

Background: The hyaluronan receptor CD44 interacts with the PDGF β-receptor and the TGFβ type I receptor.
Results: CD44, PDGF β-receptor and TGFβ type I receptor affect each other's signaling, stability and function.
Conclusion: Cross-talk between PDGF β-receptor and TGFβ type I receptor occurs in human dermal fibroblasts.
Significance: This study reveals novel modulatory mechanisms of PDGF and TGFβ signaling.
Keywords: CD44, Cell Signaling, Cell Surface Receptor, Cellular Regulation, Transforming Growth Factor β (TGF-β), PDGF
Abstract
Growth factors, such as platelet-derived growth factor BB (PDGF-BB) and transforming growth factor β (TGFβ), are key regulators of cellular functions, including proliferation, migration, and differentiation. Growth factor signaling is modulated by context-dependent cross-talk between different signaling pathways. We demonstrate in this study that PDGF-BB induces phosphorylation of Smad2, a downstream mediator of the canonical TGFβ pathway, in primary dermal fibroblasts. The PDGF-BB-mediated Smad2 phosphorylation was dependent on the kinase activities of both TGFβ type I receptor (TβRI) and PDGF β-receptor (PDGFRβ), and it was prevented by inhibitory antibodies against TGFβ. Inhibition of the activity of the TβRI kinase greatly reduced the PDGF-BB-dependent migration in dermal fibroblasts. Moreover, we demonstrate that the receptors for PDGF-BB and TGFβ interact physically in primary dermal fibroblasts and that stimulation with PDGF-BB induces internalization not only of PDGFRβ but also of TβRI. In addition, silencing of PDGFRβ by siRNA decreased the stability of TβRI and delayed TGFβ-induced signaling. We further show that the hyaluronan receptor CD44 interacts with both PDGFRβ and TβRI. Depletion of CD44 by siRNA increased signaling via PDGFRβ and TβRI by stabilizing the receptor proteins. Our data suggest that cross-talk between PDGFRβ and TβRI occurs in dermal fibroblasts and that CD44 negatively modulates signaling via these receptors.
Introduction
Platelet-derived growth factor (PDGF) isoforms potently stimulate growth, migration, and survival of cells via binding to α- and β-tyrosine kinase receptors (PDGFRα4 and PDGFRβ, respectively) (1). Ligand binding induces dimerization of the receptors followed by activation by autophosphorylation. The phosphorylated PDGF receptors provide docking sites for a wide variety of signaling molecules and adaptor proteins, including the following: Grb2, which forms a complex with the nucleotide exchange factor Sos1, leading to activation of Ras and the ERK MAPK pathway; phospholipase Cγ, which mediates activation of protein kinase C; phosphoinositide-3-kinase (PI3K), which activates the Akt kinase, the tyrosine phosphatase SHP2, the tyrosine kinase Src, and members of the STAT family (2). After ligand binding, the receptor complex is internalized and subsequently degraded, resulting in termination of the signaling (3).
TGFβ transmits its signals via formation of a heterotetrameric complex of type I (TβRI) and type II (TβRII) receptor serine/threonine kinases. Upon binding of TGFβ to TβRII, TβRI is recruited to the complex, where it is phosphorylated and activated by TβRII. Activated TβRI phosphorylates receptor-activated Smads (R-Smads), Smad2 and Smad3, which then bind to the common mediator, Smad4, and after translocation into the nucleus they regulate the expression of certain genes (4). TGFβ also induces activation of non-Smad pathways, including the ERK, JNK, and p38 MAPK pathways, as well as PI3K/Akt (5). Moreover, TβRI undergoes ligand-dependent intramembrane proteolysis, which releases its intracellular domain that, after translocation to the nucleus, drives an invasiveness program (6).
TβRI undergoes constitutive internalization even in the absence of TGFβ, but its down-regulation may be enhanced by ligand binding (7), and enhanced signaling by clathrin-mediated endocytosis has been reported (8, 9). TGFβ inhibits cell proliferation, modulates differentiation, and induces apoptosis of most epithelial, endothelial, and hematopoietic cells (10). Notably, TGFβ has a dual role during cancer development; at early stages of carcinogenesis, it acts as a tumor suppressor, although at later stages it promotes invasiveness and metastasis, e.g. through induction of epithelial-mesenchymal transition (EMT) (10–12).
Ligand access to TGFβ receptors is negatively regulated by traps that sequester the ligand and block its binding to the receptors (4). These ligand traps include the latency-associated polypeptide of the TGFβ precursor, and the small proteoglycan decorin and α2-macroglobulin. The latency-associated polypeptide is bound to the latent TGFβ-binding proteins (LTBP1, 3, 4), and dissociation from this complex is needed for activation of latent TGFβ. Integrins, proteases, thrombospondin 1, heat, and high and low pH values have been demonstrated to activate latent TGFβ (13).
CD44 is a principal receptor for the large glycosaminoglycan hyaluronan; it lacks kinase activity but influences cell behavior by several mechanisms (14, 15). First, the intracellular domain of CD44 interacts with key regulators of the actin cytoskeleton, including ankyrin, members of the ezrin, radixin, moesin (ERM) family of proteins, IQGAP (16, 17), and proteins affecting cell survival, such as the tumor suppressor protein Merlin (18). Second, CD44 can be cleaved in the transmembrane region and the intracellular part translocates to the nucleus where it binds to the cyclin D1 promoter, thereby enhancing cell proliferation (19). Third, CD44 functions as a co-receptor for several growth factor receptors, including receptors for epidermal growth factor (EGF), platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF), as well as TβRI, thereby modulating their signaling (20–26). Finally, CD44 can function as a platform for metalloproteinases, such as matrix metalloproteinase 9, which can activate latent TGFβ (27, 28). In a mouse lung metastasis model of breast carcinoma, a soluble, dominant-negative form of CD44 was shown to compromise metastasis; this could be rescued by distribution of active, but not latent, TGFβ, which is compatible with a role of membrane-bound CD44 in the activation of TGFβ (29).
Cross-talk between PDGF-BB and TGFβ signaling has been described; TGFβ induces expression of PDGF-BB in certain mesenchymal cells (30), and expression of PDGF ligand correlates with metastasis and bad prognosis in breast carcinoma (31). PDGFR expression is induced in breast epithelial cells during TGFβ-induced EMT (32, 33), and PDGF signaling maintains EMT and promotes breast cancer metastasis (34). TGFβ-mediated tumor progression in hepatocytes is also dependent on PDGF signaling (35). In a bioinformatics screen, expression of PDGFRβ was strongly associated with genes involved in TGFβ signaling and EMT in all the cohorts analyzed (33). Because CD44 interacts with both PDGFRβ and ΤβRI, we explored the possibility that CD44 simultaneously interacts with the receptors for PDGF and ΤGFβ, and facilitates cross-talk between them.
MATERIALS AND METHODS
Constructs and Vectors
The pcDNA3-PDGFRβ-HA plasmid (37) and pcDNA3.1 Hygro-CD44H-6myc (38) were generous gifts from Drs. A. Östman (Karolinska Institutet, Stockholm, Sweden) and S. Lammich (Ludwig Maximilian University, Munich, Germany), respectively. The FLAG-tagged TβRI-expressing vector has been described (39, 40). An HA-tagged truncated PDGFRβ mutant expressing only the extracellular and transmembrane parts of the receptor (40) was cloned from the pcDNA3-PDGFRβ vector by inserting a novel XhoI site 36 nucleotides into the intracellular domain using site-directed mutagenesis (Stratagene), cleavage of the truncated protein using EcoRI and XhoI, and insertion into an HA-tagged pcDNA3 vector (HA tag C-terminally located between XhoI and XbaI). As negative controls, empty pcDNA3 vectors (either untagged or tagged with HA, FLAG, or 6myc) were used. Plasmids were amplified using Qiagen® plasmid maxi kit.
Cell Culture
Cos1 (monkey kidney fibroblast-like cells; ATCC CRL-1650), primary human dermal fibroblasts from normal breast tissue (biopsies were taken after approval from patients undergoing breast reduction surgery at the Department of Plastic Surgery of University Hospital, Uppsala, Sweden (17)), and BJ-hTERT (telomerase immortalized human foreskin fibroblasts) cells (41) were routinely cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen), supplemented with 10% fetal bovine serum (FBS; Hyclone) at 37 °C in 5% CO2. Prior to stimulation, cells were starved for 24 h in DMEM supplemented with 0.1% FBS (Cos1 cells and BJ-hTERT) or 2% FBS (dermal fibroblasts) and then treated for 7 min to 24 h with TGFβ1 (PeproTech EC Ltd.; 1–5 ng/ml), PDGF-BB (Creative Biomolecules; 2–20 ng/ml), or hyaluronan (high molecular weight, Q-med; 200 μg/ml). Pretreatment with PDGFRβ inhibitor AG1296 (Calbiochem; 10 μm) or imatinib (Novartis, 5 μm), Src kinase inhibitor SU6656 (Calbiochem 1.5 μm), cycloheximide (Sigma, 20 μm), or TGFβ-neutralizing antibody (R&D Systems; 20 μg/ml) was for 1 h, and pretreatment with TβRI inhibitor GW6604 (American Custom Chemicals Corp.; 16 μm) was for 2 h. Inhibitors remained present during stimulation.
Transient Transfections
Cos1 cells (1 × 106 cells/6-cm culture dish) were transiently transfected with 0.1–2 μg each of vectors encoding PDGFRβ-HA, TβRI-FLAG, TβRII-FLAG, and/or PDGFRβ-ECTM-HA for 48 h using Lipofectamine 2000 (Invitrogen), according to the instructions of the manufacturer.
siRNA against CD44 or PDGFRβ (SmartPool, Dharmacon) was transiently transfected using SilentFECT (Bio-Rad) according to the instructions of the manufacturer. Cells were treated for 72 h with 15 nm PDGFRβ siRNA, 10 nm CD44 siRNA (Cos1 cells), or 25 nm CD44 siRNA (dermal fibroblasts and BJ-hTERT), starved for 24 h, and then stimulated with PDGF-BB or TGFβ.
Protein Extraction and Immunoprecipitation
Cells were washed in ice-cold phosphate-buffered saline (PBS) and then lysed in cell lysis buffer (0.5% Triton X-100, 0.5% sodium deoxycholate, 20 mm Tris, pH 7.4, 150 mm NaCl, 10 mm EDTA), with protease and phosphatase inhibitors (0.5 mg/ml Pefabloc, 10 μm leupeptin, 1 μm pepstatin, 100 KIU/ml aprotinin, and 1 mm sodium orthovanadate). Following centrifugation (10,000 × g, 10 min, 4 °C), the supernatants were either boiled in reducing SDS-sample buffer and analyzed by SDS-PAGE or subjected to immunoprecipitation. For immunoprecipitation, lysates were precleared with 10 μl of protein G-Sepharose beads (GE Healthcare; 50% slurry in PBS) end-over-end at 4 °C for 1 h. Following centrifugation (300 × g, 5 min, 4 °C), supernatants were incubated with 3 μg of primary antibody (polyclonal rabbit TβRI antibody sc-398, polyclonal rabbit HA antibody sc-805, monoclonal mouse c-Myc antibody sc-40, Santa Cruz Biotechnology) or monoclonal mouse FLAG-M2 antibody F-3165 (Sigma) end-over-end at 4 °C overnight. The immune complexes were captured by 25 μl of protein G-Sepharose beads with end-over-end mixing for 1 h at 4 °C. Beads were washed three times in cell lysis buffer, then one time in 0.5 m NaCl, and once more in cell lysis buffer. To elute the captured proteins, 20 μl of reducing SDS-sample buffer was added, and the samples were boiled at 95 °C for 5 min. Beads were removed by centrifugation at 300 × g for 5 min, and the supernatant was analyzed by SDS-PAGE and immunoblotting.
SDS-PAGE and Immunoblotting
Cell lysates were analyzed by SDS-PAGE, and proteins were transferred to Hybond C Extra nitrocellulose membranes (Amersham Biosciences). Membranes were blocked by incubation in 5% milk or 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS), 0.1% Tween 20 and incubated with primary antibodies (diluted in 1% BSA in TBS, 0.1% Tween 20, 0.02% NaN3) as follows: polyclonal rabbit TβRI antiserum or monoclonal mouse PY99 phosphotyrosine antibody (Santa Cruz Biotechnology); monoclonal mouse CD44 antibody (Hermes3; a kind gift from Dr. S. Jalkanen, University of Helsinki, Finland (42)); rabbit polyclonal PDGFRβ (CTβ; homemade (43)); rabbit polyclonal phospho-Smad2 (homemade (44)); rabbit polyclonal Smad2 (Epitomics) antisera; mouse monoclonal PAI1 (BD Biosciences), or GAPDH (Ambion) antibodies. Proteins were visualized by chemiluminescence and exposed to x-ray film. Between each step, the membranes were washed three times for 5 min in TBS, 0.1% Tween 20. To quantify band intensities, scanner and densitometric software (ImageJ) was used.
Proximity Ligation Assay (PLA)
Proximity ligation assay was performed with the Duolink system (Olink Bioscience) according to the instructions from the manufacturer. Briefly, BJ-hTERT fibroblasts were grown in 8-well chamber slides (BD Biosciences), starved, stimulated with 10 ng/ml PDGF-BB for 10 min, 1 ng/ml TGFβ for 1 h, or 200 μg/ml hyaluronan for 2 h, and washed in PBS. After fixation in 3% paraformaldehyde and blocking in Duolink solution, primary antibodies against PDGFRβ (B2; a kind gift from Dr. K. Rubin, Uppsala University, Sweden), TβRI (H100; Santa Cruz Biotechnology), or CD44 (Hermes3) were applied. Addition of secondary antibodies conjugated with PLA probes and ligation was followed by rolling circle amplification. Cells were counterstained with fluorescein isothiocyanate (FITC)-phalloidin (Sigma) and 4′,6-diamidino-2-phenylindole (DAPI) and mounted with Prolong Gold AntiFade (Invitrogen). Images of the cells were taken with a Zeiss Axioplan2 microscope, and signals were quantified with Duolink Image Tool software (Olink Biosciences).
Biotinylation Assay
Human dermal primary fibroblasts were transfected with siRNA against PDGFRβ, starved, and stimulated with 10 ng/ml PDGF-BB or 1 ng/ml TGFβ for up to 1 h. Cells were rinsed on ice with PBS, pH 7.4, and twice with PBS, pH 8.0. Then cell surface proteins were biotinylated by incubation with 0.3 mg/ml EZ-link Sulfo-NHS-SS-biotin (Thermo Scientific) in PBS, pH 8.0, for 20 min on ice at 4 °C. Unbound biotin was quenched by 50 mm Tris, pH 8.0, for 10 min on ice, and cells were lysed. Biotinylated proteins were captured with streptavidin-conjugated magnetic Sepharose beads (GE Healthcare) for 30 min at 4 °C, and biotinylated PDGFRβ and TβRI were visualized by SDS-PAGE and immunoblotting.
Scratch Wound Migration Assay
Confluent human dermal primary fibroblasts were starved for 24 h and wounded by scratching with a pipette tip. Following gentle washing, cells were pretreated with GW6604 (16 μm) for 2 h before cells were incubated with 2 ng/ml PDGF-BB for 24 h. Phase contrast images of wounded areas were taken at time 0 and 24 h after stimulation. Wounded areas covered by cells were quantified using T-scratch software (CSElab), and migration of cells was determined as the part of the wounded area that had been covered by cells.
Statistics
Stimulated and unstimulated samples were assumed to be paired in individual experiments; ratio paired t test was used for normally distributed data, and Wilcoxon matched pair signed rank test was used for skewed data to calculate p values in GraphPad Prism 6. p values below 0.05 were considered to be significant.
RESULTS
PDGFRβ Forms a Noninducible Complex with TβRI and TβRII
We explored the possibility that the receptors for PDGF-BB and TGFβ interact with each other in Cos1 cells overexpressing HA-tagged PDGFRβ and FLAG-tagged TβRI. Immunoprecipitation with an antibody against HA followed by immunoblotting with FLAG antibody revealed a band of the expected size of TβRI. Conversely, immunoprecipitation with a FLAG antibody followed by immunoblotting with an HA antibody revealed a band of the expected size of PDGFRβ. These observations suggest that the two receptors occur in the same complex (Fig. 1A, left panel). To investigate whether the interaction was mediated through the extracellular or intracellular domain of PDGFRβ, co-immunoprecipitation was performed with an HA-tagged PDGFRβ mutant expressing only the extracellular and transmembrane region (PDGFRβ-ECTM-HA). Immunoblotting analysis revealed that TβRI interacts also with this truncated form of PDGFRβ, suggesting that TβRI binds to the extracellular or transmembrane parts of PDGFRβ (Fig. 1B). Using the same approach, but with cells overexpressing FLAG-TβRII together with HA-PDGFRβ, an interaction between PDGFRβ and TβRII was also observed (Fig. 1A, right panel).
FIGURE 1.

PDGFRβ forms a physical complex with TβRI and -II. A and B, Cos1 cells were transiently transfected with PDGFRβ-HA (A and B), TβRI-FLAG (A, left panel), TβRII-FLAG (A, right panel), a truncated PDGFRβ (PDGFRβ-ECTM-HA) expressing only the extracellular and transmembrane parts of the receptor (B), and/or empty vectors. After 48 h, lysates were prepared and subjected to immunoprecipitation (IP) using FLAG or HA antibodies, and proteins were separated by SDS-PAGE. Total cell lysates were run in parallel. Immunoblotting (IB) was performed with FLAG and HA antibodies; β-actin was used as a loading control. C, Cos-1 cells were transiently transfected with PDGFRβ-HA, TβRI-FLAG, TβRII-His, and/or empty vectors. After 24 h, cells were starved for another 24 h and then stimulated with combinations of PDGF-BB (7 min, 10 ng/ml), PDGFRβ inhibitor imatinib (1 h, 5 μm), TGFβ (1 h, 1 ng/ml), TβRI inhibitor GW6604 (2 h, 6 μm), DMSO control (2 h), or starvation medium alone. Cells were lysed, and the lysate was subjected to immunoprecipitation with FLAG antibodies, and proteins were separated by SDS-PAGE. Total cell lysates (TCL) were run in parallel. Proteins were detected by immunoblotting with specific antibodies against the HA and FLAG tags, phosphotyrosine, phospho-Smad2, and β-actin. D, human dermal primary fibroblasts were immunoprecipitated using a TβRI antibody or IgG isotype control, followed by immunoblotting with specific PDGFRβ and TβRI antibodies. Representative data from at least three independent experiments are shown.
To gain insights into how the activation states of the receptors affect the complex formation, we cultured Cos1 cells overexpressing HA-PDGFRβ and FLAG-TβRI in the absence or presence of PDGF-BB or TGFβ, as well as inhibitors of the kinase activities of PDGFRβ (imatinib) or TβRI (GW6604). Treatments with ligands or inhibitors of the receptor kinases had no effect on the complex between PDGFRβ and TβRI, as shown by immunoprecipitation of TβRI with FLAG antibodies, followed by immunoblotting of PDGFRβ with HA antibodies (Fig. 1C). TβRII was co-transfected with the other receptors to ensure TGFβ binding to TβRI; immunoblotting with a phospho-Smad2 antiserum confirmed that Smad2 was phosphorylated in response to TGFβ stimulation and was inhibited in the presence of the TβRI kinase inhibitor. Furthermore, immunoblotting with a phosphotyrosine antiserum confirmed that PDGFRβ was phosphorylated in response to PDGF-BB stimulation and that phosphorylation was suppressed upon treatment with the PDGFRβ inhibitor imatinib (Fig. 1C).
To investigate whether the complex between PDGFRβ and TβRI could also be demonstrated in untransfected cells, we subjected human dermal primary fibroblasts to immunoprecipitation with antibodies against TβRI, followed by immunoblotting with a PDGFRβ antiserum; a band with the expected size of PDGFRβ was observed (Fig. 1D), suggesting that also endogenous TβRI and PDGFRβ form a complex. Taken together, our results show that PDGFRβ and TβRI form a complex, that the interaction is independent on ligand binding and receptor kinase activities, and that the interaction is mediated via the extracellular or transmembrane regions of PDGFRβ.
PDGFRβ Affects Stability and Downstream Signaling of TβRI
Following the observation that PDGFRβ and TβRI form a complex, we investigated whether the receptors influence each other's cell surface residency and signaling. Knockdown of PDGFRβ by siRNA resulted in decreased amounts of TβRI at the cell surface, as shown by labeling cell-surface proteins with biotin and monitoring the amount of biotinylated receptors left on the surface after stimulation with TGFβ (Fig. 2A). Furthermore, the total amount of TβRI in the cell lysate was decreased when PDGFRβ was silenced (Fig. 2A). This effect was not an unspecific off-target effect of the siRNA used, because a second siRNA directed against a single nonoverlapping sequence in PDGFRβ (Ambion) gave the same results (data not shown). In accordance with a role for PDGFRβ in stabilization of TβRI, silencing of PDGFRβ delayed TGFβ-induced phosphorylation of Smad2 (Fig. 2B). Moreover, 1 h of stimulation with PDGF-BB resulted in less TβRI on the cell surface (Fig. 2C).
FIGURE 2.

PDGFRβ affects the stability and downstream signaling of TβRI. A and B, human dermal primary fibroblasts were either transfected with siRNA against PDGFRβ or scrambled control (A and B) or left untransfected (C), then starved, and stimulated (stim) for up to 1 h with TGFβ (1 ng/ml) (A–C) and PDGF-BB (10 ng/ml) (C). A and C, cells were placed on ice to stop membrane trafficking; the cell-surface proteins were then labeled using sulfo-NHS-SS-biotin, and biotin that remained unbound was quenched. Cells were lysed, and biotinylated cell-surface proteins were precipitated using streptavidin-coupled magnetic beads. Proteins were separated by SDS-PAGE and immunoblotted for PDGFRβ and TβRI. The amount of PDGFRβ and TβRI left on the cell surface (relative to GAPDH) was quantified (C, lower panel indicates fold change of mean values with mean ± S.E. of five experiments; asterisk indicates p value <0.05 with ratio paired t test). As control, nonbiotinylated cells were treated the same way. B, cells were lysed and subjected to SDS-PAGE and immunoblotting to determine the phosphorylation status of Smad2. The blots were quantified, and phosphorylated Smad2 relative to GAPDH was determined (B, lower panel). Representative experiments of at least three independent experiments are shown. Asterisk indicates p value <0.05 using Wilcoxon matched pair signed rank test to compare siRNA control with siRNA against PDGFRβ. TCL, total cell lysates.
We then investigated whether TGFβ inversely affects PDGFRβ signaling. There was no effect of TGFβ stimulation on the tyrosine phosphorylation of PDGFRβ when up to 20 ng/ml TGFβ was used for 7 min to 4 h (data not shown). TGFβ treatment did not alter the amount of PDGFRβ on the cell surface (Fig. 2).
PDGF-BB Induces Phosphorylation of Smad2
Interestingly, we found that Smad2 was phosphorylated in human dermal fibroblasts in response not only to TGFβ but also to PDGF-BB (Fig. 3). Smad2 phosphorylation was induced about 5-fold more effectively by TGFβ than by PDGF-BB (Fig. 3A), but with similar kinetics by the two ligands, with a peak after about an hour (data not shown). PDGF-BB induced maximal Smad2-phosphorylation already at 2 ng/ml PDGF-BB, when the PDGF-BB-induced phosphorylation of PDGFRβ was only partial (data not shown).
FIGURE 3.

PDGF-BB induces phosphorylation of Smad2 as well as expression of PAI-1, and the PDGF-BB-induced Smad2 phosphorylation is dependent on the kinase activities of PDGFRβ and TβRI, as well as on Src kinase and TGFβ. A, human dermal primary fibroblasts were starved and stimulated (stim) for 1 h with 1 ng/ml TGFβ or 2 ng/ml PDGF-BB. Cells were then lysed and subjected to SDS-PAGE and immunoblotting (IB), with P-Smad2 and Smad2 antisera, and the amount of phosphorylated Smad2 relative to total Smad2 was quantified (A, lower panel indicates fold change compared with TGFβ of mean values, with S.E., of six experiments; asterisk indicates p value <0.05 with ratio paired t test; three asterisks denote p < 0.01). B, human dermal primary fibroblasts were transfected with either siRNA against PDGFRβ or a scrambled control, starved, and stimulated for 30 min or 1 h with 2 ng/ml PDGF-BB. Cells were then lysed and subjected to SDS-PAGE and immunoblotting, as stated below. C–H, human dermal primary fibroblasts were starved and pretreated for 8 h with 10 μm PDGFRβ kinase inhibitor AG1296 (C), for 2 h with 16 μm TβRI kinase inhibitor GW6604 (D and H), for 1 h with 20 μg/ml TGFβ blocking antibody (E), for 1 h with 1.5 μm Src inhibitor SU6656 (F), or for 2 h with 20 μm cycloheximide (G). Cells were starved and stimulated for indicated periods with 2 ng/ml PDGF-BB (C–H) and 1 ng/ml TGFβ (E and H), then lysed and subjected to SDS-PAGE and immunoblotting with antibodies against phospho-Smad2, Smad2, phosphotyrosine, PDGFRβ (CTβ), phospho-STAT3, PAI-1, and GAPDH (loading control). Representative experiments from at least three independent experiments are shown. CHX, cycloheximide.
The PDGF-BB-induced Smad2 phosphorylation was attenuated by specific silencing of PDGFRβ with siRNA (Fig. 3B) or by treatment with AG1296, an inhibitor of the kinase activity of PDGFRβ (Fig. 3C), indicating that PDGF-BB binding and activation of its receptor is important for Smad2 activation. Furthermore, the PDGF-BB-induced Smad2 phosphorylation was inhibited by treatment with GW6604, an inhibitor of the kinase activity of TβRI (Fig. 3D), indicating that the kinase activity of TβRI is needed for PDGF-BB-induced phosphorylation of Smad2. Moreover, we observed that antibodies that block the binding of TGFβ to TβRI abrogated PDGF-BB-induced Smad2 phosphorylation (Fig. 3E), indicating that PDGF-BB induces Smad2 via induction or activation of TGFβ. Because the kinetics of Smad2 phosphorylation was similar after PDGF-BB or TGFβ stimulation, it is likely that PDGF-BB stimulation leads to activation of latent TGFβ produced by the cells or present in the cell culture medium.
Low molecular weight kinase inhibitors were used to investigate the mechanism through which PDGF-BB induces Smad2 phosphorylation. The Src kinase inhibitor SU6656 reduced PDGF-BB-induced phosphorylation of Smad2 (Fig. 3F). The inhibitor was used at a level that inhibited Src, determined as decreased phosphorylation of the downstream substrate STAT3 (45), but not the autophosphorylation of PDGFRβ. Inhibition of ERK MAPK by the MEK inhibitor U0126 had no effect (data not shown). This observation suggests that PDGF-BB-induced Src activation is involved in the activation of TGFβ. Furthermore, PDGF-BB-induced phosphorylation of Smad2 was present also when new protein synthesis was inhibited with cycloheximide (Fig. 3G), suggesting that activation of latent TGFβ is the most likely mechanism through which PDGF-BB stimulation leads to phosphorylation of Smad2.
TGFβ is well known to induce the expression of plasminogen activator inhibitor type 1 (PAI1) in a Smad-dependent manner. PDGF-BB stimulation also led to induction of PAI-1, and this could be inhibited by the TβRI kinase inhibitor GW6604 (Fig. 3H). This indicates that PDGF-BB stimulation induces signaling via the Smad pathway.
PDGF-BB-induced Migration Is Dependent on the Kinase Activity of TβRI
To investigate whether the cross-talk between PDGF-BB and TGFβ has any functional role for the behavior of the cells, scratch wound migration assays were performed. PDGF-BB potently induced migration of dermal fibroblasts, leading to ∼70% wound closure in 24 h. In the presence of the TβRI kinase inhibitor GW6604, however, the PDGF-BB-stimulated cells showed decreased ability to migrate and only closed about 40% of the wound in 24 h, comparable with unstimulated cells (Fig. 4, presented as fold change).
FIGURE 4.
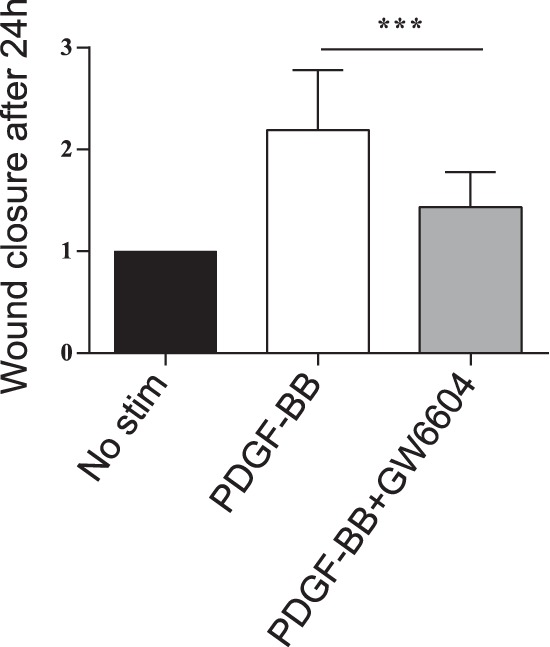
PDGF-BB-induced migration is dependent on TβRI kinase. A confluent monolayer of human dermal primary fibroblasts was starved, wounded by scratching, pretreated for 2 h with 16 μm TβRI kinase inhibitor GW6604 or DMSO, and then stimulated for 24 h with 2 ng/ml PDGF-BB. Phase contrast images of wounded areas at times 0 and 24 h were quantified by T-scratch software; migration of cells was calculated as the part of the wounded area that became covered by cells. At least eight images per condition and experiment were analyzed from three individual experiments (asterisks indicates p < 0.001 using ratio paired t test).
TβRI and CD44 Form a Complex in Fibroblasts
Given the complex between PDGFRβ and TβRI shown in Fig. 1 and the previously published data that CD44 interacts with both PDGFRβ and TβRI (22–24), we explored the possible involvement of CD44 in the cross-talk between PDGFRβ and TβRI. To investigate further the complex formation between CD44 and TβRI, we overexpressed TβRI-HA and CD44–6myc in Cos1 cells and used co-immunoprecipitation to investigate whether they can be captured together. Indeed, antibodies against TβRI or the HA tag were able to pull down CD44 (Fig. 5A), and antibodies against CD44 or the c-Myc tag precipitated TβRI (Fig. 5B), suggesting that the receptors form a complex. Furthermore, PLA was used to explore whether an endogenous complex of TβRI and CD44 is formed in fibroblasts. PLA generates signals (detected as fluorescent dots) only when two target proteins are in close enough proximity to allow rolling circle amplification of PLA probes residing on the antibodies (46, 47). PLA signals were observed in foreskin fibroblasts with antibodies against TβRI and CD44, suggesting interaction between these molecules (Fig. 5C). By omitting one of the primary antibodies, the PLA signals were lost, demonstrating the specificity of the analysis (data not shown). The endogenous complex between TβRI and CD44 was not ligand-inducible as it was seen at similar levels in the absence as well as presence of stimulation with TGFβ, PDGF-BB, and high molecular weight hyaluronan (Fig. 5C). We thus confirm the observation that CD44 and TβRI form a complex (23, 24) and show that this complex is present also in fibroblasts.
FIGURE 5.
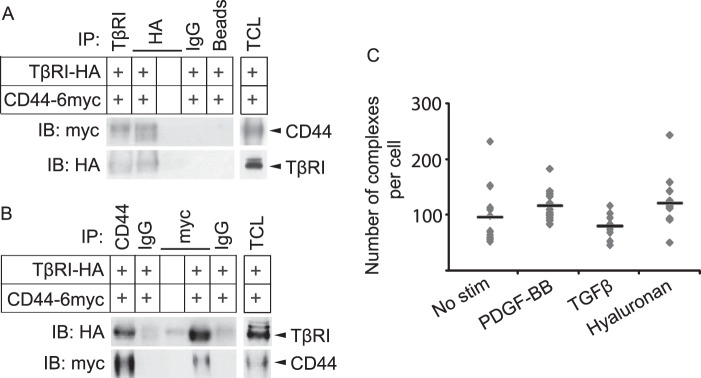
TβRI and CD44 form a ligand noninducible complex. A and B, Cos1 cells were transiently transfected with HA-tagged TβRI, 6-Myc-tagged CD44, or corresponding empty tagged vectors. After 48 h, lysates were immunoprecipitated (IP) with antibodies against TβRI or HA (A), CD44 or Myc (B), corresponding IgG isotype controls, or beads alone; proteins were separated by SDS-PAGE. Total cell lysates (TCL) were run in parallel. Immunoblotting (IB) was performed with antibodies against CD44 and TβRI. C, BJ-hTERT foreskin fibroblasts were grown in 8-well chambers, starved, and stimulated (stim) for 7 min with PDGF-BB (20 ng/ml), 2 h with hyaluronan (200 μg/ml), or 1 h with TGFβ (1 ng/ml). Cells were fixed, and PLA was performed with mouse anti-CD44 and rabbit anti-TβRI antibodies, followed by anti-mouse and anti-rabbit PLA probes conjugated with priming and nonpriming oligonucleotides. F-actin was stained with FITC-conjugated phalloidin. Individual protein-protein interactions were visualized by fluorescence microscope as red dots. PLA signals per cell were quantified using Duolink Image Tool according to the manufacturer's instructions. Average values are indicated in the graph by horizontal lines. A representative experiment out of three is shown.
PDGFRβ, TβRI, and CD44 All Interact Physically
Because we have demonstrated in Figs. 1 and 5 and in a previous publication (22) that PDGFRβ, TβRI, and CD44 interact pairwise, we examined the possibility that these three molecules are present in the same complex at the same time. Immunoprecipitations of overexpressed PDGFRβ-HA, TβRI-FLAG, and CD44-6myc in Cos1 cells in different combinations with either anti-Myc, anti-FLAG, or anti-HA antibodies demonstrated that all three receptors could be specifically pulled down with either of the antibodies, confirming that the three receptors interact pairwise in these cells (Fig. 6A). An approach with two sequential immunoprecipitations further suggested that all three receptors can form a ternary complex (data not shown).
FIGURE 6.

PDGFRβ, TβRI, and CD44 all bind each other, but the complex between PDGFRβ and TβRI is not dependent on CD44. A, Cos1 cells were transiently transfected with PDGFRβ-HA, TβRI-FLAG, and CD44–6myc or correspondingly tagged empty vectors. After 48 h, cell lysates were immunoprecipitated (IP) using FLAG, HA, or Myc antibodies. Proteins were released from beads by boiling in reducing sample buffer and subjected to SDS-PAGE and immunoblotting (IB) with antibodies against CD44, HA, and FLAG or GAPDH as loading control. Total cell lysates (TCL) were analyzed in parallel. B, Cos1 cells were transiently transfected with CD44 siRNA for 24 h and then with TβRI-FLAG and PDGFRβ-HA vectors for another 48 h. Cells were lysed and immunoprecipitated with FLAG antibodies, separated by SDS-PAGE, and detected by immunoblotting for CD44 and the HA and FLAG tags. C, BJ-hTERT foreskin fibroblasts were grown in 8-well chambers and transiently transfected with siRNA against CD44 or a scrambled control. Following starvation, cells were stimulated for 7 min with PDGF-BB (20 ng/ml), 2 h with hyaluronan (200 μg/ml), 1 h with TGFβ (1 ng/ml), or 1 h with 10% FBS. Cells were fixed and PLA was performed with mouse anti-PDGFRβ and rabbit anti-TβRI antibodies, followed by anti-mouse and anti-rabbit PLA probes conjugated with priming and nonpriming oligonucleotides. F-actin was stained with FITC-conjugated phalloidin. Single protein-protein interactions were visualized by fluorescence microscope as red dots. PLA signals per cell were quantified with Duolink Image Tool according to the manufacturer's instructions. Average is indicated in the graph by horizontal lines. D, Cos1 cells were transfected with PDGFRβ-HA and TβRI-FLAG, in combination with varying amounts of CD44-myc (0.1, 0.5, or 2 μg/sample) or empty vector. Cells were lysed, and immunoprecipitated with FLAG antibody and subjected to SDS-PAGE. The samples were then immunoblotted with specific antibodies against the FLAG and HA tags and CD44. Total cell lysates were run in parallel. Representative experiments out of three independent experiments are shown.
To investigate whether the complex between PDGFRβ-HA and TβRI-FLAG is dependent on CD44, we silenced CD44 by siRNA in Cos1 cells overexpressing PDGFRβ-HA and TβRI-FLAG. We then measured the amount of complex between PDGFRβ and TβRI, as seen by PDGFRβ pulled down by FLAG antibody, followed by immunoblotting with HA antibody. The amount of PDGFRβ pulled down was unaffected by the presence or absence of CD44 (Fig. 6B); it thus appears that even though CD44 binds to both PDGFRβ and TβRI, it is not crucial for the interaction between them. To further investigate this interaction, we performed PLA with antibodies against PDGFRβ and TβRI in foreskin fibroblasts with or without silencing of CD44 by siRNA. PLA signals were observed when the specific antibodies were used but not in the controls where one primary antibody was replaced by control IgG or a secondary antibody was omitted. Software quantification of the number of dots revealed no significant differences in the complex between PDGFRβ and TβRI in the presence or absence of CD44 (Fig. 6C). We finally investigated whether formation of the PDGFRβ-TβRI complex was affected by overexpression of CD44; no difference in the amount of complex between PDGFRβ and TβRI was observed upon transfection of Cos1 cells with increasing amounts of CD44–6myc-vector (Fig. 6D).
Depletion of CD44 Increases Signaling via PDGFRβ and TβRI by Stabilization of the Proteins
Because CD44 interacted with PDGFRβ and TβRI (Figs. 5 and 6), and has previously been reported to modulate the signaling pathways of both PDGFRβ (22) and TβRI (23, 24), we investigated the role of CD44 during signaling via PDGFβR and TβRI in fibroblast cultures. Both PDGF-BB-mediated (Fig. 7A) and TGFβ-mediated (Fig. 7B) phosphorylation of Smad2 was enhanced in cells depleted of CD44. There was also a marked increase in total Smad2 protein (Fig. 7, A and B) and TβRI (Fig. 7C) in CD44-depleted cells. Because the mRNA levels of neither Smad2 nor TβRI were increased upon CD44 knockdown (data not shown), the increase in signaling was likely due to increased stability of TβRI and Smad2. Likewise, enhanced expression of PDGFRβ protein (but not PDGFRβ mRNA; data not shown), as well as enhanced and prolonged PDGF-BB-induced phosphorylation of PDGFRβ and ERK1/2 MAPK, was observed upon CD44 silencing (Fig. 7C). CD44 thus has a negative regulatory effect on the signaling via both PDGFRβ and TβRI.
FIGURE 7.
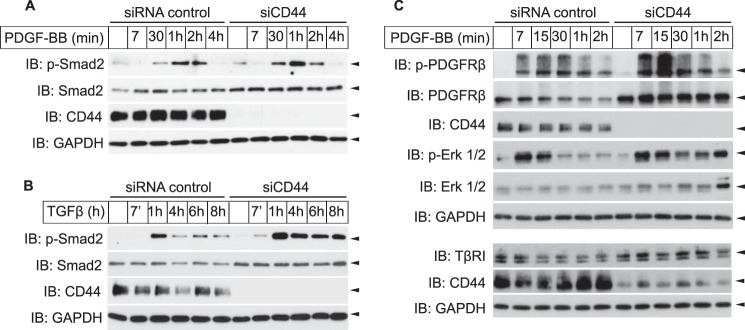
Knockdown of CD44 stabilizes the levels of PDGFRβ and Smad2. Human dermal primary fibroblasts were transfected with siRNA against CD44 or a scrambled control, starved, and stimulated for indicated time periods with either 2 ng/ml PDGF-BB (A), 1 ng/ml TGFβ (B), or 10 ng/ml PDGF-BB (C). Lysates were subjected to SDS-PAGE and immunoblotting with antibodies against phospho-Smad2, Smad2, CD44, phospho-ERK1/2, ERK1/2, and GAPDH (loading control). Representative experiments of three independent experiments are shown.
DISCUSSION
We demonstrate that the receptors for PDGF-BB and TGFβ interact with each other, as well as with CD44, and that CD44 is not essential for the interaction between PDGFRβ and TβRI. Moreover, we demonstrate that the presence of PDGFRβ correlates to increased levels of TβRI and that PDGFRβ-induced migration depends on TβRI activity.
The expressions of PDGF receptors and PDGF isoforms are enhanced during TGFβ-induced EMT (32, 34). In colorectal cancer, PDGFRβ expression correlates with poor prognosis and low overall survival, and an inhibitor of TβRI reduced PDGFRβ expression and PDGF-induced tumor cell invasion (33). This is in accordance with our finding that PDGF-BB-induced migration is inhibited by blocking TβRI kinase activity. The commonly seen induction of PDGFRβ in carcinomas opens up the possibility that the interactions between TβRI, PDGFRβ, and CD44 occur in epithelial cells undergoing EMT. Also in gliomas, TGFβ signaling through Smads activates PDGF-BB expression and secretion, and the effect on proliferation caused by TGFβ depends on PDGFRβ signaling (48). The cross-talk between PDGFRβ and TβRI described in this study may thus have widespread implications both in normal and tumor cells and in cancer metastasis.
PDGF-BB-induced Smad2 Phosphorylation
Interestingly, we found that PDGF-BB promotes signaling through the TGFβ pathway because it induces Smad2 phosphorylation and expression of the TGFβ-responsive gene PAI-1. The PDGF-BB-induced Smad2 phosphorylation was dependent on active PDGF and TGFβ receptor kinases, Src kinase, and active TGFβ. Thus, although the exact mechanism behind PDGF-BB-induced Smad2 phosphorylation remains to be elucidated, our observations support the notion that PDGF-BB, in an Src-dependent manner, promotes activation of latent TGFβ derived either from the FCS in the culture medium or secreted by the cells.
Because the phosphorylation of Smad2 by PDGF-BB occurred also when protein translation was blocked by cycloheximide, neosynthesis of TGFβ was not needed for Smad2 activation. The fast kinetics, similar to that of TGFβ itself, also makes neosynthesis of TGFβ unlikely as a mechanism. Activation of TGFβ is, however, a fast event and could be involved in the PDGF-BB-induced phosphorylation of Smad2. TGFβ can be activated from its latent form by several mechanisms, including proteolysis by e.g. matrix metalloproteinase 9, plasmin, or cathepsin (28, 49). Among these proteases, matrix metalloproteinase 9 is an especially interesting candidate for this study, because it can be localized to the plasma membrane by interaction with CD44, thereby promoting tumor invasion of mammary carcinomas (27, 28). However, a panel of general protease inhibitors did not inhibit PDGF-BB-induced Smad2 phosphorylation in our cells (data not shown); thus, it remains to be elucidated whether PDGF-BB promotes a proteolytic activation of TGFβ.
Several αv-integrins, such as αvβ1, αvβ3, αvβ5, αvβ6, and αvβ8, have also been implicated in the activation of latent TGFβ (50). Of these integrins, αvβ3 is of special interest for this study because there is a tight connection with signaling via PDGF receptors (51–53). PDGFRβ, Src, and TβRII all co-precipitate with integrin αvβ3 in several cell types, including fibroblasts (51, 53). Furthermore, Src association with αvβ3 is augmented by PDGF-BB stimulation, as well as binding to the αvβ3 ligand tenascin-C (53, 54). Interestingly, PDGF-BB regulates recycling of αvβ3 from early endosomes to the plasma membrane (52). A possible mechanism for the induction of Smad2 phosphorylation in our dermal fibroblasts is thus that PDGF-BB stimulation localizes αvβ3 to the plasma membrane, where it activates latent TGFβ. This possibility is currently under investigation.
Another possible explanation for the induction of Smad2 phosphorylation in response to PDGF-BB is that PDGF-BB binds directly to the TGFβ receptors. There is no sequence homology between PDGF and TGFβ, but there is a topological similarity (55). Although this possibility cannot be completely excluded, it is unlikely because PDGFRβ is needed for PDGF-BB-induced Smad2 phosphorylation. Another possible mechanism is that PDGF-BB activates Src, which has been demonstrated to activate TGFβ signaling by tyrosine phosphorylation of TβRII (56, 57). However, Src-induced phosphorylation of TβRII has only been demonstrated to induce non-Smad signaling through the MAPK pathway (57). Moreover, we found that TGFβ antibodies inhibit PDGF-BB-induced Smad2 phosphorylation; thus, it is unlikely that direct tyrosine phosphorylation of TβRII by Src contributes to the PDGF-BB-induced Smad2 phosphorylation in our cells.
Finally, PDGF-BB-induced phosphorylation of Smad2 could be due to inhibition of phosphatases or deubiquitinases that normally would shut off the activation of Smad2, e.g. induced by autocrine TGFβ. It is also possible that PDGFRβ, directly or indirectly, sequesters Smad2 in an unfavorable location. However, these possibilities are unlikely because the Smad2 phosphorylation was blocked by TGFβ antibodies and because we did not observe any co-immunoprecipitation of PDGFRβ and Smad2 (data not shown).
CD44 Exerts a Negative Modulatory Effect on PDGF-BB and TGFβ Signaling
We have previously reported that hyaluronan-activated CD44 mediates recruitment of a tyrosine phosphatase to PDGFRβ in foreskin fibroblasts and thus negatively regulates PDGF-BB signaling (22). Also in dermal fibroblasts, CD44 had a negative effect on signaling through PDGFRβ. In accordance with the high concentration of hyaluronan in skin, our dermal fibroblasts produced high amounts of hyaluronan even during unstimulated and starved conditions. It is thus possible that the lack of hyaluronan binding to CD44, with a concomitant loss of dephosphorylation of PDGFRβ, contributes to the increased PDGF-BB response observed upon knockdown of CD44. However, although such a dephosphorylation mechanism could contribute to the suppressive effect of CD44 on PDGFRβ and TβRI signaling, most of the suppressive effect seems to be due to destabilization of the receptors. We thus confirm the negative modulatory role of CD44 in human dermal fibroblasts and report a novel mechanism of CD44-dependent modulation of growth factor signaling.
In intestinal epithelial cells, the binding of CD44 to hyaluronan has a positive effect on the phosphorylation of PDGFRβ (58). Because this was demonstrated in epithelial cells, although this study and our previous work have used dermal fibroblasts, it is possible that the differences are cell type-dependent. In proximal renal tubular cells, hyaluronan binding to CD44 redistributes TβRI to lipid rafts, thus negatively affecting the signaling of TβRI (36). This effect is blocked by MEK inhibitors, indicating the importance of ERK MAPK signaling. Because this is in accordance with our results that PDGF-BB stimulation removes TβRI from the plasma membrane, it is possible that PDGFRβ and CD44 together regulate the localization and signaling of TβRI.
In summary, we have demonstrated cross-talk between the receptors for TGFβ and PDGF-BB and the adhesion receptor CD44. Such cross-talk could have important functions for several pathologies with dysfunctional regulation of PDGF-BB and TGFβ, such as inflammation and cancer.
Acknowledgment
We thank Aino Ruusala for technical assistance.
This work was supported in part by grants from the Swedish Cancer Society, the Agnes and Mac Rudberg Foundation, and the Gurli and Edward Brunnberg Foundation.
- PDGFR
- PDGF receptor
- TβRI
- TGFβ type I receptor
- EMT
- epithelial-mesenchymal transition
- PLA
- proximity ligation assay.
REFERENCES
- 1. Heldin C. H., Westermark B. (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79, 1283–1316 [DOI] [PubMed] [Google Scholar]
- 2. Andrae J., Gallini R., Betsholtz C. (2008) Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22, 1276–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hellberg C., Schmees C., Karlsson S., Ahgren A., Heldin C. H. (2009) Activation of protein kinase Cα is necessary for sorting the PDGFβ-receptor to Rab4a-dependent recycling. Mol. Biol. Cell 20, 2856–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y. E. (2009) Non-Smad pathways in TGF-β signaling. Cell Res. 19, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shibanaka Y., Hayashi H., Umemura I., Fujisawa Y., Okamoto M., Takai M., Fujita N. (1994) Eclosion hormone-mediated signal transduction in the silkworm abdominal ganglia: Involvement of a cascade from inositol(1,4,5)trisphosphate to cyclic GMP. Biochem. Biophys. Res. Commun. 198, 613–618 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y. G. (2009) Endocytic regulation of TGF-β signaling. Cell Res. 19, 58–70 [DOI] [PubMed] [Google Scholar]
- 8. Tsukazaki T., Chiang T. A., Davison A. F., Attisano L., Wrana J. L. (1998) SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell 95, 779–791 [DOI] [PubMed] [Google Scholar]
- 9. Di Guglielmo G. M., Le Roy C., Goodfellow A. F., Wrana J. L. (2003) Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat. Cell Biol. 5, 410–421 [DOI] [PubMed] [Google Scholar]
- 10. Feng X. H., Derynck R. (2005) Specificity and versatility in tgf-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 11. Heldin C. H., Vanlandewijck M., Moustakas A. (2012) Regulation of EMT by TGFβ in cancer. FEBS Lett. 586, 1959–1970 [DOI] [PubMed] [Google Scholar]
- 12. Moustakas A., Heldin P. (2014) TGFβ and matrix-regulated epithelial to mesenchymal transition. Biochim. Biophys. Acta 10.1016/j.bbagen.2014.02.004 [DOI] [PubMed] [Google Scholar]
- 13. Rifkin D. B. (2005) Latent transforming growth factor-β (TGF-β) binding proteins: orchestrators of TGF-β availability. J. Biol. Chem. 280, 7409–7412 [DOI] [PubMed] [Google Scholar]
- 14. Ponta H., Sherman L., Herrlich P. A. (2003) CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45 [DOI] [PubMed] [Google Scholar]
- 15. Toole B. P. (2009) Hyaluronan-CD44 interactions in cancer: paradoxes and possibilities. Clin. Cancer Res. 15, 7462–7468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bourguignon L. Y. (2008) Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 18, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Skandalis S. S., Kozlova I., Engström U., Hellman U., Heldin P. (2010) Proteomic identification of CD44 interacting proteins. IUBMB Life 62, 833–840 [DOI] [PubMed] [Google Scholar]
- 18. Morrison H., Sherman L. S., Legg J., Banine F., Isacke C., Haipek C. A., Gutmann D. H., Ponta H., Herrlich P. (2001) The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 15, 968–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J. L., Wang M. J., Chen J. Y. (2009) Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J. Cell Biol. 185, 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghatak S., Misra S., Toole B. P. (2005) Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 [DOI] [PubMed] [Google Scholar]
- 21. Kim Y., Lee Y. S., Choe J., Lee H., Kim Y. M., Jeoung D. (2008) CD44-epidermal growth factor receptor interaction mediates hyaluronic acid-promoted cell motility by activating protein kinase C signaling involving Akt, Rac1, Phox, reactive oxygen species, focal adhesion kinase, and MMP-2. J. Biol. Chem. 283, 22513–22528 [DOI] [PubMed] [Google Scholar]
- 22. Li L., Heldin C. H., Heldin P. (2006) Inhibition of platelet-derived growth factor-BB-induced receptor activation and fibroblast migration by hyaluronan activation of CD44. J. Biol. Chem. 281, 26512–26519 [DOI] [PubMed] [Google Scholar]
- 23. Bourguignon L. Y., Singleton P. A., Zhu H., Zhou B. (2002) Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor β receptor I in metastatic breast tumor cells. J. Biol. Chem. 277, 39703–39712 [DOI] [PubMed] [Google Scholar]
- 24. Ito T., Williams J. D., Fraser D., Phillips A. O. (2004) Hyaluronan attenuates transforming growth factor-β1-mediated signaling in renal proximal tubular epithelial cells. Am. J. Pathol. 164, 1979–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orian-Rousseau V., Chen L., Sleeman J. P., Herrlich P., Ponta H. (2002) CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 16, 3074–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wakahara K., Kobayashi H., Yagyu T., Matsuzaki H., Kondo T., Kurita N., Sekino H., Inagaki K., Suzuki M., Kanayama N., Terao T. (2005) Bikunin down-regulates heterodimerization between CD44 and growth factor receptors and subsequently suppresses agonist-mediated signaling. J. Cell. Biochem. 94, 995–1009 [DOI] [PubMed] [Google Scholar]
- 27. Yu Q., Stamenkovic I. (1999) Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 13, 35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu Q., Stamenkovic I. (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–176 [PMC free article] [PubMed] [Google Scholar]
- 29. Yu Q., Stamenkovic I. (2004) Transforming growth factor-β facilitates breast carcinoma metastasis by promoting tumor cell survival. Clin. Exp. Metastasis 21, 235–242 [DOI] [PubMed] [Google Scholar]
- 30. Battegay E. J., Raines E. W., Seifert R. A., Bowen-Pope D. F., Ross R. (1990) TGF-β induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 63, 515–524 [DOI] [PubMed] [Google Scholar]
- 31. Seymour L., Dajee D., Bezwoda W. R. (1993) Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res. Treat. 26, 247–252 [DOI] [PubMed] [Google Scholar]
- 32. Jechlinger M., Grunert S., Tamir I. H., Janda E., Lüdemann S., Waerner T., Seither P., Weith A., Beug H., Kraut N. (2003) Expression profiling of epithelial plasticity in tumor progression. Oncogene 22, 7155–7169 [DOI] [PubMed] [Google Scholar]
- 33. Steller E. J., Raats D. A., Koster J., Rutten B., Govaert K. M., Emmink B. L., Snoeren N., van Hooff S. R., Holstege F. C., Maas C., Borel Rinkes I. H., Kranenburg O. (2013) PDGFRB promotes liver metastasis formation of mesenchymal-like colorectal tumor cells. Neoplasia 15, 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jechlinger M., Sommer A., Moriggl R., Seither P., Kraut N., Capodiecci P., Donovan M., Cordon-Cardo C., Beug H., Grünert S. (2006) Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 116, 1561–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gotzmann J., Fischer A. N., Zojer M., Mikula M., Proell V., Huber H., Jechlinger M., Waerner T., Weith A., Beug H., Mikulits W. (2006) A crucial function of PDGF in TGF-β-mediated cancer progression of hepatocytes. Oncogene 25, 3170–3185 [DOI] [PubMed] [Google Scholar]
- 36. Ito T., Williams J. D., Fraser D. J., Phillips A. O. (2004) Hyaluronan regulates transforming growth factor-β1 receptor compartmentalization. J. Biol. Chem. 279, 25326–25332 [DOI] [PubMed] [Google Scholar]
- 37. Kovalenko M., Denner K., Sandström J., Persson C., Gross S., Jandt E., Vilella R., Böhmer F., Ostman A. (2000) Site-selective dephosphorylation of the platelet-derived growth factor β-receptor by the receptor-like protein-tyrosine phosphatase DEP-1. J. Biol. Chem. 275, 16219–16226 [DOI] [PubMed] [Google Scholar]
- 38. Lammich S., Okochi M., Takeda M., Kaether C., Capell A., Zimmer A. K., Edbauer D., Walter J., Steiner H., Haass C. (2002) Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Aβ-like peptide. J. Biol. Chem. 277, 44754–44759 [DOI] [PubMed] [Google Scholar]
- 39. Yao D., Ehrlich M., Henis Y. I., Leof E. B. (2002) Transforming growth factor-β receptors interact with AP2 by direct binding to β2 subunit. Mol. Biol. Cell 13, 4001–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Franzén P., ten Dijke P., Ichijo H., Yamashita H., Schulz P., Heldin C. H., Miyazono K. (1993) Cloning of a TGF β type I receptor that forms a heteromeric complex with the TGF β type II receptor. Cell 75, 681–692 [DOI] [PubMed] [Google Scholar]
- 41. Hahn W. C., Counter C. M., Lundberg A. S., Beijersbergen R. L., Brooks M. W., Weinberg R. A. (1999) Creation of human tumour cells with defined genetic elements. Nature 400, 464–468 [DOI] [PubMed] [Google Scholar]
- 42. Jalkanen S., Bargatze R. F., de los Toyos J., Butcher E. C. (1987) Lymphocyte recognition of high endothelium: antibodies to distinct epitopes of an 85–95 kDa glycoprotein antigen differentially inhibit lymphocyte binding to lymph node, mucosal, or synovial endothelial cells. J. Cell Biol. 105, 983–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karlsson S., Kowanetz K., Sandin A., Persson C., Ostman A., Heldin C. H., Hellberg C. (2006) Loss of T-cell protein tyrosine phosphatase induces recycling of the platelet-derived growth factor (PDGF) β-receptor but not the PDGFα-receptor. Mol. Biol. Cell 17, 4846–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piek E., Moustakas A., Kurisaki A., Heldin C. H., ten Dijke P. (1999) TGF-(β) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 112, 4557–4568 [DOI] [PubMed] [Google Scholar]
- 45. Turkson J., Bowman T., Garcia R., Caldenhoven E., De Groot R. P., Jove R. (1998) Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 18, 2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jarvius M., Paulsson J., Weibrecht I., Leuchowius K. J., Andersson A.-C., Wählby C., Gullberg M., Botling J., Sjöblom T., Markova B., Ostman A., Landegren U., Söderberg O. (2007) In situ detection of phosphorylated PDGF receptor β using a generalized proximity ligation method. Mol. Cell Proteomics 6, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 47. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 48. Bruna A., Darken R. S., Rojo F., Ocaña A., Peñuelas S., Arias A., Paris R., Tortosa A., Mora J., Baselga J., Seoane J. (2007) High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160 [DOI] [PubMed] [Google Scholar]
- 49. Lyons R. M., Keski-Oja J., Moses H. L. (1988) Proteolytic activation of latent transforming growth factor-β from fibroblast-conditioned medium. J. Cell Biol. 106, 1659–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ludbrook S. B., Barry S. T., Delves C. J., Horgan C. M. (2003) The integrin αvβ3 is a receptor for the latency-associated peptides of transforming growth factors β1 and β3. Biochem. J. 369, 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Woodard A. S., García-Cardeña G., Leong M., Madri J. A., Sessa W. C., Languino L. R. (1998) The synergistic activity of αvβ3 integrin and PDGF receptor increases cell migration. J. Cell Sci. 111, 469–478 [DOI] [PubMed] [Google Scholar]
- 52. Roberts M., Barry S., Woods A., van der Sluijs P., Norman J. (2001) PDGF-regulated rab4-dependent recycling of αvβ3 integrin from early endosomes is necessary for cell adhesion and spreading. Curr. Biol. 11, 1392–1402 [DOI] [PubMed] [Google Scholar]
- 53. Ishigaki T., Imanaka-Yoshida K., Shimojo N., Matsushima S., Taki W., Yoshida T. (2011) Tenascin-C enhances cross-talk signaling of integrin αvβ3/PDGFR-β complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J. Cell Physiol. 226, 2617–2624 [DOI] [PubMed] [Google Scholar]
- 54. Ding Q., Stewart J., Jr., Olman M. A., Klobe M. R., Gladson C. L. (2003) The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J. Biol. Chem. 278, 39882–39891 [DOI] [PubMed] [Google Scholar]
- 55. Murray-Rust J., McDonald N. Q., Blundell T. L., Hosang M., Oefner C., Winkler F., Bradshaw R. A. (1993) Topological similarities in TGF-β2, PDGF-BB, and NGF define a superfamily of polypeptide growth factors. Structure 1, 153–159 [DOI] [PubMed] [Google Scholar]
- 56. Galliher A. J., Schiemann W. P. (2007) Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 67, 3752–3758 [DOI] [PubMed] [Google Scholar]
- 57. Galliher A. J., Schiemann W. P. (2006) β3 integrin and Src facilitate transforming growth factor-β-mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 8, R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Misra S., Toole B. P., Ghatak S. (2006) Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J. Biol. Chem. 281, 34936–34941 [DOI] [PubMed] [Google Scholar]