

Background: TMEM106B polymorphisms are associated with some forms of dementia.
Results: A pathway for the sequential processing of TMEM106B on the lysosome membrane has been identified.
Conclusion: TMEM106B undergoes processing via removal of its lumenal domain, followed by intramembrane cleavage by the protease SPPL2a.
Significance: This may represent a mechanism for regulation of TMEM106B levels.
Keywords: Intramembrane Proteolysis, Lysosomal Glycoprotein, Lysosome, Neurodegenerative Disease, Shedding, Frontotemporal Lobar Degeneration, SPPL2a, TMEM106B
Abstract
The sequential processing of single pass transmembrane proteins via ectodomain shedding followed by intramembrane proteolysis is involved in a wide variety of signaling processes, as well as maintenance of membrane protein homeostasis. Here we report that the recently identified frontotemporal lobar degeneration risk factor TMEM106B undergoes regulated intramembrane proteolysis. We demonstrate that TMEM106B is readily processed to an N-terminal fragment containing the transmembrane and intracellular domains, and this processing is dependent on the activities of lysosomal proteases. The N-terminal fragment is further processed into a small, rapidly degraded intracellular domain. The GxGD aspartyl proteases SPPL2a and, to a lesser extent, SPPL2b are responsible for this intramembrane cleavage event. Additionally, the TMEM106B paralog TMEM106A is also lysosomally localized; however, it is not a specific substrate of SPPL2a or SPPL2b. Our data add to the growing list of proteins that undergo intramembrane proteolysis and may shed light on the regulation of the frontotemporal lobar degeneration risk factor TMEM106B.
Introduction
The regulated intramembrane proteolysis (RIP)2 of transmembrane proteins has emerged as a widespread and evolutionarily conserved mechanism (1). Generally, an initial proteolytic event results in the shedding of an ectodomain followed by intramembrane processing of the transmembrane stump, liberating an intracellular domain (ICD) and a small peptide corresponding to the transmembrane region between the two cleavage sites. An increasing number of functions have been being ascribed to RIP in health and disease, including signaling functions such as transcriptional regulation by ICDs liberated from amyloid precursor protein and Notch, among many others (2, 3). RIP is also implicated in the processing of MHC-I molecules by signal peptide peptidase (SPP) in the ER (4). Finally, RIP may also represent a generalized mechanism for regulating the levels of membrane proteins (5). Intramembrane cleaving proteases (iCLiPs) are a diverse group of three major protein families: the S2P-metalloproteases, the rhomboid serine proteases, and the GxGD-type aspartyl proteases, all of which are capable of cleaving proteins within the lipid bilayer (6). Within the GxGD-type aspartyl protease family, presenilin has a strong preference for cleaving type I transmembrane proteins, and the signal peptide peptidase-like (SPPL) family has a strict requirement for type II membrane proteins (7–9). Each member of the SPPL family has a specific subcellular localization and tissue distribution, suggesting that each may have unique substrate preferences (9, 10). Intramembrane cleavage of transmembrane proteins by these proteases requires an initial ectodomain shedding event for the substrate to be accessible (11–13).
TMEM106B was first identified as a genetic risk factor for frontotemporal lobar degeneration with TDP-43-positive inclusions (FTLD-TDP) caused by mutations in the progranulin gene (14, 15). TMEM106B is a type II single pass transmembrane protein residing primarily within the limiting membrane of late endosomes and lysosomes (16–18). The SNPs associated with increased risk for FTLD-TDP do not result in mutations in the TMEM106B protein but instead lead to elevated mRNA and protein levels of TMEM106B (14, 17). Increased TMEM106B levels have been shown to cause various lysosomal defects including altered morphology, impaired acidification, and reduced degradative capacity (17, 18). Furthermore, a coding variant T185S, in linkage disequilibrium with the protective allele of TMEM106B, has been proposed to be more rapidly degraded, further implicating elevated TMEM106B levels as a potential mechanism underlying FTLD risk (19). Although the risk for FTLD-TDP conferred by TMEM106B risk alleles is strongest in those with progranulin mutations, it has recently been shown to play a role in the pathogenesis of FTLD-TDP caused by C9ORF72 repeat expansions (20, 21) but not in other classes of FTLD, such as those caused by microtubule associated protein tau, MAPT mutations. Finally, the TMEM106B risk allele is associated with cognitive impairment in amyotrophic lateral sclerosis and is associated with the pathological presentation of Alzheimer's disease (22–24).
EXPERIMENTAL PROCEDURES
Pharmacological Reagents and Antibodies
The following antibodies were used in this study: mouse anti-FLAG (M2) from Sigma, mouse anti-HA (HA.11) from Covance, mouse anti-GAPDH from Proteintech Group, mouse anti-v5 from Invitrogen, and rat anti-mouse LAMP1 (1D4B) from BD Biosciences. Rabbit anti-TMEM106B was generated against the ICD as described (18). Rabbit anti-TMEM106A antibodies were generated by Pocono Rabbit Farm & Laboratory using the recombinant Gst-TMEM106A ICD region (amino acids 1–66) purified from bacteria as the antigen. 3-Methyladenine (3-MA), phorbol 12-myristate 13-acetate (PMA), leupeptin, and ammonium chloride were purchased from Sigma. TAPI-2, GM6001, BACE IV inhibitor, and (ZLL)2-ketone were from EMD Millipore.
Expression Constructs
Human TMEM106B cDNAs in the pCMV-Sport6 and pDONR223 vectors were obtained from Open Biosystems. SPPL2b, SPPL2c, TMEM106A, and TMEM106C were obtained from ORFeome Collection (kind gifts from Dr. Haiyuan Yu). SPPL2a was obtained from the DNASU Plasmid Repository (Arizona State University). All FLAG-tagged TMEM106 constructs were generated by cloning TMEM106 family cDNAs into the p3XFLAG-CMV7.1 vector (Sigma-Aldrich). All V5 tagged SPPL2 constructs were generated by cloning cDNAs into pCDNA3.1(+)-V5/HisA (Invitrogen). The SPPL2a D412A mutant and the TMEM106B Y132D, Y125D, C105A, G110A, and P118A mutants were generated by site-directed mutagenesis. HA-TNFα in the pCMV-Tag 4 vector (Stratagene) was kindly provided by Dr. Hening Lin. TMEM106B cDNA in the pQCXIP retroviral vector (Clontech) was used for generating retrovirus for stable cells.
Cell Culture, DNA Transfection, and Drug Treatment
HEK293T and mouse N2a and NSC-34 cells were maintained in Dulbecco's modified Eagle's medium (Cellgro) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Invitrogen) in a humidified incubator at 37 °C and 5% CO2. Cells were transiently transfected with polyethyleneamine as described (25). Cells were either treated with 200 nm PMA for 2 h, or with 50 μm TAPI-2, 50 μm GM6001, 20 μm BACE IV inhibitor, 15 mm ammonium chloride, 250 μm leupeptin, or 50 μm (ZLL)2-ketone for 16 h. 3-MA was used at 5 mm for 16 h. Retrovirus for stable cell generation was produced by co-transfecting pQCXIP-TMEM106B and pCMV-VSV-G in a 2:1 ratio in Phoenix HEK293T cells. Viral supernatant was collected 3 days after transfection and added to NSC-34 cells with 8 μg/ml Polybrene (Sigma) followed by treatment with 2 μg/ml puromycin (Sigma) for clonal selection of transduced cells. siRNAs were transfected in N2a cells as described (18).
Western Blot Analysis
Cells were washed with PBS 48 h post-transfection, and whole cell lysates were collected in Laemmli sample buffer with β-mercaptoethanol. Whole cell lysates were sonicated and kept on ice or heated at 95 °C for 2 min. Samples were separated on 16% Tricine gels and transferred to Immobilon-FL polyvinylidene fluoride membranes (Millipore). Membranes were blocked for 1 h with 5% nonfat milk in PBS and incubated in an equal mix of TBS with 0.1% Tween 20 (TBS-T) and Odyssey blocking buffer (LI-COR Biosciences) containing primary antibodies overnight at 4 °C. The membranes were washed three times with TBS-T and incubated with secondary antibodies conjugated to Alexa Fluor 680 (Invitrogen) or IRDye 800 (LI-COR Biosciences) for 1 h at room temperature. Membranes were washed three more times with TBS-T, then imaged, and quantified using an Odyssey Infrared imaging system (LI-COR Biosciences).
Immunofluorescence Microscopy
Cells on coverslips were washed with PBS 48 h post-transfection and fixed in 4% paraformaldehyde for 15 min at room temperature. The cells were washed three more times with PBS followed by permeabilization and blocking in blocking buffer (0.05% saponin, 3% BSA in PBS) for 1 h. Primary antibodies were incubated in blocking buffer overnight at 4 °C. The cells were washed and incubated in secondary antibodies conjugated to CF488A, CF568, or CF660C (Biotum). Cells were washed three more times, and coverslips were mounted onto slides with Fluoromount G (SouthernBiotech). Images were acquired on a CSU-X spinning disc confocal microscope (Intelligent Imaging Innovations) with an HQ2 CCD camera (Photometrics) using a 100× objective.
RESULTS
TMEM106B Is Proteolytically Processed
When lysates from HEK293T cells overexpressing TMEM106B are subjected to Western blotting with a homemade antibody against the N-terminal intracellular/cytosolic domain of TMEM106B, we observed a strong immunoreactive band at ∼14 kDa along with a much fainter band at ∼12 kDa in addition to the full-length 43-kDa band (Fig. 1A). We hypothesized that the 14-kDa band was the N-terminal transmembrane stump (the N-terminal fragment (NTF)) after cleavage of the TMEM106B lumenal domain and that the fainter 12-kDa band was a short-lived intracellular domain (ICD). Indeed, when C-terminal deletion constructs of TMEM106B were run next to the cleaved fragments of TMEM106B, the NTF ran similarly to the 1–127 fragments, which corresponds to the predicted size of NTF with the cytosolic and transmembrane region. The ICD fragment ran similarly to the 1–106 fragments, which could be the predicted size of the ICD resulting from intramembrane cleavage. Thus, it is very likely that TMEM106B is subject to RIP-mediated processing. Consistent with this hypothesis and the fact that ectodomain shedding is a prerequisite for most known instances of RIP (11–13), we found that the 1–127 fragment can be further cleaved to a smaller fragment with the same apparent size as the ICD generated by full-length TMEM106B. The 1–132 NTF fragment, on the other hand, was very poorly processed, despite similar expression levels. This suggests that the length of the remaining lumenal portion of the NTF is critical for recognition by the protease required for conversion to the ICD (Fig. 1A). The FLAG-tagged TMEM106B has significantly decreased NTF and ICD generated compared with untagged TMEM106B when overexpressed in HEK293T cells (data not shown), indicating that the N terminus of TMEM106B could affect TMEM106B processing.
FIGURE 1.

TMEM106B undergoes sequential proteolysis. A, HEK293T cells transfected with TMEM106B were treated with vehicle control or (ZLL)2-ketone for 14 h as indicated. Whole cell lysates were subject to anti-ICD Western blot, which revealed a 43-kDa monomeric TMEM106B, a 14-kDa membrane-retained NTF stub, and a 12-kDa ICD. Size standards corresponding to the first 132, 127, 106, and 101 residues of TMEM106B were run next to lysates from full-length (FL) TMEM106B expressing cells as indicated. The NTF runs closest to the 1–127 fragments, whereas the ICD runs closest to the 1–106 fragments. The asterisk indicates a nonspecific band running right below NTF. B, (ZLL)2-ketone inhibits the formation of the ICD, resulting in a relative increase in the NTF levels and a decrease in ICD. The ICD generated was quantitated by densitometry and calculated as a ratio relative to full-length and NTF TMEM106B. (n = 3 ± S.E.; ***, p < 0.001, Student's t test). C, whole cell lysates from a control NSC-34 cell expressing endogenous TMEM106B and a stable line expressing high levels of TMEM106B were blotted with anti-ICD antibodies. Treatment with 5 mm 3-MA for 16 h increases the levels of full-length TMEM106B and ICDs. D, NTFs and ICDs of TMEM106B can be detected at endogenous levels in N2a cells. N2a cells were transfected with control siRNA or siRNA against TMEM106B to determine whether these smaller fragments are derived from TMEM106B. Lysates from transfected HEK293T cells were run side by side as a control. The asterisk indicates a nonspecific band running right above NTF in N2a cells. The low molecular weight part of membrane was blotted separately with high concentration of anti-ICD antibodies to allow the detection of low levels of NTF and ICD in N2a cells. E, TMEM106B NTFs composed of the amino acids 1–127 retains its lysosomal compartment localization. F, the predicted TMEM106B ICD composed of the first 106 residues partially localizes to lysosomes, although less than full-length TMEM106B or NTF. Scale bars, 10 μm in main panels and 2 μm in insets.
Next we sought to determine whether TMEM106B processing could be detected in other cell lines without massive overexpression. Stable lines with different levels of TMEM106B expression were generated in the NSC-34 motor neuron-like cell line. In these stable lines, the appearance of NTFs is readily apparent, and the amount of NTF generated appears directly proportional to the level of TMEM106B expressed, with a clone expressing near endogenous levels of TMEM106B showing minimal NTF generation and clones expressing higher levels of full-length TMEM106B exhibiting greater levels of NTF generation (data not shown). An ICD fragment was detectable in the stable lines with higher expression levels, and its levels are increased by treatment with 3-MA, an inhibitor of the class III PI3 kinase VPS34 (Fig. 1C). 3-MA treatment prevents the induction of autophagy and increases the levels of TMEM106B (18, 26). Fragments corresponding to the sizes of NTF and ICD can also be detected in N2a cells at endogenous levels with optimized conditions for Western blot (Fig. 1D). The levels of these fragments are significantly reduced upon silencing of TMEM106B expression using siRNA (Fig. 1D), supporting that these fragments are processed products of TMEM106B.
Many ICDs generated by RIP traffic to the nucleus to regulate transcription (1). To determine whether this could be the case for TMEM106B, we examined the localization of TMEM106B 1–127 NTF and 1–106 ICD fragments. When overexpressed in N2a cells, a localization to the lysosomal compartment for both TMEM106B NTF(1–127) and ICD(1–106) could be detected, although less well so than full-length TMEM106B (Fig. 1, E and F). There are likely lumenal determinants, such as glycosylation, required for proper TMEM106B trafficking to lysosomes, which are missing in the NTF and ICD fragments (16). The fact that at least some of the NTF localizes to lysosomes and that it is apparently capable of being further processed to an ICD-like fragment (Fig. 1A) lends support to the idea that NTF conversion to the ICD occurs on the lysosomal membrane. The TMEM106B ICD(1–106) also appeared partially localized to lysosomes, suggesting that the liberated ICD can remain associated with the lysosome membrane after RIP (Fig. 1F). Several features of the ICD may explain the partial lysosomal localization observed. First, electrostatic interactions may mediate contact between negatively charged phospholipid head groups and the string of basic residues N-terminal to the transmembrane region of TMEM106B. Second, the last 10 amino acids corresponding to the partial transmembrane region predicted to be left after intramembrane cleavage are highly hydrophobic, which may facilitate insertion in to the lipid bilayer. Still, the level of lysosomal localization of the TMEM106B ICD was markedly less than the NTF and especially full-length TMEM106B, indicating that the ICD is likely capable of being liberated from the membrane after cleavage from the NTF.
The NTF Is Generated by Lysosomal Proteases
Because TMEM106B is a late endosomal/lysosomal protein (16–18), and ectodomain shedding generally refers to the extracellular release of a soluble protein domain, we henceforth refer to this analogous shedding event to generate TMEM106B NTFs as lumenal domain shedding. To narrow down the possible enzymes required for the initial lumenal domain shedding event, we screened a number of compounds that are capable of inducing or inhibiting various classes of sheddase. Members of the A disintegrin and metalloproteinase (ADAM) family are often implicated in the primary ectodomain shedding event for transmembrane proteins prior to RIP (27). Cells treated with PMA, a protein kinase C inducer that stimulates ADAM sheddase activity, failed to induce TMEM106B lumenal domain shedding as assessed by the levels of NTF produced relative to full-length protein (Fig. 2A). Treatment with the hydroxamic acid-based inhibitor TAPI-2, a broad spectrum inhibitor of sheddases in the matrix metalloproteinase (MMP) and ADAM families, failed to inhibit TMEM106B shedding (Fig. 2A). The related MMP inhibitor GM6001 also failed to decrease TMEM106B shedding (data not shown). As a positive control for PMA and TAPI-2 activity, we pretreated TNFα expressing HEK293T cells with TAPI-2 or vehicle control and then treated with PMA. As expected, PMA induced a large decrease in the levels of full-length TNFα, whereas TAPI-2 pretreatment prevented this effect (Fig. 2A). Most ADAM and MMP sheddases are primarily active outside of the cell to cleave extracellular domains of transmembrane proteins (28, 29). Our data confirms that these are unlikely candidates for TMEM106B lumenal shedding. BACE1, the sheddase for amyloid precursor protein required for β-amyloid generation, is active in endosomal and lysosomal compartments (30). However, treatment with BACE inhibitor IV also failed to inhibit NTF generation (Fig. 2A). Thus, neither the metalloprotease family (MMP and ADAM) nor the aspartic protease BACE family, which comprise the majority of known sheddases, appears to be responsible for TMEM106B lumenal domain shedding, suggesting a potentially novel mechanism.
FIGURE 2.

TMEM106B lumenal domain shedding occurs at the lysosome. A, TMEM106B lumenal domain shedding is not mediated by most known classes of sheddases including ADAMs and MMPs but is slightly reduced by inhibition of lysosomal proteases and inhibitors of lysosomal acidification. HA-TNFα blot is provided as a positive control for the effects of PMA and TAPI-2. PMA induces ectodomain shedding of full-length (FL) TNFα, which is prevented by pretreatment with TAPI-2. HEK293T cells overexpressing TMEM106B were treated with indicated chemicals for 16 h, except PMA, which was treated for 2 h. Concentrations used were 50 μm TAPI-2, 200 nm PMA, 20 μm BACE IV inhibitor, 15 mm ammonium chloride, and 250 μm leupeptin. ICD standards of different sizes show that the 106-amino acid fragment is closest in size to the ICD generated by TMEM106B. Note that the actual ICD appears as a doublet of two very closely spaced bands. B, the ratio of NTF to full-length TMEM106B was quantitated from the data presented in A (n = 6 ± S.E.; *, p < 0.05; **, p < 0.01, Student's t test). C, the TMEM106B Y132D lumenal domain shows increased levels of full-length TMEM106B and decreased levels of NTF. D, the ratio of NTF to full-length TMEM106B was quantitated from the data presented in C (n = 4 ± S.E.; *, p < 0.05; **, p < 0.01, Student's t test). E, TMEM106B Y132D co-localizes with lysosomal marker LAMP1 when expressed in N2a cells. Scale bars, 10 μm in main panels and 2 μm in insets.
Lysosomes harbor a wide range of proteases, which can be inhibited by raising the pH of the lysosome or by direct inhibition with leupeptin, a protease inhibitor (31). Treatment with leupeptin resulted in a modest decrease in the levels of NTF relative to full TMEM106B, whereas treatment with ammonium chloride significantly decreased the level of NTF relative to full-length TMEM106B. Treatment with both leupeptin and ammonium chloride further increased the significance of this effect and suggests that the TMEM106B lumenal domain shedding event may occur within the lumen of the lysosome by a resident protease (Fig. 2, A and B). We have previously shown that ammonium chloride treatment results in an increase in the levels of endogenous full-length TMEM106B, which would be expected if TMEM106B cleavage was inhibited (18).
To determine the amino acids proximal to residue 127 necessary for substrate recognition by lysosomal proteases, we used a bioinformatic approach to predict potential recognition motifs for lysosomal proteases in the cathepsin family. Potential sequences in the lumenal domain of TMEM106B were identified using SitePredict software (32). Amino acids Tyr125 and Tyr132 were identified to be in either the P1 or P1′ substrate position of at least three potential cleavage sites for cathepsins D, E, and G. These residues were mutated to aspartic acids to change the bulky hydrophobic characteristics of the substrate conferred by the tyrosines at those positions. Interestingly, the Y132D mutant had a significant reduction in the NTF levels and a concomitant increased level of full-length TMEM106B compared with wild type controls, consistent with impaired proteolytic processing of the full-length protein to the NTF (Fig. 2, C and D). Thus, Tyr132 may play an important role in the recognition or cleavage of the TMEM106B lumenal domain by resident proteases. To rule out that this is not due to mislocalization of the Y132D mutant, the localization of TMEM106B Y132D mutant was examined in N2a cells. TMEM106B Y132D strongly localized to lysosomes much like wild type TMEM106B, thus confirming that the defects in NTF formation are due to decreased proteolysis in lysosomes and not reduced substrate accessibility (Fig. 2E).
The Y125D mutant, as well as the Y125D/Y132D double mutant also showed almost completely abolished NTF formation and also increased levels of full-length TMEM106B (data not shown); however, these mutants were severely mislocalized and were retained primarily in the ER (unpublished observations). Nonetheless, these results support a model in which resident lysosomal proteases are responsible for the lumenal cleavage of TMEM106B to generate the NTF.
The TMEM106B NTF Is a Substrate for Intramembrane Proteolysis by SPPL2a and SPPL2b
The SPP family of iCLiPs has been shown to cleave a number of type II transmembrane proteins at various subcellular locations (10, 33–36). Treatment of TMEM106B-overexpressing HEK293T cells with the specific SPP class protease inhibitor (ZLL)2-ketone led to an increase in the relative amount of TMEM106B NTF generated and a concomitant decrease in the ICD, suggesting that an endogenous SPP class protease plays a role in cleaving the TMEM106B NTF in HEK293T cells (Fig. 1, A and B).
Among the SPP family members, SPPL2a and SPPL2b have recently been implicated in the intramembrane proteolysis of several type II membrane proteins. To date, five known substrates have been identified: TNFα, Fas Ligand, British dementia protein-2/ITMB2 (Bri2), Transferrin Receptor 1, and CD74 (33–41). In some cases, SPPL2a and SPPL2b appear to share a common substrate such as TNFα and Bri2. To test whether TMEM106B could be a substrate of either of these enzymes, we co-transfected TMEM106B into HEK293T cells with vector control, or expression constructs for SPPL2a, SPPL2b, and their paralog, SPPL2c. We found that both SPPL2a and SPPL2b, but not SPPL2c were capable of cleaving the TMEM106B NTF to generate smaller ICD fragments, and the activity of SPPL2a, but not SPPL2b, was inhibited by treatment with (ZLL)2-ketone (Fig. 3, A and B). Additionally, D412A, a catalytically inactive mutant of SPPL2a, failed to induce this cleavage, indicating that SPPL2a enzymatic activity is indeed required to induce cleavage within the TMEM106B NTF.
FIGURE 3.

The TMEM106B NTF is processed by SPPL2a and SPPL2b. A, SPPL2a promotes the conversion of TMEM106B NTF to the ICD. This is inhibited by (ZLL)2-ketone treatment (50 μm for 16 h). The catalytically inactive mutant of SPPL2a (D412A) also fails to induce NTF conversion to ICD. SPPL2b promotes conversion of NTF to ICD and is somewhat resistant to (ZLL)2-ketone inhibition under the conditions tested. SPPL2c fails to promote NTF to ICD conversion. Size standards of 132, 127, 106, and 101 amino acids were included as reference to highlight the relative sizes of the NTF and ICD fragments. B, the ratio of ICD to NTF and full-length (FL) TMEM106B was quantitated from the data presented in A. (n = 4–5 ± S.E.; *, p < 0.05; **, p < 0.01, Student's t test). C, processing of the TMEM106B C105A, G110A, and P118A mutants expressed in HEK293T cells. The asterisk indicates a nonspecific band running right below NTF.
To determine SPPL2a cleavage site in TMEM106B, we generated point mutants at key residues within and near the transmembrane region of TMEM106B based on studies suggesting that helix breaking residues in the transmembrane region of SPP family substrates are required for efficient catalysis of membrane proteins (42, 43). Because both cysteine and glycine have a high propensity for helix destabilization, residues Cys105 and Gly110 in the transmembrane region of TMEM106B were mutated to alanines to test their effect on TMEM106B cleavage. Proline is the most helix destabilizing amino acid, and Pro118 immediately C-terminal to the transmembrane region was also mutated to alanine (44). The G110A and P118A mutations had no effect on the ratio of ICD to NTF and full-length TMEM106B, although the P118A mutant is expressed at much lower levels (Fig. 3C). On the other hand, the C105A mutant showed a decrease in the levels of ICDs generated. Surprisingly, this mutant also exhibited a reproducible decrease in the levels of the NTFs, and there was no statistically significant change in the ratios of ICD to NTF (Fig. 3C and data not shown). Taken together, these data indicate that helix-breaking residues are not essential for the intramembrane cleavage of TMEM106B per se. However, the fact that C105A mutation results in decreased NTF formation suggests the intriguing possibility that there are transmembrane determinants for substrate recognition in the lumenal domain of TMEM106B.
SPPL2a has been reported to be primarily localized to late endosomes and lysosomes, whereas SPPL2b is primarily expressed at the cell surface (33, 45). To demonstrate the localization of these proteases in TMEM106B expressing cells, we analyzed the localization of the v5-tagged SPPL2 proteins in N2a cells along with endogenous TMEM106B. TMEM106B appears primarily distributed across the limiting membranes of LAMP1-positive vesicles. SPPL2a as well as the D412A mutant could be detected on vesicles containing both LAMP1 and TMEM106B, indicating that SPPL2a could likely access lysosomal TMEM106B as a substrate (Fig. 4, A and B). SPPL2b localizes very poorly to these TMEM106B-positive vesicles (Fig. 4C). Instead, SPPL2b accumulates in large perinuclear stacks, possibly Golgi (Fig. 4C). SPPL2b might process TMEM106B in the Golgi because a small population of TMEM106B is also observed in the Golgi at the steady state. SPPL2c shows a reticulated pattern of localization, indicative of a possible ER localization consistent with a previous report (Fig. 4D) (33). In agreement with this, SPPL2C has no activity toward TMEM106B (Fig. 3, A and B).
FIGURE 4.

SPPL2a localizes to lysosomes along with TMEM106B and its NTF. A, SPPL2a-V5 co-localizes with endogenous TMEM106B on LAMP1-positive vesicles in N2a cells. B, a catalytically inactive D412A mutant of SPPL2a also localizes with TMEM106B on LAMP1-positive vesicles. C, SPPL2b-V5 localizes poorly with TMEM106B on LAMP1-positive vesicles. Most SPPL2b appears accumulated within ER and Golgi compartments. D, SPPL2c-V5 does not localize with co-expressed TMEM106B. The majority of SPPL2c localizes in the ER. Scale bars, 10 μm in main panels and 2 μm in inset.
The TMEM106B Paralogs TMEM106A and TMEM106C Are Not Subject to Intramembrane Proteolysis
To test the specificity of TMEM106B as a substrate for SPPL2a-mediated intramembrane proteolysis, we decided to explore the localization and cleavage patterns of two paralogs of TMEM106B: TMEM106A and TMEM106C. N-terminally FLAG-tagged TMEM106A and TMEM106C constructs were used to determine intracellular localization. Like TMEM106B, TMEM106A is primarily localized to lysosomes in N2a cells (Fig. 5A). TMEM106C does not localize to lysosomes but rather shows a reticulated pattern indicative of an ER localization (Fig. 5B). To determine whether TMEM106A and TMEM106C can be cleaved by SPPL2a, untagged TMEM106A construct and FLAG-TMEM106C constructs were co-transfected with SPPL2a in HEK293T cells. No specific degradation products consistent with a size expected of a homologous NTF or ICD were detected for TMEM106A with SPPL2a overexpression (Fig. 5C). TMEM106C exhibits a series of closely spaced bands at ∼37 kDa, possibly because of different levels of glycosylation. Additionally, specific bands of ∼21 and ∼18 kDa were detected. These bands could be degradation products and could conceivably correlate with an NTF and ICD product. However, SPPL2a had no effect on TMEM106C processing (Fig. 5D). SPPL2b and SPPL2c also had no effect on TMEM106A or TMEM106C NTF processing (data not shown). These results suggest that SPPL2a cleavage of TMEM106B is relatively specific, because even a close homolog, TMEM106A, expressed at similar levels and localized to the same subcellular compartment, fails to be cleaved by SPPL2a under our experimental conditions. Although iCLiPs can have a large array of potential substrates, they do appear to display some selectivity even for potentially very similar substrates. This modality is supported by experiments by Martin et al. (12) in which they demonstrate that SPPL2b efficiently cleaves the Bri2 protein, but not the highly homologous Bri3, even after it is artificially truncated to mimic the NTF of Bri2.
FIGURE 5.
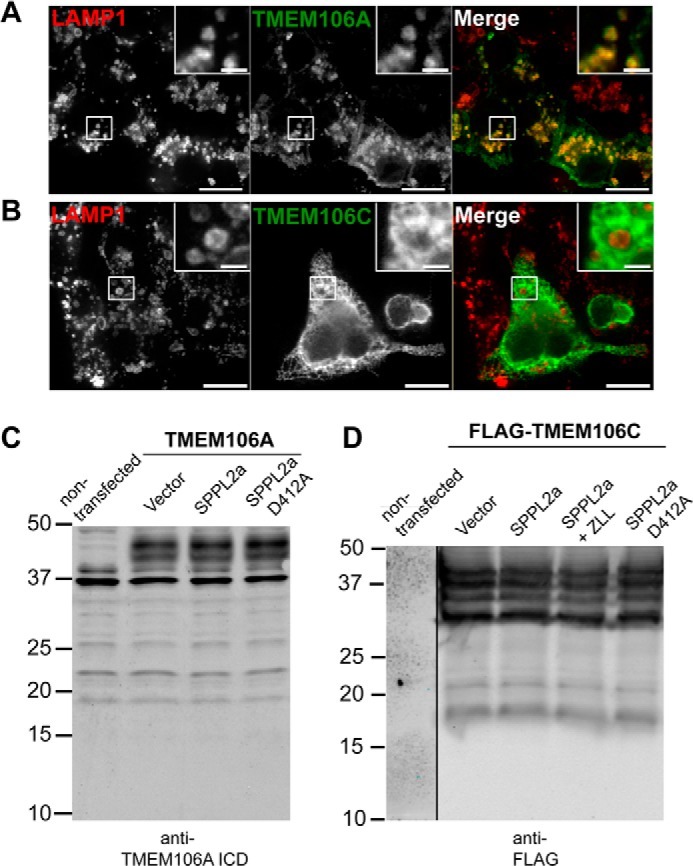
The TMEM106B homolog TMEM106A localizes to lysosomes but is not a substrate for SPPL2a. A, FLAG-TMEM106A expressed in N2a cells is localized primarily to lysosomes, similar to TMEM106B. B, FLAG-TMEM106C expressed in N2a cells does not localize to lysosomes and resides in the ER. C, HEK293T cells are transfected with untagged TMEM106A and SPPL2a constructs as indicated. The lysates were subjected to Western blot with anti-TMEM106A ICD antibody. D, HEK293T cells are transfected with FLAG-tagged TMEM106C and SPPL2a constructs as indicated and treated with 3-MA for 16 h. Neither TMEM106A or TMEM106C is specifically cleaved by co-expression of SPPL2a-v5. Scale bars, 10 μm in main panels and 2 μm in inset.
Model of TMEM106B Processing and RIP
A cartoon schematic of the predicted major proteolytic events in TMEM106B processing is proposed in Fig. 6 (A and B). An initial series of shedding events occurs in the lysosomal lumen by resident proteases to generate a stable TMEM106B NTF of ∼127 amino acids. This NTF is subsequently cleaved by SPPL2a on lysosome membranes, yielding a short-lived and highly unstable ICD product.
FIGURE 6.

Model for TMEM106B processing. A, schematic illustration of lumenal domain cleavage and regulated intramembrane proteolysis of TMEM106B, yielding a released lumenal domain and a membrane-retained NTF that is further processed into an ICD and a small predicted peptide by SPPL2a. B, locations of predicted cleavage sites of TMEM106B by an as of yet unidentified sheddase at a lumenal juxtamembrane region near amino acid 127 and within the transmembrane region centered around amino acid 106. The transmembrane region is shown as a blue cylinder. C, sequence alignment of the TMEM106B transmembrane region and proximal lumenal domain from various species shows a high degree of conservation. The transmembrane residues are indicated by a line above the relevant residues, whereas the remaining residues correspond to the adjacent lumenal domain. The sequence alignment was performed using Clustal Omega (52).
Close examination of the ICDs generated by TMEM106B reveals the appearance of two or more extremely closely spaced bands, suggesting that the ICDs could actually result from at least two different cleavage events close to amino acid 106 (Figs. 1A, 2A, and 3A). This may suggest some degree of degeneracy in the cleavage site by SPPL2a, which has been reported for the other SPPL2a substrate TNFα (37). It has been argued that these multiple cleavage sites are required for the liberation of the TNFα ICD into the cytosol (7). This lack of absolute specificity of the intramembrane cleavage site may be a more general feature of the GxGD family of iCLiPs with certain substrates, because similar patterns of intramembrane cleavage are seen by proteases such as presenilin (46).
TMEM106B is a highly conserved protein, detected throughout the vertebrate lineage. Sequence alignment shows that the transmembrane region and membrane-proximal lumenal domain region is absolutely conserved in most mammals and is largely identical even in Danio rerio and Xenopus laevis (Fig. 6C). Similarly, intramembrane proteolysis is a ubiquitous mechanism across all domains of life. This points to the prospect that RIP of TMEM106B may also be an evolutionarily conserved event.
DISCUSSION
In this study we demonstrate the selective processing of the lysosomal membrane protein TMEM106B via the sequential actions of lumenal domain shedding and RIP. This TMEM106B processing bears a striking resemblance to the processing of CD74 on the late endosomes/lysosomes of B cells, in which CD74 is sequentially cleaved by lumenal cathepsin S followed by intramembrane cleavage by SPPL2a. Knock-out of SPPL2a causes an accumulation of intermediate sized fragments of CD74 between that of the full-length protein and NTF, suggesting multiple processing steps occur in the acidified lumen (35, 40).
We showed that inhibition of lysosomal hydrolases with either ammonium chloride or leupeptin reduces lumenal domain shedding of TMEM106B. The tyrosine at position 132 appears to play a role in TMEM106B lumenal shedding. Mutation of this residue results in impaired lumenal shedding, although not a complete block. It is conceivable that the TMEM106B lumenal shedding event may be mediated by one or more soluble lysosomal proteases such as cathepsins present within the lumen. Alternatively, this event may be mediated by an as yet unidentified sheddase present within the lysosome membrane. Lysosomal proteases such as the cathepsins have highly redundant substrate specificities, and it is likely that multiple residues are implicated in substrate recognition. Further experiments with a more systematic series of mutations and more specific cathepsin inhibitors or in specific cathepsin knock-out backgrounds may help clarify the exact role of different proteases involved in lumenal domain shedding. It will be also interesting to understand whether this shedding happens constitutively in response to elevated levels of TMEM106B or whether it is regulated by other factors.
We show that the GxGD proteases SPPL2a and SPPL2b are capable of cleaving TMEM106B when overexpressed; however, SPPL2a appears to be more specifically inhibited by the SPP family inhibitor (ZLL)2-ketone, and it co-localizes much better with TMEM106B on the lysosomes, in agreement with previous reports showing that SPPL2a is predominantly trafficked to endosomes and lysosomes (33, 45). SPPL2b localizes to the cell surface and is also observed to accumulate intracellularly when expressed in N2a cells. We speculate that this overexpression may have saturated the machinery normally required for SPPL2b trafficking to the cell surface, causing it to accumulate in the secretory pathway where it may mediate the constitutive cleavage of newly synthesized TMEM106B. Microarray studies performed by Friedmann et al. (9) show that SPPL2b is expressed at low levels in most tissues except the adrenal cortex and mammary glands; SPPL2a, on the other hand, is expressed at high levels in a large number of tissues, with the highest levels detected in the brain. Expressed sequence tag profiles of TMEM106B also show TMEM106B expression in a large number of tissues, including the brain (47). Because of these considerations, we predict SPPL2a, and not SPPL2b, to be the major physiologically relevant iCLiP responsible for processing the TMEM106B NTF in vivo.
RIP generation of soluble ICDs has been proposed to mediate a large variety of signaling events both in the cytosol and in the nucleus to regulate transcription (48–50). However, because of the extremely short-lived nature of the vast majority of ICDs generated by RIP, many have yet to be detected under endogenous conditions. We see no evidence of the TMEM106B ICD fragments in the nucleus when overexpressed. Instead, the TMEM106B ICD seems somewhat lysosomally localized (Fig. 1F). A recent study reported that lysosomal TMEM106B may act as a brake against retrograde dendritic trafficking through its interaction with the microtubule-associated protein MAP6 (51). It will be interesting to see whether the cleavage of TMEM106B by RIP could serve as a mechanism to further fine tune lysosomal trafficking. Although we cannot rule out that processing of TMEM106B by RIP has a signaling function, another possibility is that RIP processing of TMEM106B serves as a membrane protein quality control mechanism allowing the efficient removal of excess TMEM106B and controlling TMEM106B levels, which might be essential for proper lysosomal function (17, 18). Given the accumulating evidence linking elevated TMEM106B levels to FTLD risk, identifying pathways that regulate TMEM106B levels may represent an important avenue in developing strategies for therapeutic intervention.
Acknowledgments
We thank Dr. Haiyuan Yu and Dr. Hening Lin for kind gifts of cDNAs and plasmids and Xiaochun Wu for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant R21 NS081357-01 (to F. H.). This work was also supported by funding from the Weill Institute for Cell and Molecular Biology and from the Association of Frontotemporal Dementia, Alzheimer's Association (to F. H.).
- RIP
- regulated intramembrane proteolysis
- ICD
- intracellular domain
- SPP
- signal peptide peptidase
- iCLiP
- intramembrane cleaving protease
- SPPL
- SPP-like
- 3-MA
- 3-methyladenine
- PMA
- phorbol myristate acetate
- FTLD
- frontotemporal lobar degeneration
- NTF
- N-terminal fragment
- ADAM
- a disintegrin and metalloproteinase
- MMP
- matrix metalloproteinase
- Bri2
- British dementia protein-2/ITMB2
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Lemberg M. K. (2011) Intramembrane proteolysis in regulated protein trafficking. Traffic 12, 1109–1118 [DOI] [PubMed] [Google Scholar]
- 2. Cao X., Südhof T. C. (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120 [DOI] [PubMed] [Google Scholar]
- 3. Mumm J. S., Schroeter E. H., Saxena M. T., Griesemer A., Tian X., Pan D. J., Ray W. J., Kopan R. (2000) A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of Notch1. Mol Cell 5, 197–206 [DOI] [PubMed] [Google Scholar]
- 4. Lemberg M. K., Bland F. A., Weihofen A., Braud V. M., Martoglio B. (2001) Intramembrane proteolysis of signal peptides: an essential step in the generation of HLA-E epitopes. J. Immunol. 167, 6441–6446 [DOI] [PubMed] [Google Scholar]
- 5. Kopan R., Ilagan M. X. (2004) γ-Secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 5, 499–504 [DOI] [PubMed] [Google Scholar]
- 6. Lichtenthaler S. F., Haass C., Steiner H. (2011) Regulated intramembrane proteolysis: lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796 [DOI] [PubMed] [Google Scholar]
- 7. Fluhrer R., Steiner H., Haass C. (2009) Intramembrane proteolysis by signal peptide peptidases: a comparative discussion of GXGD-type aspartyl proteases. J. Biol. Chem. 284, 13975–13979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weihofen A., Binns K., Lemberg M. K., Ashman K., Martoglio B. (2002) Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218 [DOI] [PubMed] [Google Scholar]
- 9. Friedmann E., Lemberg M. K., Weihofen A., Dev K. K., Dengler U., Rovelli G., Martoglio B. (2004) Consensus analysis of signal peptide peptidase and homologous human aspartic proteases reveals opposite topology of catalytic domains compared with presenilins. J. Biol. Chem. 279, 50790–50798 [DOI] [PubMed] [Google Scholar]
- 10. Krawitz P., Haffner C., Fluhrer R., Steiner H., Schmid B., Haass C. (2005) Differential localization and identification of a critical aspartate suggest non-redundant proteolytic functions of the presenilin homologues SPPL2b and SPPL3. J. Biol. Chem. 280, 39515–39523 [DOI] [PubMed] [Google Scholar]
- 11. Struhl G., Adachi A. (2000) Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 6, 625–636 [DOI] [PubMed] [Google Scholar]
- 12. Martin L., Fluhrer R., Haass C. (2009) Substrate requirements for SPPL2b-dependent regulated intramembrane proteolysis. J. Biol. Chem. 284, 5662–5670 [DOI] [PubMed] [Google Scholar]
- 13. Shah S., Lee S. F., Tabuchi K., Hao Y. H., Yu C., LaPlant Q., Ball H., Dann C. E., 3rd, Südhof T., Yu G. (2005) Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447 [DOI] [PubMed] [Google Scholar]
- 14. Van Deerlin V. M., Sleiman P. M., Martinez-Lage M., Chen-Plotkin A., Wang L. S., Graff-Radford N. R., Dickson D. W., Rademakers R., Boeve B. F., Grossman M., Arnold S. E., Mann D. M., Pickering-Brown S. M., Seelaar H., Heutink P., van Swieten J. C., Murrell J. R., Ghetti B., Spina S., Grafman J., Hodges J., Spillantini M. G., Gilman S., Lieberman A. P., Kaye J. A., Woltjer R. L., Bigio E. H., Mesulam M., Al-Sarraj S., Troakes C., Rosenberg R. N., White C. L., 3rd, Ferrer I., Lladó A., Neumann M., Kretzschmar H. A., Hulette C. M., Welsh-Bohmer K. A., Miller B. L., Alzualde A., Lopez de Munain A., McKee A. C., Gearing M., Levey A. I., Lah J. J., Hardy J., Rohrer J. D., Lashley T., Mackenzie I. R., Feldman H. H., Hamilton R. L., Dekosky S. T., van der Zee J., Kumar-Singh S., Van Broeckhoven C., Mayeux R., Vonsattel J. P., Troncoso J. C., Kril J. J., Kwok J. B., Halliday G. M., Bird T. D., Ince P. G., Shaw P. J., Cairns N. J., Morris J. C., McLean C. A., DeCarli C., Ellis W. G., Freeman S. H., Frosch M. P., Growdon J. H., Perl D. P., Sano M., Bennett D. A., Schneider J. A., Beach T. G., Reiman E. M., Woodruff B. K., Cummings J., Vinters H. V., Miller C. A., Chui H. C., Alafuzoff I., Hartikainen P., Seilhean D., Galasko D., Masliah E., Cotman C. W., Tunon M. T., Martinez M. C., Munoz D. G., Carroll S. L., Marson D., Riederer P. F., Bogdanovic N., Schellenberg G. D., Hakonarson H., Trojanowski J. Q., Lee V. M. (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 42, 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van der Zee J., Van Langenhove T., Kleinberger G., Sleegers K., Engelborghs S., Vandenberghe R., Santens P., Van den Broeck M., Joris G., Brys J., Mattheijssens M., Peeters K., Cras P., De Deyn P. P., Cruts M., Van Broeckhoven C. (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134, 808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lang C. M., Fellerer K., Schwenk B. M., Kuhn P. H., Kremmer E., Edbauer D., Capell A., Haass C. (2012) Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J. Biol. Chem. 287, 19355–19365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen-Plotkin A. S., Unger T. L., Gallagher M. D., Bill E., Kwong L. K., Volpicelli-Daley L., Busch J. I., Akle S., Grossman M., Van Deerlin V., Trojanowski J. Q., Lee V. M. (2012) TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J, Neurosci. 32, 11213–11227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brady O. A., Zheng Y., Murphy K., Huang M., Hu F. (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum. Mol. Genet. 22, 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nicholson A. M., Finch N. A., Wojtas A., Baker M. C., Perkerson R. B., 3rd, Castanedes-Casey M., Rousseau L., Benussi L., Binetti G., Ghidoni R., Hsiung G. Y., Mackenzie I. R., Finger E., Boeve B. F., Ertekin-Taner N., Graff-Radford N. R., Dickson D. W., Rademakers R. (2013) TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J. Neurochem. 126, 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gallagher M. D., Suh E., Grossman M., Elman L., McCluskey L., Van Swieten J. C., Al-Sarraj S., Neumann M., Gelpi E., Ghetti B., Rohrer J. D., Halliday G., Van Broeckhoven C., Seilhean D., Shaw P. J., Frosch M. P., Alafuzoff I., Antonell A., Bogdanovic N., Brooks W., Cairns N. J., Cooper-Knock J., Cotman C., Cras P., Cruts M., De Deyn P. P., DeCarli C., Dobson-Stone C., Engelborghs S., Fox N., Galasko D., Gearing M., Gijselinck I., Grafman J., Hartikainen P., Hatanpaa K. J., Highley J. R., Hodges J., Hulette C., Ince P. G., Jin L. W., Kirby J., Kofler J., Kril J., Kwok J. B., Levey A., Lieberman A., Llado A., Martin J. J., Masliah E., McDermott C. J., McKee A., McLean C., Mead S., Miller C. A., Miller J., Munoz D. G., Murrell J., Paulson H., Piguet O., Rossor M., Sanchez-Valle R., Sano M., Schneider J., Silbert L. C., Spina S., van der Zee J., Van Langenhove T., Warren J., Wharton S. B., White C. L., 3rd, Woltjer R. L., Trojanowski J. Q., Lee V. M., Van Deerlin V., Chen-Plotkin A. S. (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 127, 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Blitterswijk M., Mullen B., Nicholson A. M., Bieniek K. F., Heckman M. G., Baker M. C., DeJesus-Hernandez M., Finch N. A., Brown P. H., Murray M. E., Hsiung G. Y., Stewart H., Karydas A. M., Finger E., Kertesz A., Bigio E. H., Weintraub S., Mesulam M., Hatanpaa K. J., White C. L., 3rd, Strong M. J., Beach T. G., Wszolek Z. K., Lippa C., Caselli R., Petrucelli L., Josephs K. A., Parisi J. E., Knopman D. S., Petersen R. C., Mackenzie I. R., Seeley W. W., Grinberg L. T., Miller B. L., Boylan K. B., Graff-Radford N. R., Boeve B. F., Dickson D. W., Rademakers R. (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 127, 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu R. C., Wang H., Tan M. S., Yu J. T., Tan L. (2014) TMEM106B and APOE polymorphisms interact to confer risk for late-onset Alzheimer's disease in Han Chinese. J. Neural Transm. 121, 283–287 [DOI] [PubMed] [Google Scholar]
- 23. Rutherford N. J., Carrasquillo M. M., Li M., Bisceglio G., Menke J., Josephs K. A., Parisi J. E., Petersen R. C., Graff-Radford N. R., Younkin S. G., Dickson D. W., Rademakers R. (2012) TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79, 717–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vass R., Ashbridge E., Geser F., Hu W. T., Grossman M., Clay-Falcone D., Elman L., McCluskey L., Lee V. M., Van Deerlin V. M., Trojanowski J. Q., Chen-Plotkin A. S. (2011) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 121, 373–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vancha A. R., Govindaraju S., Parsa K. V., Jasti M., González-García M., Ballestero R. P. (2004) Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol. 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petiot A., Ogier-Denis E., Blommaart E. F., Meijer A. J., Codogno P. (2000) Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275, 992–998 [DOI] [PubMed] [Google Scholar]
- 27. Edwards D. R., Handsley M. M., Pennington C. J. (2008) The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu Q., Stamenkovic I. (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–176 [PMC free article] [PubMed] [Google Scholar]
- 29. Seals D. F., Courtneidge S. A. (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30 [DOI] [PubMed] [Google Scholar]
- 30. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 31. Tanaka R. D., Li A. C., Fogelman A. M., Edwards P. A. (1986) Inhibition of lysosomal protein degradation inhibits the basal degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Lipid Res. 27, 261–273 [PubMed] [Google Scholar]
- 32. Verspurten J., Gevaert K., Declercq W., Vandenabeele P. (2009) SitePredicting the cleavage of proteinase substrates. Trends Biochem. Sci 34, 319–323 [DOI] [PubMed] [Google Scholar]
- 33. Friedmann E., Hauben E., Maylandt K., Schleeger S., Vreugde S., Lichtenthaler S. F., Kuhn P. H., Stauffer D., Rovelli G., Martoglio B. (2006) SPPL2a and SPPL2b promote intramembrane proteolysis of TNFα in activated dendritic cells to trigger IL-12 production. Nat Cell Biol. 8, 843–848 [DOI] [PubMed] [Google Scholar]
- 34. Kirkin V., Cahuzac N., Guardiola-Serrano F., Huault S., Lückerath K., Friedmann E., Novac N., Wels W. S., Martoglio B., Hueber A. O., Zörnig M. (2007) The Fas ligand intracellular domain is released by ADAM10 and SPPL2a cleavage in T-cells. Cell Death Differ. 14, 1678–1687 [DOI] [PubMed] [Google Scholar]
- 35. Schneppenheim J., Dressel R., Hüttl S., Lüllmann-Rauch R., Engelke M., Dittmann K., Wienands J., Eskelinen E. L., Hermans-Borgmeyer I., Fluhrer R., Saftig P., Schröder B. (2013) The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J. Exp. Med. 210, 41–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zahn C., Kaup M., Fluhrer R., Fuchs H. (2013) The transferrin receptor-1 membrane stub undergoes intramembrane proteolysis by signal peptide peptidase-like 2b. FEBS J. 280, 1653–1663 [DOI] [PubMed] [Google Scholar]
- 37. Fluhrer R., Grammer G., Israel L., Condron M. M., Haffner C., Friedmann E., Böhland C., Imhof A., Martoglio B., Teplow D. B., Haass C. (2006) A γ-secretase-like intramembrane cleavage of TNFα by the GxGD aspartyl protease SPPL2b. Nat. Cell Biol. 8, 894–896 [DOI] [PubMed] [Google Scholar]
- 38. Martin L., Fluhrer R., Reiss K., Kremmer E., Saftig P., Haass C. (2008) Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J. Biol. Chem. 283, 1644–1652 [DOI] [PubMed] [Google Scholar]
- 39. Beisner D. R., Langerak P., Parker A. E., Dahlberg C., Otero F. J., Sutton S. E., Poirot L., Barnes W., Young M. A., Niessen S., Wiltshire T., Bodendorf U., Martoglio B., Cravatt B., Cooke M. P. (2013) The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J. Exp. Med. 210, 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bergmann H., Yabas M., Short A., Miosge L., Barthel N., Teh C. E., Roots C. M., Bull K. R., Jeelall Y., Horikawa K., Whittle B., Balakishnan B., Sjollema G., Bertram E. M., Mackay F., Rimmer A. J., Cornall R. J., Field M. A., Andrews T. D., Goodnow C. C., Enders A. (2013) B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8- dendritic cells require the intramembrane endopeptidase SPPL2A. J. Exp. Med. 210, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schneppenheim J., Hüttl S., Mentrup T., Lüllmann-Rauch R., Rothaug M., Engelke M., Dittmann K., Dressel R., Araki M., Araki K., Wienands J., Fluhrer R., Saftig P., Schröder B. (2014) The intramembrane proteases signal Peptide peptidase-like 2a and 2b have distinct functions in vivo. Mol. Cell. Biol. 34, 1398–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lemberg M. K., Martoglio B. (2002) Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol. Cell 10, 735–744 [DOI] [PubMed] [Google Scholar]
- 43. Fluhrer R., Martin L., Klier B., Haug-Kröper M., Grammer G., Nuscher B., Haass C. (2012) The α-helical content of the transmembrane domain of the British dementia protein-2 (Bri2) determines its processing by signal peptide peptidase-like 2b (SPPL2b). J. Biol. Chem. 287, 5156–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pace C. N., Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Behnke J., Schneppenheim J., Koch-Nolte F., Haag F., Saftig P., Schröder B. (2011) Signal-peptide-peptidase-like 2a (SPPL2a) is targeted to lysosomes/late endosomes by a tyrosine motif in its C-terminal tail. FEBS Lett. 585, 2951–2957 [DOI] [PubMed] [Google Scholar]
- 46. Steiner H., Fluhrer R., Haass C. (2008) Intramembrane proteolysis by γ-secretase. J. Biol. Chem. 283, 29627–29631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sayers E. W., Barrett T., Benson D. A., Bolton E., Bryant S. H., Canese K., Chetvernin V., Church D. M., Dicuccio M., Federhen S., Feolo M., Fingerman I. M., Geer L. Y., Helmberg W., Kapustin Y., Krasnov S., Landsman D., Lipman D. J., Lu Z., Madden T. L., Madej T., Maglott D. R., Marchler-Bauer A., Miller V., Karsch-Mizrachi I., Ostell J., Panchenko A., Phan L., Pruitt K. D., Schuler G. D., Sequeira E., Sherry S. T., Shumway M., Sirotkin K., Slotta D., Souvorov A., Starchenko G., Tatusova T. A., Wagner L., Wang Y., Wilbur W. J., Yaschenko E., Ye J. (2012) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 40, D13–D25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X., Sato R., Brown M. S., Hua X., Goldstein J. L. (1994) SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77, 53–62 [DOI] [PubMed] [Google Scholar]
- 49. Ye J., Rawson R. B., Komuro R., Chen X., Davé U. P., Prywes R., Brown M. S., Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 50. Selkoe D., Kopan R. (2003) Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu. Rev. Neurosci. 26, 565–597 [DOI] [PubMed] [Google Scholar]
- 51. Schwenk B. M., Lang C. M., Hogl S., Tahirovic S., Orozco D., Rentzsch K., Lichtenthaler S. F., Hoogenraad C. C., Capell A., Haass C., Edbauer D. (2014) The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 33, 450–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]