

Background: Constitutive localization of RH-RhoGEFs to the plasma membrane elevates levels of active RhoA.
Results: Acute localization of RH-RhoGEFs activates RhoA to extents that are comparable with those observed during hormone signaling.
Conclusion: Regulated localization is a viable mechanism for controlling the activity of RhoGEFs.
Significance: Multiple membrane-associated binding partners of RH-RhoGEFs could mediate robust localized signaling by anchoring the RhoGEFs in the proximity of their substrate.
Keywords: Cell Signaling; G Protein-coupled receptor (GPCR); Phospholipid Vesicle; Protein Translocation; Ras Homolog Gene Family, Member A (RhoA); Regulator of G Protein Signaling (RGS); Signal Transduction; G13; RhoGEF; Localization
Abstract
The regulator of G protein signaling homology (RH) Rho guanine nucleotide exchange factors (RhoGEFs) (p115RhoGEF, leukemia-associated RhoGEF, and PDZ-RhoGEF) contain an RH domain and are specific GEFs for the monomeric GTPase RhoA. The RH domains interact specifically with the α subunits of G12 heterotrimeric GTPases. Activated Gα13 modestly stimulates the exchange activity of both p115RhoGEF and leukemia-associated RhoGEF but not PDZ-RhoGEF. Because all three RH-RhoGEFs can localize to the plasma membrane upon expression of activated Gα13, cellular localization of these RhoGEFs has been proposed as a mechanism for controlling their activity. We use a small molecule-regulated heterodimerization system to rapidly control the localization of RH-RhoGEFs. Acute localization of the proteins to the plasma membrane activates RhoA within minutes and to levels that are comparable with activation of RhoA by hormonal stimulation of G protein-coupled receptors. The catalytic activity of membrane-localized RhoGEFs is not dependent on activated Gα13. We further show that the conserved RH domains can rewire two different RacGEFs to activate Rac1 in response to a traditional activator of RhoA. Thus, RH domains act as independent detectors for activated Gα13 and are sufficient to modulate the activity of RhoGEFs by hormones via mediating their localization to substrate, membrane-associated RhoA.
Introduction
The Rho2 family of small Ras-like GTPases contains the Rho, Rac, and Cdc42 subfamilies. These GTPases are best known for their regulation of the actin cytoskeleton and are necessary for processes such as cell division and migration (1). In many cells, overexpression of Rho, Rac, and Cdc42 proteins causes formation of stress fibers, lamellipodia, and filopodia, respectively, although the activities of these GTPases are likely coordinated during normal cellular signaling events and not mutually exclusive (2, 3).
The activity of Rho proteins is determined by cycling between their active GTP- and inactive GDP-bound states. Regulators of these proteins include GTPase-activating proteins, which enhance GTP hydrolysis and inactivation of Rho, and guanine nucleotide exchange factors (GEFs), which promote binding of GTP and, thus, activation of Rho (4).
Over 60 different RhoGEFs have been identified in mammalian genomes (5). One subfamily of these exchange factors, the regulator of G-protein signaling homology RhoGEFs (RH-RhoGEFs), consists of three family members: p115RhoGEF, leukemia-associated RhoGEF (LARG), and PDZ-RhoGEF (PRG). Besides the RH domain, these proteins contain the classical tandem Dbl homology (DH)/pleckstrin homology (PH) domains that are responsible for exchange activity toward Rho GTPases and a C-terminal coiled coil region. Both PRG and LARG also contain an N-terminal PDZ domain (6).
The RH domains of these three exchange factors interact specifically with the α subunits of the heterotrimeric G12 and G13 proteins (7, 8). In the case of p115RhoGEF and LARG, the exchange activity of the RhoGEF can be stimulated in vitro by the α subunit of the heterotrimeric G13 protein (8, 9). This stimulation of p115RhoGEF activity requires the interaction of the activated α subunit with dual sites on the RH domain and the backside of the DH domain (10). Additionally, the RH regions of p115RhoGEF and LARG act as GTPase-activating proteins in vitro toward Gα12 and Gα13 (7, 8), potentially creating a feedback loop for bidirectional regulation. PRG differs from the first two family members. Its RH domain binds to the activated α subunits of Gα12 and Gα13, but it does not act as a GTPase-activating protein (11), and the exchange activity of PRG is not modulated in vitro by the presence of activated Gα13 (11). The latter observation, combined with the modest 2- to 4-fold activation of p115RhoGEF and LARG by Gα13, indicates that an additional mechanism likely exists to regulate the activity of the RH-RhoGEFs in vivo.
Because the RH-RhoGEFs have intrinsic basal activity, a second putative mechanism for promoting exchange activity is localization of the RhoGEFs to sites of their substrates, membrane-associated RhoA that is free of Rho guanine nucleotide dissociation inhibitor. Overexpression of constitutively activated Gα12 in Madin-Darby canine kidney cells induced relocalization of GFP-tagged p115RhoGEF, LARG, and PRG from the cytosol to the plasma membrane (12). In HEK293 cells, which stably overexpressed thromboxane A2 receptors, stimulation with the agonist U44619 facilitated the relocalization of endogenous p115RhoGEF to the plasma membrane (13). However, it remains unclear whether the induced plasma membrane localization of these RhoGEFs is a cause or an effect of the activation of the proteins.
Overexpression of LARG-containing mutations in the PH domain that cause mislocalization of the protein induces smaller increases in active RhoA in HEK293T cells than overexpression of wild-type LARG. The functional output of the overexpressed mutant LARG can be restored by chimeric addition of tandem PH domains from phospholipase Cδ1 that are known to interact with phospholipids (14). Finally, expression of a modified p115RhoGEF that is constitutively localized to the plasma membrane drives cell rounding in PC12 cells (15). These observations all suggest that localization to the plasma membrane can drive activity of the RH-RhoGEFs. However, these results were obtained with overexpression of proteins that are constitutively localized to the membrane. It is not known whether acute localization of RH-RhoGEFs to the plasma membrane can account for the rapid activation of RhoA observed with hormone signaling.
In this study, we utilize a translocation system that can be regulated by a small molecule to acutely localize the RH-RhoGEFs to the plasma membrane. We show that acute localization is sufficient to initiate activation of RhoA to concentrations that are comparable in both magnitude and kinetics with those observed with hormone-induced receptor signaling. Using chimeric proteins containing RH domains and RacGEF domains, we define the RH domain as a hormone-responsive module that can rewire the Rac pathway to respond to traditional activators of RhoA.
EXPERIMENTAL PROCEDURES
Plasmids
The ARGENT heterodimerization kit containing pC4-RHE (encoding FRB) and pC4M-F2E (encoding tandem FKBPs) was obtained from Ariad Pharmaceuticals Inc. Vectors are now available commercially (Clontech). All constructs were inserted into either pC4-RHE or Myc2pCMV5 for mammalian expression and pGEX-KG-TEV (16) for bacterial expression. Constructs encoded full-length proteins unless specified otherwise. Truncated constructs of human p115RhoGEF included p115-RH-DHPH encoding amino acids (aa) 1–760, p115-DHPH encoding aa 248–760, and the p115-RH domain encoding aa 1–252. Truncated constructs of human PRG (long isoform) included PRG-RH-DHPH encoding aa 330–1125 and PRG-RH encoding aa 330–532 of PRG. PRex2-DHPH codes for aa 1–366 of human PRex2. Trio-DHPH (aa 1226–1535 of human Trio RhoGEF) was provided by Dr. John Sondek. PRex2 and TRIO constructs were cloned into the vectors mentioned above alone or 3′ to the various RH domains in combination with a GSGTGSGIDGTGTGTG linker. pEF-myc-Rac1 was provided by Dr. Helen Yin for mammalian expression. A His6 tag was added to the C terminus of full-length RhoA, and a His9 tag was added to Rac1 in place of the C-terminal CAAX box. The tagged GTPases were expressed via the pGEX-KG-TEV vector and used in vesicle-based nucleotide exchange assays. Vectors encoding Clostridium botulinum C3 transferase (17) and pRL-TK coding for Renilla reniformis luciferase under the thymidine kinase promoter were provided by Dr. Melanie Cobb (18). Vector coding for firefly luciferase under the SRE.L promoter was constructed as described previously (19).
Protein Expression and Purification
Gα13 with an N-terminal His6 tag was expressed and purified from insect cells as described previously (20). All other purified proteins were expressed in Escherichia coli strain BL21 (DE3) cultured in Luria broth medium overnight at 22 °C in the presence of 100 μm isopropyl-β-d-thiogalactopyranoside. Cells were lysed at 4 °C in 20 mm NaHEPES (pH 8.0), 5 mm β-mercaptoethanol, protease inhibitors (1 μg/ml pepstatin A, 21 μg/ml phenylmethylsulfonyl fluoride, 21 μg/ml Nα-p-tosyl-L-lysine chloromethyl ketone, 21 μg/ml tosylphenylalanyl chloromethyl ketone, and 21 μg/ml Nα-p-tosyl-L-arginine methyl ester), and 1 mg/ml lysozyme for 1 h. MgCl2 (5 mm), 100 mm NaCl, and 10 μg/ml DNase were added for 1 h, and cell debris was removed by centrifugation at 35,000 × g for 30 min. Glutathione-Sepharose 4B resin (Amersham Biosciences) was used to affinity-purify GST-tagged proteins. TEV protease was used to cleave proteins for release from the resin. Proteins were further purified by chromatography with a Mono Q anion exchange column and by size exclusion with tandem Superdex 200/70 gel filtration columns (Amersham Biosciences).
Tissue Culture
Two sets of HeLa cells were used, the Tet-On line from Clontech and the Tet-On line stably expressing human Edg5 receptors. Tet-On HeLa cells were cultured in high-glucose DMEM (Invitrogen) containing 0.1 mg/ml G418 and 10% FBS (Benchmark). Tet-On HeLa cells expressing Edg5 receptors were cultured in the above medium with the addition of 0.1 mg/ml hygromycin. PC3 cells were cultured in RPMI containing 10% FBS. All cells were maintained in a humidified incubator at 37 °C and 5% CO2.
Cell Fractionation
HeLa cells were seeded onto 100-mm tissue culture plates, cultured for 20 h, and then transfected using the FuGENE HD transfection reagent (Promega). After 24 h, transfected cells were harvested with ice-cold hypotonic lysis buffer (20 mm NaHEPES (pH 7.5), 1 mm EDTA, 1 mm DTT, 10 mm NaCl, 1.5 mm MgCl2, and protease inhibitors) and lysed by passage (10 times) through a 25-gauge needle. Nuclei and unbroken cells were removed by centrifugation at 500 × g for 5 min. Membranes were separated from the cytosol by centrifugation at 100,000 × g for 15 min. Pellets were resuspended in Laemmli buffer. All samples were separated by SDS-PAGE, transferred overnight to PVDF membranes, and immunoblotted using anti-p115-RH and anti-PRG-RH antibodies (U2760 (19) and 1186, respectively). Antiserum 1186 was prepared by Capralogics Inc. against the purified RH domain (aa 286–539) of the PRG rat homolog E48.
SRE.L Transcriptional Reporter Assay
HeLa or PC3 cells were seeded onto 48-well tissue culture plates, grown for 20 h, and transfected with SRE.L, pRL-TK, and various combinations of the indicated plasmids using the FuGENE HD transfection reagent (Promega). After 4 h of transfection, cells were placed in Opti-MEM medium (Invitrogen) for 20 h of culture without serum. Cells were then stimulated with the indicated ligands and incubated for 5 h. Cells were harvested and analyzed using the SRE.L transcriptional reporter assay kit from Promega and a Promega Glomax 20/20 luminometer. Luciferase activity is expressed as the ratio of RhoA responsive firefly luciferase to constitutively expressed R. reniformis luciferase. Fold activity is the ratio of ligand-stimulated luciferase activity to control-stimulated luciferase activity of cells transfected with vector alone.
Assays for Activated Rho GTPases
HeLa cells were cultured on 100-mm tissue culture plates. Twenty hours after seeding, cells were transfected with plasmids as indicated for 4 h and cultured in Opti-MEM medium for 20 h without serum. Cells were then stimulated at 37 °C as indicated, placed on ice, rinsed once with 10 ml of ice-cold PBS, and lysed with 600 μl of lysis buffer (50 mm TrisCl (pH 7.6), 10 mm MgCl2, 500 mm NaCl, 2% IGEPAL CA-630, and protease inhibitors). Lysates were spun at 16,000 × g for 2 min. Small aliquots of the supernatant were removed for protein assays, and the remaining supernatant was frozen rapidly in liquid N2 and stored at −80 °C. Protein assays were performed using Precision Red advanced protein assay reagent #2 from Cytoskeleton. Samples were adjusted to equivalent protein amounts for analysis. Thawed soluble lysates were mixed with affinity beads, rotated for 30 min at 4 °C, and washed with 500 μl of wash buffer (25 mm TrisCl (pH 7.6), 30 mm MgCl2, and 40 mm NaCl) prior to elution. Eluates were separated by SDS-PAGE and transferred overnight to PVDF membranes for Western blotting. Bands were quantified using ImageJ. The total integrated density was used to normalize individual experiments for combined analysis of response profiles.
Activated RhoA from 450 μg of lysate protein was isolated with agarose beads (15 μl) containing bound GST-RBD (50 μg, Rho binding domain of rhotekin) (Cytoskeleton). Beads were eluted with 1.5× Laemmli buffer. Eluates were separated as described and visualized by Western blotting with anti-RhoA antibodies (Cytoskeleton).
Activated Rac1 was isolated with the Cdc42/Rac binding domain of p21 activated kinase 1 (PBD) (21). Purified GST-PBD was incubated with glutathione-agarose beads for 15 min and rinsed twice with lysis buffer. 40 μl of suspended beads containing 1 μg/μl GST-PBD were used to isolate activated myc-Rac1 from 450 μg of lysate protein. Beads were eluted with 2× Laemmli buffer. Eluates were separated as described and visualized by Western blotting with anti-myc antibodies (Santa Cruz Biotechnology, catalog no. sc-40).
Preparation of Unilamellar Phospholipid Vesicles
All lipids were obtained from Avanti. 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, and 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) (DGS-Ni-NTA) were mixed in a mole ratio of 4.75:5:0.25, respectively, dried with a steady stream of N2 gas for 30 min, and placed under a vacuum overnight. Lipids were then suspended to 10 mm in 1 ml of buffer (20 mm NaHEPES (pH 7.5), 200 mm NaCl, 2 mm MgCl2, and 5 mm β-mercaptoethanol), subjected to five freeze-thaw cycles using an ethanol/dry ice bath, and then passed 21 times through an Avanti mini extruder using a 100-nm polycarbonate membrane.
Nucleotide Exchange Assay
Binding of N-methylanthraniloyl-GDP (mant-GDP, Invitrogen) or N-methylanthraniloyl-GTP (mant-GTP, Invitrogen) was measured with a Fluorolog-3 spectrofluorometer at 25 °C at λexcitation, 356 nm; λemission, 445 nm, and slits, 1/1 nm. Vesicles (0.5 mm phospholipid) containing DGS-Ni-NTA were mixed with 2 μm RhoA-His6 or 2 μm Rac1-His9 in reaction buffer (20 mm NaHEPES (pH 7.5), 200 mm NaCl, 2 mm MgCl2, and 5 mm β-mercaptoethanol). Mant nucleotides (5 μm) were added, and reactions were started with the addition of RhoGEFs as indicated. When included, His6-Gα13 was added to vesicles with the other GTPases.
RESULTS
Regulated Localization of RH-RhoGEFs to the Plasma Membrane with a Heterodimerization System
The ARGENT regulated heterodimerization system from Ariad Pharmaceuticals uses the rapamycin analog AP21967 (Rapalog) to acutely control plasma membrane localization of proteins of interest. The system consists of two binding partners, an FKBP that is constitutively localized to the plasma membrane with an N-terminal myristoylation (myr) sequence, and a modified FRB that localizes to the cytosol. Interaction of FKBP and FRB can be induced by the addition of Rapalog, leading to effective translocation of FRB from the cytosol to the plasma membrane. Only the modified FRB domains, but not the WT domains of endogenous proteins, respond to Rapalog (22). We constructed chimeras of FRB and various RH-RhoGEFs to allow the regulated localization of exogenously expressed RH-RhoGEFs to the plasma membrane in the presence of Rapalog.
Localization of FRB-PRG and FRB-p115 in the presence and absence of Rapalog was verified using crude fractionation of HeLa cells (Fig. 1). Both proteins localize to the cytosolic fraction in the absence of Rapalog. Levels of FRB-PRG and FRB-p115 in the cytosolic fraction decrease upon addition of Rapalog and increase concordantly in the membrane fraction, verifying that the addition of Rapalog induces translocation of the proteins. In contrast, endogenous p115-RhoGEF does not redistribute to the membrane fraction upon addition of Rapalog.
FIGURE 1.

Regulated localization of RhoGEFs to the plasma membrane. A, diagram of constructs utilized with the heterodimerization system. See text for details. B, Rapalog induces translocation of RH-RhoGEFs from cytosolic to membrane fractions. HeLa cells were transfected with plasmids encoding myrFKBP and either FRB-p115 or FRB-PRG. After 24 h, cells were incubated in the presence or absence of 500 nm Rapalog for 5 min and lysed hypotonically. Lysates were fractionated by centrifugation and analyzed by Western blotting.
Translocation of RH-RhoGEFs Is Sufficient to Activate RhoA
The small molecule-regulated heterodimerization system was used to investigate whether induced membrane localization of FRB-PRG was capable of activating RhoA. As expected, the expression of increasing amounts of FRB-PRG in HeLa cells raised the basal activation of RhoA on the basis of the SRE.L transcriptional reporter assay for detection of active RhoA (Fig. 2A). Importantly, the addition of Rapalog increased the reported activity ∼3-fold at lower levels of FRB-PRG expression. These results indicate that localization of PRG to the plasma membrane is sufficient to stimulate the signaling pathway. Transcription of the reporter plasmid was abolished by the expression of C3 transferase, verifying that the activation of RhoA was responsible for regulation of the reporter (Fig. 2B). Induced translocation of FRB-PRG was also shown to be an effective activator of RhoA in PC3 cells, demonstrating that this is not a unique phenomenon of HeLa cells (Fig. 2C).
FIGURE 2.

Translocation of RH-RhoGEFs activates RhoA. A, membrane localization of FRB-PRG stimulates a RhoA-responsive transcriptional reporter. HeLa cells were transfected with plasmids encoding the SRE.L reporter, myrFKBP, and increasing amounts of FRB-PRG. Cells were then stimulated with either 0.05% EtOH (Control) or 500 nm Rapalog for 5 h, processed, and analyzed for luciferase activity as described. B, activation of the transcriptional reporter is dependent on RhoA activity. HeLa cells were transfected with plasmids encoding myrFKBP and FRB-PRG either alone or in combination with C3 transferase. Cells were stimulated with 0.05% EtOH and 0.1 mg/ml BSA (Control), 1 μm S1P, or 500 nm Rapalog as indicated and then processed and analyzed as described. C, activation of RhoA by membrane localization of FRB-PRG is not cell line-specific. PC3 cells were transfected with plasmids encoding the SRE.L reporter, myrFKBP, and increasing amounts of FRB-PRG. Cells were then stimulated, processed, and analyzed as stated in A. D, membrane localization of FRB-p115 activates RhoA. HeLa cells were transfected with plasmids encoding the SRE.L reporter, myrFKBP, and increasing amounts of FRB-p115. Cells were then stimulated, processed, and analyzed as stated in A. The experiments shown are representative of at least three independent experiments. Error bars represent mean ± S.E. within the experiment shown.
The intrinsic exchange activity of p115RhoGEF can be stimulated in vitro by the addition of activated Gα13, but only modestly (8, 9). Localization to the plasma membrane could be an additional mechanism contributing to activation of p115RhoGEF in vivo. Similar to FRB-PRG, Rapalog-mediated translocation of FRB-p115 to the plasma membrane was sufficient to stimulate the reporter of RhoA activity ∼5-fold (Fig. 2D). Again, constitutive activation of RhoA was observed at higher levels of FRB-p115 expression, even in the absence of Rapalog, underscoring the necessity for the tight regulation of endogenous RhoGEFs.
Gα13 Is Not Required for the Activity of Translocated RH-RhoGEFs
Although Gα13 does not activate PRG in vitro, overexpression of PRG in combination with Gα12 and Gα13 has been reported to synergistically activate RhoA in cells (8). Therefore, we tested whether hormone activation of Gα13 would increase the activity of FRB-PRG translocated to the plasma membrane by the addition of Rapalog. In HeLa cells, sphingosine-1-phosphate (S1P) stimulates G13 via receptors of the Edg family. Treatment of cells with 1 μm S1P stimulated the transcription of the reporter for RhoA ∼5-fold. The simultaneous addition of both S1P and Rapalog produced an essentially additive response, indicating that activated Gα13 does not further enhance the catalytic activity of translocated FRB-PRG (Fig. 3A).
FIGURE 3.

Gα13 is not required for catalytic activity of translocated RH-RhoGEFs. A, stimulation of Gα13 with S1P does not enhance the activity of translocated FRB-PRG. HeLa cells were transfected with plasmids encoding the SRE.L reporter, myrFKBP, and FRB-PRG. Cells were stimulated for 5 h with 0.05% EtOH and 0.1 mg/ml BSA (Control), 500 nm Rapalog, 1 μm S1P, or both Rapalog and S1P, as indicated, and then processed and analyzed as described. B, stimulation of Gα13 with S1P does not enhance the activity of translocated FRB-p115. HeLa cells were transfected with plasmids encoding the SRE.L reporter, myrFKBP, and FRB-p115. Cells were stimulated, processed, and analyzed as stated in A. C and D, attenuation of Gα13 activity by p115-RH does not affect activation of RhoA in response to FRB-RH-RhoGEF translocation. HeLa cells were transfected with plasmids encoding the SRE.L reporter, myrFKBP, and FRB-p115 or FRB-PRG either alone or in combination with the p115-RH domain as indicated. Cells were stimulated, processed, and analyzed as stated in A. Experiments shown are representative of at least three independent experiments. Error bars represent mean ± S.E. within the experiment shown.
Surprisingly, the simultaneous stimulation of cells expressing FRB-p115 with both S1P and Rapalog also resulted in additive synthesis of the reporter. Together, these results show that Gα13 activated by hormone does not enhance the catalytic activities of the translocated FRB-RhoGEFs. Although this may represent an inability to further stimulate the enzymes on the membrane, it is also possible that Gα13 activated by hormone and the myrFKBP targeting domain used in these experiments inhabit separate regions of the plasma membrane, thus abrogating contact between the FRB-RhoGEFs and activated Gα13.
Although the translocated RhoGEFs may be insensitive to Gα13 activated by hormone, it is still possible that basal levels of active Gα13 contribute to their activity. To test for such dependence, the FRB-RhoGEFs were translocated in the presence of the overexpressed RH domain of p115RhoGEF, a potent inactivator of both Gα12 and Gα13 proteins (7). Although stimulation of the SRE.L reporter by S1P was almost completely abolished by overexpression of the RH domain, activation because of Rapalog-induced translocation of both FRB-PRG and FRB-p115 was not affected (Fig. 3, C and D). This clearly demonstrates that activated Gα13 is not required for the RH-RhoGEFs to maintain their intrinsic catalytic activity and supports the hypothesis that recruitment of these GEFs to the plasma membrane, a site of concentrated substrate, is a feasible mechanism for their regulation. In the case of the RH-RhoGEFs, this recruitment, in response to hormones, could be driven by binding of their RH domains to activated Gα13 or Gα12.
Activation Induced by Translocation and Activation Stimulated by Receptors Are Comparable in Both Magnitude and Time
Induction of the transcriptional reporter occurs over hours, whereas activation of RhoA by hormones occurs in minutes. Therefore, the ability of regulated translocation of the RH-RhoGEFs to acutely activate RhoA was directly assessed by measuring RhoA-GTP via its specific association with the RhoA binding domain of rhotekin (Fig. 4). Stimulation of HeLa cells by S1P produced a robust elevation in active RhoA within 1 min, which began declining by 3 min. Similarly, the addition of Rapalog generated a significant activation of RhoA within 1 min, but the levels of active RhoA continued to rise, reaching a plateau between 3–5 min. This level of activation could be sustained for at least 30 min (data not shown). A well established mechanism for down-regulation of G protein-coupled signaling pathways is desensitization and/or internalization of GPCRs, which may account for the rapid decline in activation of RhoA by S1P. In contrast, translocation of the RhoGEF by Rapalog bypasses receptor and G protein activation and produces more prolonged stimulation by stably maintaining the RhoGEF at the plasma membrane. These results verify that translocation of endogenous RH-RhoGEFs is capable of significantly activating the RhoA signaling pathway within minutes, analogous to hormonal activation of GPCRs.
FIGURE 4.
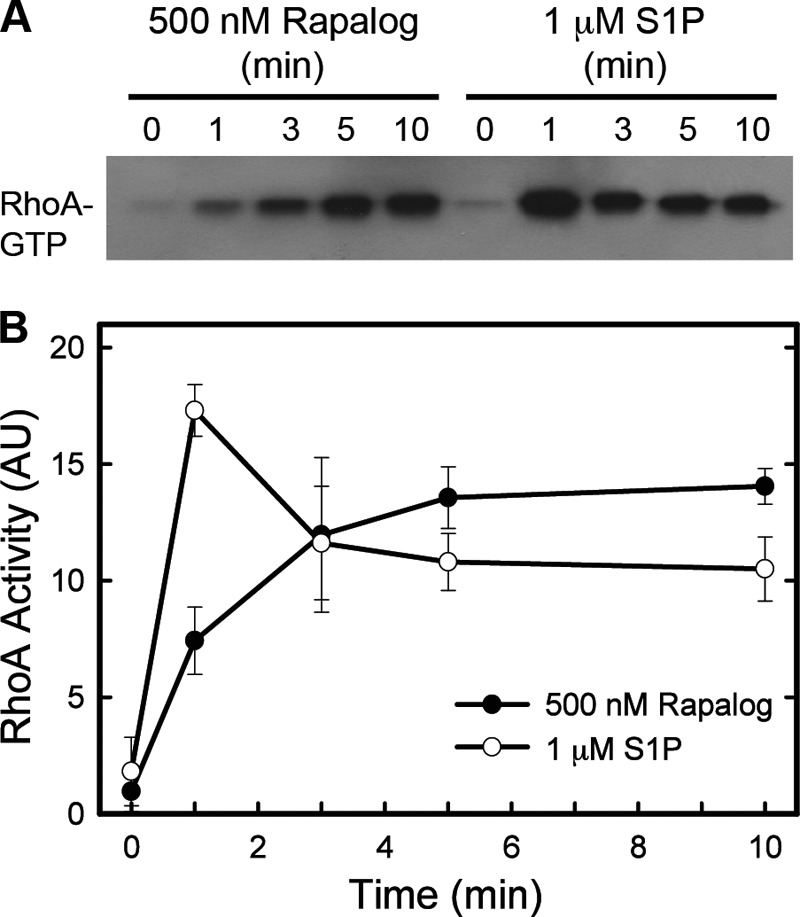
The magnitude and timing of RhoA activation induced by translocation of RhoGEFs is comparable with activation mediated by hormonal stimulation. HeLa cells were transfected with myrFKBP and FRB-p115 plasmids. Cells were stimulated with 1 μm S1P or 500 nm Rapalog for the lengths of time shown and then lysed. Active RhoA was isolated from lysates using GST-RBD bound to agarose beads and then analyzed by Western blotting. A representative blot is shown in A. Blots were quantitated with the ImageJ program, and data from three independent experiments were normalized and combined in B; AU, integrated density in arbitrary units. Error bars represent the mean ± S.D. throughout combined experiments.
RH Domains Act as Recruiting Modules
The most logical mechanism for recruitment of RH-RhoGEFs to the plasma membrane is interaction of the RH domain with membrane-associated Gα12 and Gα13. The RH domains of these GEFs interact specifically with the active forms of the α subunits, which remain associated with the plasma membrane through N-terminal acylation (23). We used defined phospholipid vesicles to test in vitro whether this mechanism of recruitment could regulate the RH-RhoGEFs. Stable association of proteins with the vesicle surface was achieved through inclusion of DGS-Ni-NTA, a synthetic lipid that contains a nickel-chelating head group that binds polyhistidine tags. This mediates the localization of Rho GTPases with C-terminal His tags and Gα13 with an N-terminal His6 tag to the surface of vesicles, where they act as anchored substrates and as a regulator, respectively. In this paradigm, the activity of RH-RhoGEFs on the sequestered substrate should be regulated by the presence or absence of activated Gα13. This is shown in Fig. 5, which uses the binding of mant-GDP to RhoA to measure the exchange activity of the RhoGEF. Although the presence of inactive Gα13 on vesicles had little effect on the activity of p115-RH-DHPH, addition of AlF4− to activate Gα13 produced a large increase in exchange activity (Fig. 5B). The fold change for initial rates of RhoA exchange under different conditions is shown in Fig. 5C. Addition of the p115-RH-DHPH to vesicles that contain associated RhoA increased the basal exchange on RhoA ∼2.5-fold. The presence of activated Gα13 increased the rate to >80-fold above the basal exchange levels. This >30-fold net increase induced by activated Gα13 was completely dependent on the RH domain (Fig. 5C). Importantly, the activity of p115-DHPH and p115-RH-DHPH toward free RhoA in solution was identical (Fig. 5D). The lack of significant stimulation with inactive Gα13 suggests that the RH domain of p115RhoGEF can act as a detector of Gα13 activation and function to recruit p115RhoGEF to the plasma membrane.
FIGURE 5.

RH domains function as recruiting modules. A, diagram of exchange factors used in nucleotide exchange assays. L, linker region. B, activated Gα13 enhances nucleotide exchange on membrane-delimited RhoA. RhoA-His6 (2 μm) bound to vesicles was incubated with 5 μm mant-GDP either alone, with 0.5 μm His6-Gα13, or with 0.5 μm His6-Gα13-AlF4−. Exchange reactions were started by addition of 30 nm p115-RH-DHPH. AU, arbitrary units. C, RH domains localize RH-RhoGEFs to activated Gα13, promoting nucleotide exchange on RhoA. RhoA-6His (2 μm) bound to vesicles was incubated with 5 μm mant-GDP either alone, with 0.5 μm His6-Gα13, or with 0.5 μm His6-Gα13-AlF4−. Exchange reactions were started by addition of the indicated RhoGEFs (30 nm). D, purified p115-DHPH and p115-RH-DHPH proteins have the same intrinsic catalytic activity. RhoA- His6 (2 μm) was incubated free in solution with 5 μm mant-GDP. Exchange reactions were started by addition of RhoGEFs. Error bars represent the mean ± S.D. of the initial rates from at least three separate experiments.
Similarly, PRG-RH-DHPH exhibited the greatest effect on exchange rates in the presence of vesicle-associated Gα13-AlF4− (Fig. 5C). Because Gα13-AlF4− does not stimulate the intrinsic catalytic activity of PRG, the enhancement of initial rate can be solely attributed to localization via the RH domain and concentration of the enzyme in proximity with its substrate.
Activated Gα13 can modestly stimulate the intrinsic catalytic activity of p115RhoGEF toward RhoA that is free in solution. To directly compare the effect of vesicle-associated and soluble Gα13-AlF4− on exchange rates induced by p115-RH-DHPH, we measured the activation of RhoA in the presence of vesicles without or with DGS-Ni-NTA in combination with His-tagged or non-tagged Gα13-AlF4− (Fig. 6, A and B). As expected, both forms of activated Gα13 stimulate the exchange activity of p115-RH-DHPH ∼2-fold in the absence of localization to vesicles. In contrast, His- Gα13-AlF4− stimulates the activity of the RhoGEF >30-fold in the presence of DGS-Ni-NTA vesicles, whereas non-tagged Gα13-AlF4− retained a 2-fold stimulus. Thus, any stimulation of intrinsic catalytic activity has only a minor role in the robust activation observed with the localization to the substrate.
FIGURE 6.

Activity of p115RhoGEF is modulated through localization with the substrate. A and B, Gα13 associated with vesicles greatly enhances nucleotide exchange on membrane delimited RhoA. RhoA- His6 (2 μm) in the presence of vesicles either without or with DGS-Ni-NTA was incubated with 5 μm mant-GDP either alone, with 0.5 μm Gα13-AlF4−, or with 0.5 μm His6-Gα13-AlF4−. Exchange reactions were started by addition of 30 nm p115-RH-DHPH. The initial rates in B are the mean ± S.D. of eight experiments. The significance for stimulation of the RhoGEF by Gα13 was <0.001 in all cases (standard two-tailed Student's t test). C, stimulation of intrinsic catalytic activity of p115RhoGEF by Gα13-AlF4− does not contribute to the enhancement of exchange activity on vesicle-associated RhoA. RhoA- His6 (2 μm) bound to vesicles was incubated with 5 μm mant-GDP either alone or with 0.5 μm His6-Gα13-AlF4−. Exchange reactions were started by addition of 30 nm p115-RH-DHPH or the W507E mutant of p115-RH-DHPH.
To further examine any contribution of stimulation of intrinsic catalytic activity of p115RhoGEF to the 30-fold increase seen by localization of the RhoGEF to vesicle-delimited RhoA, we used the W507E mutant of p115RhoGEF, which retains basal levels of catalytic activity but lacks enhancement of activity in the presence of activated Gα13 (10). When this mutant was compared with wild-type p115-RH-DHPH, the initial rates of exchange activity were the same either in the absence or presence of activated vesicular Gα13 (Fig. 6C). These results prove that activated Gα13 can regulate both RH-RhoGEFs through simple localization with the substrate on a membrane surface. However, in the context of the cellular environment, activation of the intrinsic exchange activity of p115RhoGEF by Gα13 may play an important role.
The RH region forms a stable individual domain that should be capable of functioning as a detector of active Gα13 independent of the DHPH domains of the RH-RhoGEFs. To test this, the RH domains from p115RhoGEF and PRG were placed in front of the N-terminal DHPH domains of Trio and the DHPH domains of PRex2. Both GEFs are specific for activation of Rac rather than RhoA (24–26). Fig. 7A shows that the activity of PRex2-DHPH on vesicular Rac1 could be increased significantly by the combination of a chimeric PRG-RH domain and vesicular G13α. Stimulation was not observed when the activated α subunit was not localized to the vesicular surface. Identical behavior was observed with chimeric Trio-DHPH that was linked to the RH domains of either p115 or PRG (Fig. 7B). These data validate the RH domains as independent units capable of responding to activation of Gα13 by localizing associated enzymatic modules.
FIGURE 7.

Chimeric RH-Trio-DHPH and RH-PRex2-DHPH facilitate activation of Rac1 by G13. A, vesicle-associated His6-Gα13-AlF4− stimulates the exchange activity of PRG-RH-PRex-DHPH. Rac1-His9 (2 μm) bound to vesicles was incubated with 5 μm mant-GTP either alone, with His6-Gα13-AlF4− (50 nm), or with soluble Gα13-AlF4− (50 nm). Reactions were started by addition of either 50 nm RH-PRex or 12.5 nm PRex (concentrations at which activities toward Rac1 were equivalent in the absence of Gα13). AU, arbitrary units. B, RH domains localize Trio to activated vesicular G13α, promoting nucleotide exchange on Rac1. Rac1-His9 (2 μm) bound to vesicles was incubated with 5 μm mant-GTP either alone, with 0.5 μm His6-Gα13, or with 0.5 μm His6-Gα13-AlF4−. Exchange reactions were started by addition of 100 nm RacGEFs. Error bars represent the mean ± S.D. of the initial rates from at least three separate experiments.
The RH Domain of PRG Can Mediate the Hormonal Activation of Rac by G12/13
Can the RH domains of the RhoGEFs function as detectors of hormonal activation and drive signaling via simple localization in living cells? Stimulation of HeLa cells with S1P uses a G12/13-dependent mechanism to activate RhoA but not Rac1 (Fig. 8A). If RH domains are capable of localizing GEFs in response to hormone, the addition of RH domains to Rac-specific DHPH domains should rewire RacGEFs to respond to the G12/13 pathway and allow S1P to drive activation of Rac1. Fig. 8B shows the expected rapid activation of RhoA by S1P in Edg5-HeLa cells, which also express the PRG-RH-PRex2-DHPH chimera. In contrast to HeLa cells that do not express the chimera (Fig. 8A), S1P now also stimulates activation of Rac1 (Fig. 8C). The activation of Rac occurs on a similar time scale to RhoA, which is consistent with the same kinetic mechanism. This effect is not dependent on overexpressed Edg5 receptors because expression of the chimeric PRG-RH-Trio-DHPH in HeLa cells facilitates activation of Rac1 in response to endogenous S1P receptors (Fig. 8D). Although the overexpression of the RacGEFs raises basal levels of activated Rac1, enhancement of this activation by S1P requires the chimeric RH domains. This illustrates that the RH domain is sufficient to detect the hormonal activation of Gα13 in the plasma membrane and facilitate the localization of RhoGEFs to drive their respective signaling pathways.
FIGURE 8.

RH domains rewire RacGEFs to respond to S1P. A, S1P stimulates RhoA but not Rac1. HeLa cells were stimulated with 0.1 mg/ml BSA, 1 μm S1P, or 50 ng/ml EGF for 2 min and then lysed. GST-PBD and GST-RBD bound to agarose beads were used to isolate active Rac1 and active RhoA, respectively, from lysates. Active Rac1 and RhoA were then analyzed by Western blotting. B–D, the RH domain rewires Trio-DHPH and PRex2-DHPH to activate Rac1 in response to S1P. Edg5-HeLa cells were transfected with myc-Rac1 and either PRex-DHPH (D) or PRG-RH-PRex-DHPH (B–D) plasmids. HeLa cells were transfected with myc-Rac1 and either Trio-DHPH or PRG-RH-TrioDHPH plasmids (D), as indicated. Cells were stimulated with 1 μm S1P for the indicated times (B and C) or for 2 min (D) and lysed. Active RhoA (B) and myc-Rac1 (C and D) were isolated with GST-RBD and GST-PBD, respectively, and then analyzed by Western blotting and quantitated as described. Representative results are shown in the top panels, and quantitation of at least three separate experiments was normalized for total signals and is shown in the bottom panels. Error bars represent the mean ± S.D. for combined data. Significance was determined with a standard two-tailed Student's t test for stimulation by S1P over basal. *, p < 0.01; **, p < 0.001.
DISCUSSION
The action of RH-RhoGEFs can potentially be driven through multiple mechanisms, including direct stimulation of intrinsic exchange activity. The substrate for the RhoGEFs, RhoA free of Rho guanine nucleotide dissociation inhibitor, resides in membranes. Therefore, a second mechanism is recruitment of the RH-RhoGEFs to membranes that are enriched in available RhoA (27). Evidence exists to support both mechanisms. Gα13 stimulates the exchange activity of p115RhoGEF and LARG in vitro, and all three RhoGEFs have been suggested to function synergistically with Gα13 in cells (8, 9, 28). However, all three RH-RhoGEFS do exhibit significant basal exchange activity in vitro, and all three have been shown to localize to the plasma membrane in cells upon addition of activated upstream regulators (12, 13). In addition, constitutive expression of a membrane-localized mutant of LARG induced higher levels of activated RhoA within cells than similar amounts of cytosolic protein (14). The activity studies all utilized prolonged overexpression of proteins and reporter assays that could lead to indirect responses or proceed through mechanisms that do not account for the acute effects of hormones.
Here we used three experimental approaches to show that the regulated localization of the RH-RhoGEFs to the membrane is sufficient to account for the acute activation of RhoA observed upon treatment of cells with hormones. An in vitro approach utilized phospholipid vesicles and tethered Rho GTPases to simulate the plasma membrane in cells. In these experiments, the action of the RH-RhoGEFs on RhoA was low but could be stimulated >30-fold when Gα13, also tethered to the vesicles, was activated. These results demonstrate that activated Gα13 can localize the RH-RhoGEFs to the membrane surface to facilitate the processive activation of RhoA. Further, this result indicates that the interaction between Gα13 and the RH domain is oriented to facilitate interaction of the rest of the RhoGEF with its substrate in the membrane, as predicted by previous models (10).
Two approaches were used in living cells. First, a regulated heterodimerization system was used to allow controlled translocation of the RH-RhoGEFs to the plasma membrane in the absence of receptor-mediated activation of G proteins. We found that induced membrane localization of RH-RhoGEFs was indeed capable of activating RhoA within 1 min, a time scale compatible with that observed during hormonal stimulation of RhoA. Additionally, both induced localization of RH-RhoGEFs as well as hormone addition produced similar levels of activated RhoA. These data indicate that basal exchange rates of the RH-RhoGEFs are sufficient for driving the activation of the RhoA pathway and that simple localization of the proteins to their membrane-associated substrates can explain the activities observed with hormones.
The specificity in this system is defined by the interaction of the activated α subunits of G12/13 with the RH domains of these RhoGEFs. To demonstrate the importance of this domain, we constructed chimeric proteins consisting of RH domains along with the RacGEF domains of either Trio or PRex2. Both in vitro and in living cells, we show that inclusion of the RH domain is sufficient to drive the regulation of Rac1 via pathways evolved to regulate RhoA.
It will be interesting to see which other regions of these proteins may also play a role in controlling their cellular localization. The PDZ domains of PRG and LARG are known to interact with various GPCRs of the Edg family as well as membrane-spanning plexins (29–31). More recently, the PH domain of PRG has been shown to interact with activated RhoA (16). This observation was extended to all seven members of the Lbc RhoGEF family along with evidence that this mediates a positive feedback mechanism for these RhoGEFs (32). It is likely that all three domains contribute to membrane anchoring, perhaps in a cooperative manner in the context of multiple binding sites. Under this scenario, high-affinity binding between the RH domain and activated Gα13 would act as an initial, diffusion-controlled detector of hormone signaling. When the RH domain attracts the RhoGEF to the plasma membrane, the lower-affinity binding sites between the PDZ domain and receptors could provide effective secondary anchoring sites. Following activation of a small amount of RhoA, the PH domains could interact with active RhoA and facilitate positive feedback regulation by reinforcing localization of the RhoGEF at the membrane. On the basis of this speculation, it is likely that efficient signaling through these pathways is dependent upon the cooperative interaction of these multiplex binding sites that anchor the RhoGEFs at the plasma membrane and, thus, facilitate robust localized responses. In addition, this cooperative binding may be needed to compete with other interactions of the RhoGEFs with sites such as actin and microtubules that may sequester them away from the plasma membrane and prevent their activity under basal conditions (33, 34).
Acknowledgments
We thank Jana Hadas for technical assistance and Dr. Zhe Chen for discussions.
This work was supported, in whole or in part, by National Institute of Health Grant GM31954 (to P. C. S.) and by National Institutes of Health Pharmacological Sciences Training Grant GM007062 (to A. M. C.). This work was also supported by Robert A. Welch Foundation Grant I-1262 (to P. C. S.) and by the Alfred and Mabel Gilman Chair in Molecular Pharmacology (to P. C. S.).
- Rho
- Ras homology
- GEF
- guanine nucleotide exchange factor
- RH
- regulator for G protein signaling homology
- LARG
- leukemia-associated Rho guanine nucleotide exchange factor
- PRG
- PDZ- Rho guanine nucleotide exchange factor
- PH
- pleckstrin homology
- DH
- Dbl homology
- FRB
- FKBP12-rapamycin binding domain
- aa
- amino acids
- TEV
- tobacco etch virus
- RBD
- Rho binding domain of rhotekin
- PBD
- Rac binding domain of p21 protein kinase
- DGS
- 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl]
- Ni-NTA
- nickel-nitrilotriacetic acid
- mant-GTP
- N-methylanthraniloyl-GTP
- mant-GDP
- N-methylanthraniloyl-GDP
- FKBP
- FK506 binding protein
- S1P
- sphingosine-1-phosphate
- myr
- myristoylation.
REFERENCES
- 1. Schmidt A., Hall A. (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 2. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 3. Machacek M., Hodgson L., Welch C., Elliott H., Pertz O., Nalbant P., Abell A., Johnson G. L., Hahn K. M., Danuser G. (2009) Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Aelst L., D'Souza-Schorey C. (1997) Rho GTPases and signaling networks. Genes Dev. 11, 2295–2322 [DOI] [PubMed] [Google Scholar]
- 5. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 6. Sternweis P. C., Carter A. M., Chen Z., Danesh S. M., Hsiung Y. F., Singer W. D. (2007) Regulation of Rho guanine nucleotide exchange factors by G proteins. Adv. Protein Chem. 74, 189–228 [DOI] [PubMed] [Google Scholar]
- 7. Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., Sternweis P. C. (1998) p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science 280, 2109–2111 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki N., Nakamura S., Mano H., Kozasa T. (2003) Gα12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. U.S.A. 100, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hart M. J., Jiang X., Kozasa T., Roscoe W., Singer W. D., Gilman A. G., Sternweis P. C., Bollag G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science 280, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 10. Chen Z., Guo L., Hadas J., Gutowski S., Sprang S. R., Sternweis P. C. (2012) Activation of p115-RhoGEF requires direct association of Gα13 and the Dbl homology domain. J. Biol. Chem. 287, 25490–25500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wells C. D., Liu M. Y., Jackson M., Gutowski S., Sternweis P. M., Rothstein J. D., Kozasa T., Sternweis P. C. (2002) Mechanisms for reversible regulation between G13 and Rho exchange factors. J. Biol. Chem. 277, 1174–1181 [DOI] [PubMed] [Google Scholar]
- 12. Meyer B. H., Freuler F., Guerini D., Siehler S. (2008) Reversible translocation of p115-RhoGEF by G12/13-coupled receptors. J. Cell. Biochem. 104, 1660–1670 [DOI] [PubMed] [Google Scholar]
- 13. Bhattacharyya R., Wedegaertner P. B. (2003) Characterization of Gα13-dependent plasma membrane recruitment of p115RhoGEF. Biochem. J. 371, 709–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aittaleb M., Gao G., Evelyn C. R., Neubig R. R., Tesmer J. J. (2009) A conserved hydrophobic surface of the LARG pleckstrin homology domain is critical for RhoA activation in cells. Cell. Signal. 21, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhattacharyya R., Banerjee J., Khalili K., Wedegaertner P. B. (2009) Differences in Gα12- and Gα13-mediated plasma membrane recruitment of p115-RhoGEF. Cell Signal 21, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Z., Medina F., Liu M. Y., Thomas C., Sprang S. R., Sternweis P. C. (2010) Activated RhoA binds to the pleckstrin homology (PH) domain of PDZ-RhoGEF, a potential site for autoregulation. J. Biol. Chem. 285, 21070–21081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alberts A. S., Geneste O., Treisman R. (1998) Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell 92, 475–487 [DOI] [PubMed] [Google Scholar]
- 18. Frost J. A., Swantek J. L., Stippec S., Yin M. J., Gaynor R., Cobb M. H. (2000) Stimulation of NFκB activity by multiple signaling pathways requires PAK1. J. Biol. Chem. 275, 19693–19699 [DOI] [PubMed] [Google Scholar]
- 19. Wells C. D., Gutowski S., Bollag G., Sternweis P. C. (2001) Identification of potential mechanisms for regulation of p115 RhoGEF through analysis of endogenous and mutant forms of the exchange factor. J. Biol. Chem. 276, 28897–28905 [DOI] [PubMed] [Google Scholar]
- 20. Chen Z., Singer W. D., Danesh S. M., Sternweis P. C., Sprang S. R. (2008) Recognition of the activated states of Gα13 by the rgRGS domain of PDZRhoGEF. Structure 16, 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benard V., Bokoch G. M. (2002) Assay of Cdc42, Rac, and Rho GTPase activation by affinity methods. Methods Enzymol. 345, 349–359 [DOI] [PubMed] [Google Scholar]
- 22. Spencer D. M., Wandless T. J., Schreiber S. L., Crabtree G. R. (1993) Controlling signal transduction with synthetic ligands. Science 262, 1019–1024 [DOI] [PubMed] [Google Scholar]
- 23. Bhattacharyya R., Wedegaertner P. B. (2000) Gα13 requires palmitoylation for plasma membrane localization, Rho-dependent signaling, and promotion of p115-RhoGEF membrane binding. J. Biol. Chem. 275, 14992–14999 [DOI] [PubMed] [Google Scholar]
- 24. Debant A., Serra-Pagès C., Seipel K., O'Brien S., Tang M., Park S. H., Streuli M. (1996) The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate Rac-specific and Rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. U.S.A. 93, 5466–5471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Donald S., Hill K., Lecureuil C., Barnouin R., Krugmann S., John Coadwell W., Andrews S. R., Walker S. A., Hawkins P. T., Stephens L. R., Welch H. C. (2004) P-Rex2, a new guanine-nucleotide exchange factor for Rac. FEBS Lett. 572, 172–176 [DOI] [PubMed] [Google Scholar]
- 26. Rosenfeldt H., Vázquez-Prado J., Gutkind J. S. (2004) P-REX2, a novel PI-3-kinase sensitive Rac exchange factor. FEBS Lett. 572, 167–171 [DOI] [PubMed] [Google Scholar]
- 27. DerMardirossian C., Bokoch G. M. (2005) GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 15, 356–363 [DOI] [PubMed] [Google Scholar]
- 28. Mao J., Yuan H., Xie W., Wu D. (1998) Guanine nucleotide exchange factor GEF115 specifically mediates activation of Rho and serum response factor by the G protein α subunit Gα13. Proc. Natl. Acad. Sci. U.S.A. 95, 12973–12976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamada T., Ohoka Y., Kogo M., Inagaki S. (2005) Physical and functional interactions of the lysophosphatidic acid receptors with PDZ domain-containing Rho guanine nucleotide exchange factors (RhoGEFs). J. Biol. Chem. 280, 19358–19363 [DOI] [PubMed] [Google Scholar]
- 30. Swiercz J. M., Kuner R., Behrens J., Offermanns S. (2002) Plexin-B1 directly interacts with PDZ-RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron 35, 51–63 [DOI] [PubMed] [Google Scholar]
- 31. Hirotani M., Ohoka Y., Yamamoto T., Nirasawa H., Furuyama T., Kogo M., Matsuya T., Inagaki S. (2002) Interaction of plexin-B1 with PDZ domain-containing Rho guanine nucleotide exchange factors. Biochem. Biophys. Res. Commun. 297, 32–37 [DOI] [PubMed] [Google Scholar]
- 32. Medina F., Carter A. M., Dada O., Gutowski S., Hadas J., Chen Z., Sternweis P. C. (2013) Activated RhoA is a positive feedback regulator of the Lbc family of Rho guanine nucleotide exchange factor proteins. J. Biol. Chem. 288, 11325–11333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Banerjee J., Fischer C. C., Wedegaertner P. B. (2009) The amino acid motif L/IIxxFE defines a novel actin-binding sequence in PDZ-RhoGEF. Biochemistry 48, 8032–8043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Longhurst D. M., Watanabe M., Rothstein J. D., Jackson M. (2006) Interaction of PDZRhoGEF with microtubule-associated protein 1 light chains: link between microtubules, actin cytoskeleton, and neuronal polarity. J. Biol. Chem. 281, 12030–12040 [DOI] [PubMed] [Google Scholar]