemia. Like the proband in our family, nearly all of the previously described probands with GPIHBP1 mutations had severe hypertriglyceridemia and associated phenotypes (e.g. pancreatitis, eruptive xanthomas) (11–15). Together, these reports have created the impression that GPIHBP1 deficiency typically results in severe hypertriglyceridemia along with all of the expected comorbidities. The fact that two of the homozygotes in this study had only moderately elevated plasma triglyceride levels raises the possibility that the view that GPIHBP1 mutations invariably cause severe hypertriglyceridemia could be the result of ascertainment bias and that broader screening efforts might eventually identify GPIHBP1 deficiency as a cause of mild to moderate hypertriglyceridemia. Ascertainment bias has been well documented in the case of LDL receptor mutations and familial hypercholesterolemia. The plasma cholesterol levels in familial hypercholesterolemia heterozygotes identified by the presence of ischemic heart disease and tendon xanthomas are higher than those in familial hypercholesterolemia heterozygotes identified through family or population studies (36).
This work was supported, in whole or in part, by National Institutes of Health Grants HL094732, HL090553, and HL087228. This work was also supported by the Asahi Glass Foundation, the Thailand Government Research Budget (Years 2556 and 2557), the Ratchadapiseksompotch Research Fund, Chulalongkorn University grants, and by Leducq Transatlantic Network Grant 12CVD04.
A. P. Beigneux, unpublished observations.
- GPIHBP1
- glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1
- Bis-Tris
- bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane
- LPL
- lipoprotein lipase
- Ly6
- lymphocyte antigen 6
- PIPLC
- phosphatidylinositol-specific phospholipase C
- uPAR
- urokinase-type plasminogen activator receptor.
REFERENCES
- 1. Davies B. S., Beigneux A. P., Barnes R. H., 2nd, Tu Y., Gin P., Weinstein M. M., Nobumori C., Nyrén R., Goldberg I., Olivecrona G., Bensadoun A., Young S. G., Fong L. G. (2010) GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 12, 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinstein M. M., Goulbourne C. N., Davies B. S., Tu Y., Barnes R. H., 2nd, Watkins S. M., Davis R., Reue K., Tontonoz P., Beigneux A. P., Fong L. G., Young S. G. (2012) Reciprocal metabolic perturbations in the adipose tissue and liver of GPIHBP1-deficient mice. Arterioscler. Thromb. Vasc. Biol. 32, 230–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ginzinger D. G., Clee S. M., Dallongeville J., Lewis M. E., Henderson H. E., Bauje E., Rogers Q. R., Jensen D. R., Eckel R. H., Dyer R., Innis S., Jones B., Fruchart J. C., Hayden M. R. (1999) Lipid and lipoprotein analysis of cats with lipoprotein lipase deficiency. Eur. J. Clin. Invest. 29, 17–26 [DOI] [PubMed] [Google Scholar]
- 4. Ploug M., Gårdsvoll H., Jørgensen T. J., Lønborg Hansen L., Danø K. (2002) Structural analysis of the interaction between urokinase-type plasminogen activator and its receptor: a potential target for anti-invasive cancer therapy. Biochem. Soc. Trans. 30, 177–183 [DOI] [PubMed] [Google Scholar]
- 5. Mallya M., Campbell R. D., Aguado B. (2006) Characterization of the five novel Ly-6 superfamily members encoded in the MHC, and detection of cells expressing their potential ligands. Protein Sci. 15, 2244–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fry B. G., Wüster W., Kini R. M., Brusic V., Khan A., Venkataraman D., Rooney A. P. (2003) Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 57, 110–129 [DOI] [PubMed] [Google Scholar]
- 7. Galat A. (2008) The three-fingered protein domain of the human genome. Cell. Mol. Life Sci. 65, 3481–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gårdsvoll H., Gilquin B., Le Du M. H., Ménèz A., Jørgensen T. J., Ploug M. (2006) Characterization of the functional epitope on the urokinase receptor. J. Biol. Chem. 281, 19260–19272 [DOI] [PubMed] [Google Scholar]
- 9. Gårdsvoll H., Ploug M. (2007) Mapping of the vitronectin-binding site on the urokinase receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 282, 13561–13572 [DOI] [PubMed] [Google Scholar]
- 10. Mertens H. D., Kjaergaard M., Mysling S., Gårdsvoll H., Jørgensen T. J., Svergun D. I., Ploug M. (2012) A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 287, 34304–34315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rios J. J., Shastry S., Jasso J., Hauser N., Garg A., Bensadoun A., Cohen J. C., Hobbs H. H. (2012) Deletion of GPIHBP1 causing severe chylomicronemia. J. Inherit. Metab. Dis. 35, 531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beigneux A. P., Franssen R., Bensadoun A., Gin P., Melford K., Peter J., Walzem R. L., Weinstein M. M., Davies B. S., Kuivenhoven J. A., Kastelein J. J., Fong L. G., Dallinga-Thie G. M., Young S. G. (2009) Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol. 29, 956–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Charrière S., Peretti N., Bernard S., Di Filippo M., Sassolas A., Merlin M., Delay M., Debard C., Lefai E., Lachaux A., Moulin P., Marçais C. (2011) GPIHBP1 C89F neomutation and hydrophobic C-Terminal domain G175R mutation in two pedigrees with severe hyperchylomicronemia. J. Clin. Endocrinol. Metab. 96, E1675–E1679 [DOI] [PubMed] [Google Scholar]
- 14. Coca-Prieto I., Kroupa O., Gonzalez-Santos P., Magne J., Olivecrona G., Ehrenborg E., Valdivielso P. (2011) Childhood-onset chylomicronaemia with reduced plasma lipoprotein lipase activity and mass: identification of a novel GPIHBP1 mutation. J. Intern. Med. 270, 224–228 [DOI] [PubMed] [Google Scholar]
- 15. Franssen R., Young S. G., Peelman F., Hertecant J., Sierts J. A., Schimmel A. W., Bensadoun A., Kastelein J. J., Fong L. G., Dallinga-Thie G. M., Beigneux A. P. (2010) Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects. Circ. Cardiovasc. Genet. 3, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olivecrona G., Ehrenborg E., Semb H., Makoveichuk E., Lindberg A., Hayden M. R., Gin P., Davies B. S., Weinstein M. M., Fong L. G., Beigneux A. P., Young S. G., Olivecrona T., Hernell O. (2010) Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J. Lipid Res. 51, 1535–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Surendran R. P., Visser M. E., Heemelaar S., Wang J., Peter J., Defesche J. C., Kuivenhoven J. A., Hosseini M., Péterfy M., Kastelein J. J., Johansen C. T., Hegele R. A., Stroes E. S., Dallinga-Thie G. M. (2012) Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J. Intern. Med. 272, 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamamoto H., Onishi M., Miyamoto N., Oki R., Ueda H., Ishigami M., Hiraoka H., Matsuzawa Y., Kihara S. (2013) Novel combined GPIHBP1 mutations in a patient with hypertriglyceridemia associated with CAD. J. Atheroscler. Thromb. 20, 777–784 [DOI] [PubMed] [Google Scholar]
- 19. Voss C. V., Davies B. S., Tat S., Gin P., Fong L. G., Pelletier C., Mottler C. D., Bensadoun A., Beigneux A. P., Young S. G. (2011) Mutations in lipoprotein lipase that block binding to the endothelial cell transporter GPIHBP1. Proc. Natl. Acad. Sci. U.S.A. 108, 7980–7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peterson J., Fujimoto W. Y., Brunzell J. D. (1992) Human lipoprotein lipase: relationship of activity, heparin affinity, and conformation as studied with monoclonal antibodies. J. Lipid Res. 33, 1165–1170 [PubMed] [Google Scholar]
- 21. Beigneux A. P., Davies B. S., Tat S., Chen J., Gin P., Voss C. V., Weinstein M. M., Bensadoun A., Pullinger C. R., Fong L. G., Young S. G. (2011) Assessing the role of the glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) three-finger domain in binding lipoprotein lipase. J. Biol. Chem. 286, 19735–19743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gin P., Beigneux A. P., Davies B., Young M. F., Ryan R. O., Bensadoun A., Fong L. G., Young S. G. (2007) Normal binding of lipoprotein lipase, chylomicrons, and apo-AV to GPIHBP1 containing a G56R amino acid substitution. Biochim. Biophys. Acta 1771, 1464–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beigneux A. P., Gin P., Davies B. S., Weinstein M. M., Bensadoun A., Fong L. G., Young S. G. (2009) Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J. Biol. Chem. 284, 30240–30247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gårdsvoll H., Hansen L. V., Jørgensen T. J., Ploug M. (2007) A new tagging system for production of recombinant proteins in Drosophila S2 cells using the third domain of the urokinase receptor. Protein Expr. Purif. 52, 384–394 [DOI] [PubMed] [Google Scholar]
- 25. Gårdsvoll H., Kriegbaum M. C., Hertz E. P., Alpízar-Alpízar W., Ploug M. (2013) The urokinase receptor homolog haldisin is a novel differentiation marker of stratum granulosum in squamous epithelia. J. Histochem. Cytochem. 61, 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ben-Zeev O., Mao H. Z., Doolittle M. H. (2002) Maturation of lipoprotein lipase in the endoplasmic reticulum: concurrent formation of functional dimers and inactive aggregates. J. Biol. Chem. 277, 10727–10738 [DOI] [PubMed] [Google Scholar]
- 27. Gårdsvoll H., Jacobsen B., Kriegbaum M. C., Behrendt N., Engelholm L., Østergaard S., Ploug M. (2011) Conformational regulation of urokinase receptor function: impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J. Biol. Chem. 286, 33544–33556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu X., Gårdsvoll H., Yuan C., Lin L., Ploug M., Huang M. (2012) Crystal structure of the urokinase receptor in a ligand-free form. J. Mol. Biol. 416, 629–641 [DOI] [PubMed] [Google Scholar]
- 29. Beigneux A. P., Davies B. S., Gin P., Weinstein M. M., Farber E., Qiao X., Peale F., Bunting S., Walzem R. L., Wong J. S., Blaner W. S., Ding Z. M., Melford K., Wongsiriroj N., Shu X., de Sauvage F., Ryan R. O., Fong L. G., Bensadoun A., Young S. G. (2007) Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 5, 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hobbs H. H., Russell D. W., Brown M. S., Goldstein J. L. (1990) The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu. Rev. Genet. 24, 133–170 [DOI] [PubMed] [Google Scholar]
- 31. Nevo Y., Ben-Zeev B., Tabib A., Straussberg R., Anikster Y., Shorer Z., Fattal-Valevski A., Ta-Shma A., Aharoni S., Rabie M., Zenvirt S., Goldshmidt H., Fellig Y., Shaag A., Mevorach D., Elpeleg O. (2013) CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood 121, 129–135 [DOI] [PubMed] [Google Scholar]
- 32. Haritunians T., Boulter J., Hicks C., Buhrman J., DiSibio G., Shawber C., Weinmaster G., Nofziger D., Schanen C. (2002) CADASIL Notch3 mutant proteins localize to the cell surface and bind ligand. Circ. Res. 90, 506–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fletcher A., Bryant J. A., Gardner B., Judson P. A., Spring F. A., Parsons S. F., Mallinson G., Anstee D. J. (1992) New monoclonal antibodies in CD59: use for the analysis of peripheral blood cells from paroxysmal nocturnal haemoglobinuria (PNH) patients and for the quantitation of CD59 on normal and decay accelerating factor (DAF)-deficient erythrocytes. Immunology 75, 507–512 [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J., Hegele R. A. (2007) Homozygous missense mutation (G56R) in glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPI-HBP1) in two siblings with fasting chylomicronemia (MIM 144650). Lipids Health Dis. 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weinstein M. M., Yin L., Beigneux A. P., Davies B. S., Gin P., Estrada K., Melford K., Bishop J. R., Esko J. D., Dallinga-Thie G. M., Fong L. G., Bensadoun A., Young S. G. (2008) Abnormal patterns of lipoprotein lipase release into the plasma in GPIHBP1-deficient mice. J. Biol. Chem. 283, 34511–34518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tybjaerg-Hansen A., Jensen H. K., Benn M., Steffensen R., Jensen G., Nordestgaard B. G. (2005) Phenotype of heterozygotes for low-density lipoprotein receptor mutations identified in different background populations. Arterioscler. Thromb. Vasc. Biol. 25, 211–215 [DOI] [PubMed] [Google Scholar]
- 37. Gin P., Beigneux A. P., Voss C., Davies B. S., Beckstead J. A., Ryan R. O., Bensadoun A., Fong L. G., Young S. G. (2011) Binding preferences for GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. Arterioscler. Thromb. Vasc. Biol. 31, 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]