

Background: The IRF6 transcription factor is critical for epithelial barrier function; however, a role for IRF6 in signaling by Toll-like receptors has not been addressed.
Results: The IRAK1-mediated activation of IRF6 promotes TLR2-dependent CCL5 chemokine gene expression in epithelial cells.
Conclusion: IRF6 differentially regulates TLR2 inflammatory responses in epithelial cells.
Significance: Our results reveal an additional immune-related function for IRF6.
Keywords: Chemokine, Epithelial Cell, Inflammation, Toll-like Receptor (TLR), Transcription Factor, Interferon Regulatory Factor (IRF), Interleukin Receptor-associated Kinase
Abstract
Epidermal and mucosal epithelial cells are integral to host defense. They not only act as a physical barrier but also utilize pattern recognition receptors, such as the Toll-like receptors (TLRs), to detect and respond to pathogens. Members of the interferon regulatory factor (IRF) family of transcription factors are key components of TLR signaling as they impart specificity to downstream responses. Although IRF6 is a critical regulator of epithelial cell proliferation and differentiation, its role in TLR signaling has not previously been addressed. We show here that IRF6 is activated by IRAK1 as well as by MyD88 but not by TRIF or TBK1. Co-immunoprecipitation experiments further demonstrated that IRF6 can interact with IRAK1. Gene silencing in epithelial cells along with gene promoter reporter assays showed that IRAK1 mediates TLR2-inducible CCL5 gene expression at least in part by promoting IRF6 activation. Conversely, IRAK1 regulated CXCL8 gene expression independently of IRF6, thus identifying a molecular mechanism by which TLR2 signaling differentially regulates the expression of specific chemokines in epithelial cells. Bioinformatics analysis and mutagenesis-based experiments identified Ser-413 and Ser-424 as key regulatory sites in IRF6. Phosphomimetic mutation of these residues resulted in greatly enhanced IRF6 dimerization and trans-activator function. Collectively, our findings suggest that, in addition to its importance for epithelial barrier function, IRF6 also contributes to host defense by providing specificity to the regulation of inflammatory chemokine expression by TLR2 in epithelial cells.
Introduction
Epidermal and mucosal epithelial cells are positioned at the interface between the host and the environment and thus play pivotal roles in host defense. Although one of their primary functions is to provide a physical barrier to pathogen invasion (1, 2), they also express pattern recognition receptors (e.g. Toll-like receptors), thereby enabling them to actively participate in host defense by functioning as immune sentinels (2–5). For example, the production of inflammatory cytokines, chemokines, and type I interferons (IFNs)5 by epithelial cells serves to recruit and activate different leukocyte cell populations. However, the dysregulated production of such factors can lead to pathologic states of chronic inflammation, as occurs in inflammatory bowel disease, psoriasis, and chronic periodontitis (6–8). Chronic mucosal inflammation is also an important factor in some cancers (e.g. gastric cancer) (9).
Toll-like receptors (TLRs) are fundamental to the detection and subsequent host response to pathogens (10, 11). The specificity of TLR signaling is determined, in part, by differential use of adapter proteins, for example, MyD88 and TRIF. Accordingly, TLR signaling can be broadly divided into the MyD88-dependent and TRIF-dependent pathways. The former pathway is used by all TLRs with the exception of TLR3 and the latter only by TLR3 and TLR4 (10, 11). The MyD88-dependent pathway employs the protein kinase IL-1 receptor-associated kinase-1 (IRAK1) to trigger the activation of various transcription factors, including NF-κB and members of the interferon regulatory factor family (e.g. IRF5 and IRF7), resulting in inflammatory gene expression. The induction of inflammatory gene expression by the TRIF-dependent pathway occurs in response to the activation of IRF3 by TBK1 along with TAK1-mediated NF-κB activation (10–12).
In addition to regulating type I IFN (e.g. IFNβ) gene expression, IRFs also play important roles in regulating the expression of other inflammatory proteins, including chemokines (e.g. CCL5, CXCL8, and CXCL10) (12–17). Consequently, the differential regulation of inflammatory gene expression by IRFs allows them to appropriately shape the immune response by imparting signaling specificity to TLRs. The activation of IRF3 by TLR3 and TLR4 signaling, for instance, enables these receptors to initiate TLR3- and TLR4-specific gene expression responses (18, 19). Analogously, IRF5 and IRF7 induce specific gene expression responses downstream of MyD88-dependent TLRs (20–22).
Prior studies have largely focused on the roles of IRFs in mediating TLR-elicited responses in leukocyte cell populations (e.g. macrophages and dendritic cells). Although IRF3 has also been shown to be important for TRIF-dependent TLR responses in epithelial cells (23–25), the IRF(s) that mediates MyD88-dependent responses in these cells is less clear. In contrast to other IRFs, IRF6 expression appears for the most part to be limited to epithelial cells (26–30), where at least one function is to regulate cell proliferation and differentiation (27–29, 31, 32). Notably, IRF6-deficient mice exhibit defective epidermal barrier function due to impaired keratinocyte differentiation; they also die perinatally (28, 29). Given the key roles of other IRFs in orchestrating the TLR-elicited inflammatory responses of leukocytes (12, 13), we investigated whether IRF6 was similarly important for specific TLR responses in epithelial cells. Our findings here link IRF6 to IRAK1-dependent TLR2 responses (e.g. CCL5 expression) in epithelial cells, thus revealing an additional immune-related function for IRF6 in these cells.
EXPERIMENTAL PROCEDURES
Reagents
Cell culture medium and supplements, fetal calf serum (FCS), SuperScript III reverse transcriptase, random primers, deoxyribonucleotide triphosphates, TaqMan Universal Master Mix II, Lipofectamine RNAiMAX, precast 10% NuPAGE gels, mouse anti-V5 antibodies (HRP-conjugated and unconjugated), Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody, and ProLong® Gold Antifade reagent (containing 4′,6-diamidino-2-phenylindole) were from Invitrogen. Recombinant human CSF-1 was generously provided by Chiron. Restriction enzymes were from New England Biolabs, whereas Pfu DNA polymerase, Passive Lysis Buffer, and the Dual-GloTM luciferase assay system were from Promega. PCR primers were synthesized by GeneWorks. FuGENE 6TM and CompleteTM protease inhibitors were supplied by Roche Applied Science. The ON-TARGETplus IRF6 and IRAK1 siRNAs as well as the control non-targeting siRNA were from Dharmacon. TLR ligands were from InvivoGen. The rabbit anti-IRF6 antibody and HRP-conjugated mouse anti-HA antibody were from Cell Signaling, and the mouse anti-HSP90 antibody was from BD Biosciences.
Cell Culture
OKF6/TERT-2 cells (33) were cultured in keratinocyte serum-free medium supplemented with 25 μg/ml bovine pituitary extract, 2 ng/ml EGF, 0.4 mm CaCl2, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-1TM. AGS and MKN28 cells were cultured in RPMI 1640 supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-1TM. HT-29 cells were cultured in McCoy's 5A medium supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-1TM. HEK293T cells were cultured in Dulbecco's-modified Eagle's medium supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-TM. Human monocytes were purified from buffy coats (Red Cross Blood Bank, Australia) using a RosetteSep antibody mixture (Stem Cell Technologies) followed by Ficoll-Paque density gradient centrifugation. The monocytes were cultured overnight in RPMI 1640 supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm GlutaMax-TM. Monocyte-derived macrophages were generated by culturing monocytes in the presence of CSF-1 (2500 units/ml) for 6–8 days (34). All cells were cultured at 37 °C in a humidified atmosphere of 5% CO2.
Real-time PCR
Total RNA was purified using an RNAeasy Mini kit (Qiagen). RNA (1 μg) was reverse-transcribed into cDNA using random primers and SuperScript III reverse transcriptase. Real-time PCR was performed (in triplicate) using an Applied Biosystems Prism 7900HT sequence detection system and pre-developed TaqMan assays (Invitrogen) for the following genes: CCL5 (Hs00174575_m1), CXCL8 (Hs00174103_m1), IFNβ (Hs02621180_s1), IRAK1 (Hs01018347_m1), IRAK2 (Hs00176394_m1), IRF3 (Hs00155574_m1), IRF5 (Hs00158114_m1), IRF6 (Hs00196213_m1), IRF7 (Hs00185375_m1), TLR1 (Hs00413978_m1), TLR2 (Hs00152932_m1), TLR3 (Hs01551078_m1), TLR4 (Hs01060206_m1), TLR5 (Hs00152825_m1), TLR6 (Hs00271977_s1), TLR7 (Hs01933259_s1), TLR8 (Hs00607866_mH), and TLR9 (Hs00370913_s1). Messenger RNA levels, relative to those of the endogenous control gene, HPRT, were calculated using the ΔCt (cycle threshold) method.
Expression Vectors and Site-directed Mutagenesis
The human IRF6 expression vector, pCMV6-XL6-IRF6, was purchased from Origene. The expression vector, pEF-HA-IRF6 (expresses an N-terminal HA-tagged version of IRF6), was created by PCR using the primer pair F1 (5′-CG ACG CGT GCC CTC CAC CCC CGC AGA GTC CGG CTA AAG-3′) and R1 (5′-CG ACG CGT TTA CTG GGG AGG CAG GGC AGG GGG CAG TTG-3′) and pCMV6-XL6-IRF6 as the template. The PCR product was digested with MluI and cloned into the expression vector, pEF-HA. The expression vector, pEF-V5-IRF6 (expresses an N-terminal V5-tagged version of IRF6), was created by excising the cDNA insert from pEF-HA-IRF6 with MluI and cloning it into pEF-V5. The expression vector, pEF-HA-IRF6 S413A (Ser-413 replaced by alanine), was created by overlapping PCR using the primer pairs F1 and R2 (5′-ACT GCC ACT ATC AAA GGC TCG TGT GAA ATC ACC-3′) and F2 (5′-GGT GAT TTC ACA CGA GCC TTT GAT AGT GGC AGT-3′) and R1 and pCMV6-XL6-IRF6 as the template. The expression vector, pEF-HA-IRF6 S424A (Ser-424 replaced by alanine), was created using the primer pairs F1 and R3 (5′-CTT GAT GTC TGG GGT TGC GAT CTG CAG GCG GAC-3′) and pairs F3 (5′-GTC CGC CTG CAG ATC GCA ACC CCA GAC ATC AAG-3′) and R1 and pCMV6-XL6-IRF6 as the template. The expression vector, pEF-HA-IRF6 S413A/S424A (Ser-413 and Ser-424 replaced by alanine), was created using the primer pairs F1 and R3 and pairs F3 and R1 and pEF-HA-IRF6 S413A as the template. The expression vector, pEF-HA-IRF6 S413E (Ser-413 replaced by glutamic acid), was created using the primer pairs F1 and R4 (5′-ACT GCC ACT ATC AAA TTC TCG TGT GAA ATC ACC-3′) and pairs F4 (5′-GGT GAT TTC ACA CGA GAA TTT GAT AGT GGC AGT-3′) and R1 and pCMV6-XL6-IRF6 as the template. The expression vector, pEF-HA-IRF6 S424E (Ser-424 replaced by glutamic acid), was created using the primer pairs F1 and R5 (5′-CTT GAT GTC TGG GGT TTC GAT CTG CAG GCG GAC-3′) and pairs F5 (5′-GTC CGC CTG CAG ATC GAA ACC CCA GAC ATC AAG-3′) and R1 and pCMV6-XL6-IRF6 as the template. The expression vector, pEF-HA-IRF6 S413E/S424E (Ser-413 and Ser-424 replaced by glutamic acid), was created using the primer pairs F1 and R5 and pairs F5 and R1 and pEF-HA-IRF6 S413E as the template. The expression vector, pEF-V5-IRF6 S413E/S424E (expresses an N-terminal V5-tagged version of IRF6 S413E/S424E), was created by excising the cDNA insert from pEF-HA-IRF6 S413E/S424E with MluI and cloning it into pEF-V5. The IRAK1 expression vectors, pEF-V5-IRAK1 and pEF-V5-IRAK1 K239A (express V5-tagged versions of wild type and kinase-dead IRAK1, respectively), are as previously described (35, 36). The IRAK2 expression vector was a generous gift from Dr. Luke O'Neill (Trinity College, Ireland), whereas the MyD88, TBK1, and TRIF expression vectors were kindly provided by Dr. Ashley Mansell (Monash Institute of Medical Research, Australia).
Gene Promoter Reporter Assays
HEK293T cells were seeded in 12-well tissue culture plates at a density of 3 × 105 cells per well and transfected (in duplicate) the next day using FuGENE 6TM transfection reagent. The total amount of plasmid in each transfection was kept constant by using empty vector where required. The cells were lysed 24 h post-transfection with Passive Lysis Buffer and assayed for firefly and Renilla luciferase activities using the Dual-Glo™ luciferase assay system. Renilla luciferase activity was used to normalize transfection efficiencies. The luciferase-based CCL5 (37), CXCL8 (38), and IFNβ and IFNα4 gene promoter reporter plasmids were generously provided by Drs. Paula Pitha (Johns Hopkins University), Allan Brasier (University of Texas Medical Branch), and Ashley Mansell (Monash Institute of Medical Research, Australia), respectively. The Renilla luciferase reporter plasmid, pRL-TK, was from Promega.
Silencing of IRAK1 and IRF6 Expression
A reverse-transfection protocol was used for siRNA transfections. Briefly, the IRAK1- and IRF6-targeting siRNAs as well as the control non-targeting siRNA were diluted to 120 nm with 100 μl of Opti-MEM I-reduced serum medium (Invitrogen). The diluted siRNA was mixed with 100 μl of Opti-MEM I-reduced serum medium containing 1.0 μl of Lipofectamine RNAiMAX transfection reagent and incubated at room temperature for 20 min. OKF6/TERT-2 cells (2 × 105 cells in 1.0 ml of antibiotic-free growth medium) were plated into 12-well plates, and the transfection mixture then added. The medium was replaced 24 h later, and the cells were analyzed or stimulated 48 h post-transfection.
Cell Lysis and Western Blotting
Cells were washed twice with ice-cold PBS and then lysed (20 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 10% glycerol, 10 mm β-glycerol phosphate, 10 mm NaF, and CompleteTM protease inhibitors) on ice for 60 min. The lysates were clarified by centrifugation (13,000 × g for 10 min at 4 °C), and the protein concentrations measured using a protein assay kit (Bio-Rad). Cell lysates were subjected to electrophoresis on 10% NuPAGE gels followed by Western blotting according to standard protocols. Immunoreactive bands were visualized using ECL reagents (Millipore) and a LAS-3000 Imager (Fujifilm) or by exposure to x-ray film (Fujifilm). Films were scanned using a GS-800 Calibrated Imaging Densitometer (Bio-Rad).
In Vitro Protein Dephosphorylation Assay
Transfected HEK293T cells were lysed as above, except phosphatase inhibitors were omitted from the lysis buffer. Protein dephosphorylation was carried out in 50-μl reactions consisting of 50 μg of cell protein and 10 units of calf intestinal phosphatase. The reactions were incubated at 37 °C for 30 min followed by SDS-PAGE and Western blotting.
Immunoprecipitation Assays
V5-tagged IRAK1 and IRF6 were immunoprecipitated from transfected HEK293T cells by incubating 1 mg of cell lysate (in 1 ml lysis buffer) with 1 μg of anti-V5 antibody and 20 μl of Protein G-Sepharose for 4 h at 4 °C with constant mixing. The beads were washed 4 times with lysis buffer and then subjected to electrophoresis on 10% NuPAGE gels followed by Western blotting.
Immunofluorescent Staining and Confocal Microscopy
HEK293T cells, which had been seeded onto glass coverslips prior to transfection, were fixed with 4% paraformaldehyde (30 min), solubilized with 0.1% Triton X-100 (5 min), and then blocked in 5% goat serum (60 min), all at room temperature. The cells were subsequently stained overnight (at 4 °C) with a rabbit anti-IRF6 antibody. After three washes with PBS, the cells were probed with an Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody for 60 min (at room temperature). The cells were washed 3 times with PBS and finally mounted on glass microscope slides using ProLong® Gold Antifade reagent containing DAPI. Mounted coverslips were allowed to cure for 24 h in the dark before images of the cells being acquired on an Olympus FV1000 scanning confocal microscope. No anti-IRF6 staining was apparent in HEK293T cells transfected with empty vector only.
Statistical Analysis
Data combined from three or more independent experiments are given as the means ± S.E. Statistical analyses were performed using GraphPad Prism software Version 6.01 (GraphPad Software, La Jolla, CA). Differences between two groups were evaluated using Student's t test. For multiple comparisons, statistical analysis was performed using a one-way analysis of variance and then the Sidak's or Dunnett's test as a post-hoc test. A p value <0.05 was considered to be statistically significant.
RESULTS
Activation of IRF6 Trans-activator Function by IRAK1
Prior studies have demonstrated that IRAK1 mediates, either directly or indirectly, IRF5 and IRF7 activation in response to MyD88-dependent TLR signaling in myeloid cells (39–42). Given that phylogenetic analysis had also revealed that IRF6 was most closely related to IRF5 (43), we investigated the ability of IRAK1 to activate IRF6. IRF6 activity was assayed using an IFNβ gene promoter reporter plasmid, which was activated in a concentration-dependent manner by IRF6 (Fig. 1A). IRF6 trans-activator function was strongly potentiated in a kinase-dependent manner by IRAK1 (Fig. 1B). Its trans-activator function was also potentiated by IRAK2 (Fig. 1C) and MyD88 (Fig. 1D) but not by TBK1 (Fig. 1E) or TRIF (data not shown). As an indicator of some promoter specificity, IRF6, either alone or when co-expressed with IRAK1, did not trans-activate the promoter from the IFNα4 gene in this assay (Fig. 1F), although IRF3 robustly synergized with TBK1 in activating the IFNα4 reporter.
FIGURE 1.

Activation of IRF6 by IRAK1. A, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid in the presence of increasing amounts of an expression vector encoding HA-tagged IRF6. Gene reporter activity was measured 24 h post-transfection and is shown as the -fold increase over cells transfected with empty vector. Data from n = 3 experiments are presented as the mean ± S.E. B–E, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid together with expression vectors encoding the indicated proteins. Gene reporter activity was measured 24 h post-transfection and is shown as the -fold increase over cells transfected with empty vector. Data from n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01). F, HEK293T cells were transfected with an IFNα4 gene promoter reporter plasmid together with expression vectors encoding the indicated proteins. Gene reporter activity was measured 24 h post-transfection and is shown as the -fold increase over cells transfected with empty vector. Data from n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01). G, lysates of HEK293T cells transiently expressing HA-IRF6 were incubated in the absence and presence of calf intestinal phosphatase (CIP) followed by Western blotting with an anti-HA antibody. Data are representative of n = 4 experiments. H, lysates of HEK293T cells transiently expressing the indicated proteins were subjected to Western blotting. The IRF6 doublet represents phosphorylated (upper band, denoted by p) and non-phosphorylated (lower band, denoted by np) forms. I, V5-tagged wild type and kinase-dead IRAK1 were immunoprecipitated from the cell lysates in H followed by Western blotting. Data are representative of n = 3 experiments.
The ability of IRF6 to interact with IRAK1 was also investigated through co-immunoprecipitation assays. An IRF6 doublet was detected when lysates of transfected HEK293T cells were subjected to Western blotting (Fig. 1, G and H), with the upper band reported to arise from the cell cycle-dependent phosphorylation of IRF6 (27, 31). That the upper band is due to IRF6 phosphorylation was also confirmed here by an in vitro dephosphorylation assay (Fig. 1G). As previously shown (44), several electrophoretically distinct forms of wild type, but not kinase-dead (KD), IRAK1 were also apparent (Fig. 1H, bottom panel). IRF6 interacted with the IRAK1 KD mutant, either directly or as part of a complex (Fig. 1I). In contrast, the co-immunoprecipitation of IRF6 with wild type IRAK1 was often largely undetectable, suggesting that IRF6 interacted only transiently with active IRAK1. IRF6 did not co-immunoprecipitate with ectopically expressed MyD88 (data not shown). Collectively, these data suggest that IRF6 may mediate IRAK1-dependent inflammatory gene expression in response to MyD88-dependent TLR activation in epithelial cells.
IRF6 and TLR2-inducible Chemokine Gene Expression in Epithelial Cells
To investigate whether IRF6 may mediate IFNβ or chemokine gene expression in response to IRAK1-dependent TLR signaling in epithelial cells, we screened several human cell lines for IRF6 expression. IRF6 mRNA was detected in the non-transformed oral epithelial cell line, OKF6/TERT-2 (hereafter referred to as OKF6 cells) (Fig. 2A); lower levels were also detected in the gastric cancer epithelial cell lines, AGS and MKN28, and the intestinal cancer epithelial cell line, HT-29 (Fig. 2A). Consistent with the concept that, by comparison to other IRF family members, IRF6 is preferentially expressed in epithelial cells (26–30), IRF6 mRNA was not detected in human blood monocytes or in monocyte-derived macrophages (MDM) (Fig. 2A). IRF6 protein expression in OKF6 cells and its absence in HEK293T cells was confirmed by Western blotting (Fig. 2A, inset); additionally, phosphorylated and non-phosphorylated forms of IRF6 were detected. IRF6 expression levels in OKF6 cells were also compared with those of IRF3, IRF5, and IRF7 (Fig. 2B). IRF6 mRNA levels were 5–10-fold higher than those of IRF3 and up to 100-fold higher than those of IRF5 and IRF7. A similar analysis revealed that IRAK1 was expressed at levels up to 10-fold higher than IRAK2 (Fig. 2C).
FIGURE 2.

Expression analysis of specific TLR signaling proteins and chemokine gene induction in epithelial cells. A, IRF6 mRNA expression levels in the indicated cells were measured by real-time PCR. Expression levels are relative to those of the endogenous control gene, HPRT. Data from n = 3 experiments are presented as the mean ± S.E. (ND = not detected). Inset, lysates of HEK293T and OKF6 cells were subjected to Western blotting with an anti-IRF6 antibody. Phosphorylated (upper band, denoted by p) and non-phosphorylated (lower band, denoted by np) forms of IRF6 are indicated. B–D, the expression levels of IRF3, IRF5, IRF6, and IRF7 (B), IRAK1 and IRAK2 (C), and TLR1–9 (D) in OKF6 cells were measured by real-time PCR. Expression levels are relative to those of HPRT. Data from n = 3 experiments are presented as the mean ± S.E. E and F, OKF6 cells were stimulated with FSL-1 (100 ng/ml) for the times indicated. CCL5 (E) and CXCL8 (F) mRNA levels were measured by real-time PCR and are shown as the -fold increase relative to mock-treated cells. Data from n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01).
OKF6 cells expressed mRNA for TLR1–9, except TLR8 (Fig. 2D). TLR2, which recognizes structural components of both Gram-positive and Gram-negative bacteria, including lipopeptides and lipoteichoic acid (10, 11), was most abundantly expressed, at least at the mRNA level. Therefore, we tested the ability of the lipopeptide, FSL-1, to induce IFNβ and chemokine gene expression. IFNβ gene expression was not induced by FSL-1 (data not shown), nor was it induced by ligands for TLR7 (imiquimod) or TLR9 (CpG DNA) (data not shown). However, FSL-1 did robustly up-regulate CCL5 (Fig. 2E) and CXCL8 gene expression (Fig. 2F). FSL-1-inducible CCL5 gene expression was markedly more sustained than CXCL8 expression.
IRAK1 Is Required for TLR2-inducible CCL5 and CXCL8 Gene Expression in Epithelial Cells
The importance of IRAK1 for the induction of CCL5 and CXCL8 gene expression by TLR2 signaling in OKF6 cells was examined by gene silencing. Transfection of the cells with IRAK1-targeting siRNAs markedly reduced IRAK1 expression (Fig. 3A) without affecting IRAK2 (data not shown). Knock-down of IRAK1 expression greatly inhibited (>70%) FSL-1-inducible CCL5 gene expression (Fig. 3B). Similarly, the induction of CXCL8 gene expression was also strongly inhibited by IRAK1 knock-down (Fig. 3C). These data, therefore, establish IRAK1 as an essential mediator of TLR2-inducible CCL5 and CXCL8 gene expression in OKF6 cells.
FIGURE 3.

IRAK1-dependent regulation of CCL5 and CXCL8 gene expression by TLR2. A–C, OKF6 cells were transfected with a control non-targeting (−) siRNA or two separate IRAK1-targeting (+) siRNAs. Forty-eight hours post-transfection IRAK1 mRNA levels were measured by real-time PCR (A). IRAK1 mRNA levels in cells transfected with the control siRNA were arbitrarily given a value of 100%. B–C, the cells were stimulated with FSL-1 (100 ng/ml) for the times indicated. CCL5 (B) and CXCL8 (C) mRNA levels were measured by real-time PCR and are shown as -fold increase relative to mock-treated cells. Data from n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01; * = p < 0.05).
IRF6 Is Required for TLR2-inducible CCL5 Gene Expression in Epithelial Cells
We next used the same approach to determine the importance of IRF6 in the FSL-1-mediated induction of CCL5 and CXCL8 gene expression. Transfection of an IRF6-targeting siRNA reduced levels of IRF6 mRNA (Fig. 4A) and protein (Fig. 4B) in OKF6 cells without significantly affecting IRF3, IRF5, and IRF7 mRNA levels (data not shown). Knock-down of IRF6 expression inhibited FSL-1-inducible CCL5 gene expression by >50% (Fig. 4C). In contrast, the induction of CXCL8 gene expression by FSL-1 was not significantly affected by IRF6 knock-down (Fig. 4D).
FIGURE 4.

IRF6-dependent regulation of CCL5 gene expression by TLR2. A–D, OKF6 cells were transfected with a control non-targeting (−) or IRF6-targeting (+) siRNA. Forty-eight hours post-transfection IRF6 mRNA levels were measured by real-time PCR (A). IRF6 mRNA levels in cells transfected with the control siRNA were arbitrarily given a value of 100%. B, cell lysates were subjected to Western blotting with anti-IRF6 and anti-HSP90 (loading control) antibodies. C and D, the cells were stimulated with FSL-1 (100 ng/ml) for the times indicated. CCL5 (C) and CXCL8 (D) mRNA levels were measured by real-time PCR and are shown as -fold increase relative to mock-treated cells. Data from n = 3 experiments are presented as the mean ± S.E. (* = p < 0.05). E and F, HEK293T cells were transfected with a CCL5 (E) or CXCL8 (F) gene promoter reporter plasmid together with expression vectors encoding the indicated proteins. Gene reporter activity was measured 24 h post-transfection and is shown as -fold increase over cells transfected with empty vector. Data from n = 4 experiments are presented as the mean ± S.E. (** = p < 0.01).
Co-immunoprecipitation experiments failed to detect an FSL-1-inducible interaction between endogenous IRF6 and IRAK1 in OKF6 cells (data not shown). This was not unexpected given that the data in Fig. 1I suggest IRF6 and IRAK1 interact only transiently. Therefore, the potential for IRF6 to mediate the IRAK1-dependent regulation of CCL5 gene expression was further assessed through gene promoter reporter assays. Ectopic IRF6 expression was not sufficient to activate the CCL5 reporter (Fig. 4E). However, IRAK1 co-expression resulted in the robust trans-activation of the reporter by IRF6; IRAK1 only weakly activated the reporter in the absence of IRF6 (Fig. 4E). In contrast, IRAK1 alone strongly activated the CXCL8 reporter, and this effect was only modestly increased by the co-expression of IRF6 (Fig. 4F). Taken together, these findings are consistent with IRF6 mediating a sub-set of TLR2/IRAK1-dependent inflammatory responses in epithelial cells.
Identification of Ser-413 and Ser-424 as Putative Regulatory Phosphorylation Sites in IRF6
Little is known about how the trans-activator function of IRF6 is regulated. The inducible-phosphorylation of specific serine residues has been shown to play a critical role in regulating the trans-activator functions of other IRFs (14, 45–48). To identify potential regulatory phosphorylation sites in IRF6, the regions of IRF3, IRF5, and IRF7 that contain such sites (e.g. Ser-437 and Ser-446 in IRF5) were aligned with the corresponding region in IRF6 (Fig. 5A). Ser-413 and Ser-424 in the C-terminal domain of IRF6 were conserved in IRF3, IRF5, and IRF7 and were also conserved in the mouse, rat, bovine, chicken, and zebrafish IRF6 orthologs (data not shown).
FIGURE 5.

Identification of regulatory phosphorylation sites in IRF6. A, schematic representation of IRF6 (DBD = DNA binding domain; IAD = IRF association domain; CTD = C-terminal domain). Partial amino acid sequence alignment of human IRF6 with IRF3, IRF5, and IRF7 is shown. Serine residues that are conserved between all four IRFs are shown in bold. B, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid together with expression vectors encoding the indicated proteins (IRF6 S413A, Ser-413 replaced by alanine; IRF6 S424A, Ser-424 replaced by alanine; IRF6 2SA, Ser-413 and Ser-424 replaced by alanine). Gene reporter activity was measured 24 h post-transfection and is shown as -fold increase over cells transfected with empty vector. Data from n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01). C, HEK293T cells transiently expressing the indicated proteins were lysed 24 h post-transfection. Wild type and kinase-dead IRAK1 were immunoprecipitated (IP) from the lysates using anti-V5 antibodies followed by Western blotting with anti-HA and anti-V5 antibodies. Phosphorylated (upper band, denoted by p) and non-phosphorylated (lower band, denoted by np) forms of IRF6 are indicated. Cell lysates were subjected to Western blotting with an anti-HA antibody. Data are representative of n = 3 experiments.
The importance of Ser-413 and Ser-424 for the regulation of IRF6 trans-activator function by IRAK1 was investigated by mutating, either individually or together, these residues to alanine. As shown in Fig. 5B, the mutation of Ser-413 to alanine partially abrogated IRAK1-mediated IRF6 activation. By contrast, IRF6 S424A and the IRF6 mutant in which both Ser-413 and Ser-424 had been replaced by alanine, IRF6 S413A/S424A, were completely inactive in this assay (Fig. 5B). Co-immunoprecipitation experiments revealed that the IRF6 serine-to-alanine mutants, IRF6 S413A and IRF6 S413A/S424A, still interacted with IRAK1 at comparable levels to wild type IRF6 (Fig. 5C). Therefore, although Ser-413 and Ser-424 do not appear to be important for the interaction of IRF6 with IRAK1, they are critical for optimal IRAK1-mediated IRF6 activation.
As mentioned earlier, cell cycle-dependent phosphorylation of IRF6 results in the detection of an IRF6 doublet in Western blots of cell lysates (27, 31). Significantly, an IRF6 doublet was still detected after the mutation of Ser-413 and/or Ser-424 to alanine (Fig. 5C, bottom panel), thus suggesting these residues are distinct from the site(s) that is phosphorylated in response to growth factors.
Mutation of Ser-413 and Ser-424 to Glutamic Acid Results in Constitutive IRF6 Activation
The involvement of Ser-413 and Ser-424 in the regulation of IRF6 trans-activator function was next addressed by mutating, either individually or together, the two residues to the phosphomimetic, glutamic acid. Mutation of Ser-413, Ser-424, or both Ser-413 and Ser-424 to glutamic acid increased IRF6 activity toward the IFNβ gene promoter reporter 15-, 5-, and 40-fold, respectively (Fig. 6A). The ability of IRAK1 to increase further the activities of these IRF6 gain-of-function mutants was also tested. The activity of the IRF6 S413E mutant was enhanced by IRAK1 co-expression, whereas those of IRF6 S424E and IRF6 S413E/S424E were not (Fig. 6B). Notably, in the absence of IRAK1, the activity exhibited by the IRF6 S413E mutant was 3-fold higher than that when wild type IRF6 was co-expressed with IRAK1, whereas the activity of the IRF6 S424E mutant was comparable to that exhibited by wild type IRF6 after co-expression with IRAK1 (Fig. 6B).
FIGURE 6.
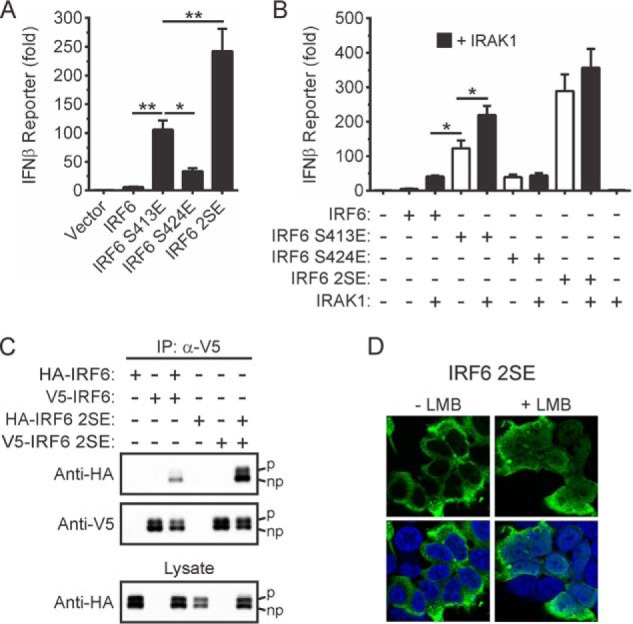
Phosphomimetic-mediated activation of IRF6. A and B, HEK293T cells were transfected with an IFNβ gene promoter reporter plasmid together with expression vectors encoding the indicated proteins (IRF6 S413E, Ser-413 replaced by glutamic acid; IRF6 S424E, Ser-424 replaced by glutamic acid; IRF6 2SE, Ser-413 and Ser-424 replaced by glutamic acid). Gene reporter activity was measured 24 h post-transfection and is shown as -fold increase over cells transfected with empty vector. Data from at least n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01, * = p < 0.05). C, HEK293T cells transiently expressing the indicated proteins were lysed 24 h post-transfection. V5-IRF6 and V5-IRF6 S413E/S424E were immunoprecipitated (IP) using anti-V5 antibodies followed by Western blotting with anti-HA and anti-V5 antibodies. Phosphorylated (upper band, denoted by p) and non-phosphorylated (lower band, denoted by np) forms of IRF6 are indicated. Cell lysates were subjected to Western blotting with an anti-HA antibody. Data are representative of n = 3 experiments. D, HEK293T cells were transiently transfected with an expression vector encoding IRF6 S413E/S424E. Twenty-four hours later the cells were treated with 20 ng/ml leptomycin B (+LMB) for 60 min, or left untreated (−LMB). The cells were stained with an anti-IRF6 antibody (green staining); nuclei were stained with DAPI (blue staining). Data are representative of n = 5 experiments.
As was the case for wild type IRF6 and the IRF6 S413A/S424A mutant (Fig. 5C), an IRF6 S413E/S424E doublet was detected by Western blotting (Fig. 6C, bottom panel). The more slowly migrating form of the IRF6 S413E/S424E mutant was absent when the cell lysates were treated with calf intestinal phosphatase before electrophoresis (data not shown).
Given that dimerization is a key step in the activation and nuclear translocation of IRFs (12, 13, 45, 47, 49), the effects of mutating Ser-413 and Ser-424 to glutamic acid on IRF6 dimerization were assessed. V5- and HA-tagged versions of wild type IRF6 and IRF6 S413E/S424E were ectopically expressed, and dimerization was then evaluated through co-immunoprecipitation experiments. The IRF6 S413E/S424E mutant exhibited increased levels of spontaneous dimerization (Fig. 6C), consistent with its greatly enhanced trans-activator function (Fig. 6A). Despite its strong trans-activator function, consistent nuclear localization of IRF6 S413E/S424E was not observed (Fig. 6D). To test whether this may have been due to its rapid nuclear export, the cells were treated with the nuclear export inhibitor, leptomycin B (50). Treatment with the inhibitor resulted in partial IRF6 S413E/S424E nuclear localization (Fig. 6D). This not only suggests that IRF6 nuclear translocation is highly dynamic, but it may also involve mechanisms in addition to IRF6 phosphorylation.
Ser-413 and Ser-424 Are Important for the IRAK1-mediated Trans-activation of the CCL5 Promoter by IRF6
The creation of loss-of-function and gain-of-function IRF6 mutants (i.e. IRF6 S413A/S424A and IRF6 S413E/S424E, respectively) allowed us to further investigate the mechanism underlying the IRAK1-mediated regulation of CCL5 gene expression by IRF6. Ser-413 and Ser-424 were required for the synergistic activation of the CCL5 gene promoter reporter by IRF6 and IRAK1 (Fig. 7A). The IRF6 S413E/S424E mutant only weakly activated the CCL5 reporter in the absence of co-expressed IRAK1 (Fig. 7B), which differed from its robust and IRAK1-independent activation of the IFNβ gene promoter reporter (Fig. 6B). Therefore, phosphorylation of Ser-413 and Ser-424 is likely to be necessary, but not sufficient, for the optimal induction of CCL5 gene expression by IRF6.
FIGURE 7.

Effects of mutating Ser-413 and Ser-424 on the trans-activation of the CCL5 promoter by IRF6. A and B, HEK293T cells were transfected with a CCL5 gene promoter reporter plasmid together with expression vectors encoding the indicated proteins (IRF6 2SA, Ser-413 and Ser-424 replaced by alanine; IRF6 2SE, Ser-413 and Ser-424 replaced by glutamic acid). Gene reporter activity was measured 24 h post-transfection and is shown as -fold increase over cells transfected with empty vector. Data from n = 3 experiments are presented as the mean ± S.E. (** = p < 0.01).
DISCUSSION
IRF6 is a critical regulator of epithelial cell proliferation and differentiation (27–29, 31, 32) and is important for epithelial barrier function (28, 29). Data presented herein also positions IRF6 as a key regulator of TLR2-inducible chemokine (CCL5) gene expression in epithelial cells. Our findings thus reveal new insights into the molecular mechanisms through which epithelial cells actively contribute to the host immune response to pathogens.
A role for IRF6 in MyD88-dependent TLR signaling was suggested by our finding that IRAK1, as well as MyD88, but not TBK1 or TRIF, strongly potentiated IRF6 trans-activator function. Furthermore, the ability of IRF6 to form a complex, either directly or indirectly, with IRAK1 suggests that the entry point for IRF6 in the MyD88-dependent TLR signaling pathway is likely to be at the level of IRAK1. IRF5 and IRF7, which similarly operate downstream of IRAK1 in the MyD88-dependent pathway, are key mediators of the inflammatory responses elicited by TLRs in myeloid cells (20–22). Not only does IRF6 expression appear to be restricted to epithelial cells (26–30), but more widespread analysis of gene expression data via the BioGPS portal suggested that IRF6 is the only epithelial-restricted IRF family member. As such, IRF6 may uniquely regulate specific MyD88-dependent TLR responses in epithelial cells.
Accordingly, we investigated a possible functional relationship between IRF6 and IRAK1 in the context of TLR2 signaling in human oral epithelial cells (e.g. OKF6 cells). TLR2 is a key mediator of host defense as it recognizes conserved molecular patterns associated with Gram-positive and Gram-negative bacteria (11, 51–53). Significantly, although IRF6 and IRAK1 were important for the up-regulation of chemokine gene expression by TLR2, their contributions were chemokine-specific, at least for CCL5 and CXCL8. IRAK1 was required for the induction of both CCL5 and CXCL8 gene expression. In contrast, whereas TLR2-inducible CCL5 gene expression was also heavily reliant on IRF6, CXCL8 expression was induced independently of IRF6. In line with these findings, the CCL5 promoter was robustly activated by IRF6 and IRAK1 co-expression, whereas IRAK1 strongly activated the CXCL8 promoter independently of IRF6. Hence, IRAK1 likely mediates TLR2-inducible CCL5 gene expression, at least in part, by promoting the activation of IRF6. The differing effect of IRF6 in regulating CCL5 versus CXCL8 gene expression provides a molecular mechanism for the differential regulation of specific chemokines downstream of TLR2, thereby enabling distinct responses to be elicited. CXCL8 is a key regulator of neutrophil trafficking (54), whereas CCL5 mediates the trafficking and homing of various leukocyte cell populations, including T-cells, macrophages, and eosinophils (55). Our findings, therefore, place IRF6 in the signaling framework as a likely mediator of CCL5-dependent leukocyte recruitment to sites of epithelial infection.
IRF6 also activated the IFNβ promoter, suggesting that this gene may also be an IRF6 target. Although we found that MyD88-selective TLR agonists, including those of TLR7 and TLR9, did not induce IFNβ expression in OKF6 cells, it remains possible that this response does occur in other epithelial cell types as well as in vivo. Similarly, a role for IRF6 in regulating IFNα gene expression cannot be excluded.
The inducible phosphorylation of serine residues has been shown to play a critical role in regulating the activation of several IRFs (14, 45–48, 56). A two-step, sequential phosphorylation model has been proposed for IRF3 activation (57). In this model, the phosphorylation of serine residues in “site 2” (e.g. Ser-396 in IRF3) is needed to alleviate autoinhibition and allow interaction with the co-activator, CBP/p300; it also facilitates the phosphorylation of residues in “site 1” (e.g. Ser-386 in IRF3), which is required for IRF dimerization (57). We show here that Ser-413 and Ser-424, which correspond, respectively, to Ser-386 and Ser-396 in IRF3, are important for IRF6 function. Although not formally demonstrated, the marked increase in IRF6 trans-activator function by the phosphomimetic mutation of Ser-413 and Ser-424 to glutamic acid argues that they are regulatory phosphorylation sites. Thus, the trans-activator function of IRF6 may likewise be regulated by a two-step, sequential phosphorylation mechanism in which Ser-424 serves as a “gatekeeper” phosphorylation site.
Importantly, our data strongly suggest that Ser-424 is an IRAK1-regulated phosphorylation site, although it has not yet been established if IRAK1 directly regulates the phosphorylation of Ser-424 or does so by activating another kinase. Although Ser-413 is likewise required for maximal IRAK1-mediated IRF6 activation, IRAK1 did not further enhance the trans-activator function of the IRF6 S424E mutant. Nonetheless, phosphorylation of Ser-413 would be expected to be important for maximal IRF6 activity in view of the activating effect of its phosphomimetic mutation on IRF6 trans-activator function. The activation of IRF7 by TLR7/9 signaling appears to be mediated by both IRAK1 and IKKα (inhibitor of nuclear factor κB kinase α) (39–41, 58). By analogy, IRF6 may be regulated in a similar manner, with IRAK1 specifically regulating, either directly or indirectly, the phosphorylation of Ser-424 and another kinase then phosphorylating Ser-413.
The phosphomimetic mutation of Ser-424 and Ser-413 strongly enhanced IRF6 dimerization and trans-activator function; however, it did not result in demonstrable nuclear localization. Nuclear translocation of the IRF6 S413E/S424E mutant was apparent after treatment with the nuclear export inhibitor, leptomycin B. This would suggest that IRF6 is subject to tightly regulated cytoplasmic-nuclear shuttling. Consequently, additional posttranslational modifications (e.g. ubiquitination) may also be necessary for sustained IRF6 nuclear translocation.
The cooperation between IRFs and NF-κB is necessary for the optimal expression of some inflammatory genes (59–61). This may also be the case for the regulation of CCL5 gene expression by IRF6. Despite the IRF6 S413E/S424E mutant being highly active, it was largely ineffective in trans-activating the CCL5 promoter; the co-expression of IRAK1, which can also activate endogenous NF-κB, was necessary for robust activation of the CCL5 promoter. Virus-induced CCL5 gene expression in alveolar epithelial cells was reported to require both IRF and NF-κB activity (15). Therefore, optimal induction of CCL5 gene expression by IRAK1-mediated TLR2 signaling in epithelial cells may require functional cooperation between IRF6 and NF-κB.
In addition to its trans-activator function, phosphorylation also appears to regulate the cell cycle-dependent degradation of IRF6 (31). However, neither Ser-413 nor Ser-424 is likely to be the phosphorylation site(s) that targets IRF6 for proteasomal degradation as their mutation did not affect IRF6 protein levels. The phosphorylation-mediated regulation of IRF6 trans-activator function and degradation are thus likely to be regulated in a stimulus-dependent manner by distinct signaling pathways.
In summary, this study has uncovered a non-redundant role for IRF6 in differentially regulating TLR2-elicited chemokine responses in epithelial cells. Given its role in also regulating epithelial cell proliferation and differentiation, IRF6 may act as a pivotal nexus for distinct signaling pathways and regulate both the barrier and inflammatory functions of epithelial cells.
This work was supported by National Health and Medical Research Council Project Grant 628769 and the Oral Health CRC.
- IFN
- interferon
- TLR
- Toll-like receptor
- IRAK1
- IL-1 receptor-associated kinase-1
- IRF
- interferon regulatory factor.
REFERENCES
- 1. Proksch E., Brandner J. M., Jensen J. M. (2008) The skin: an indispensable barrier. Exp. Dermatol 17, 1063–1072 [DOI] [PubMed] [Google Scholar]
- 2. Goto Y., Kiyono H. (2012) Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol. Rev. 245, 147–163 [DOI] [PubMed] [Google Scholar]
- 3. Miller L. S. (2008) Toll-like receptors in skin. Adv. Dermatol. 24, 71–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saenz S. A., Taylor B. C., Artis D. (2008) Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 226, 172–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abreu M. T. (2010) Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 10, 131–144 [DOI] [PubMed] [Google Scholar]
- 6. Gribar S. C., Anand R. J., Sodhi C. P., Hackam D. J. (2008) The role of epithelial Toll-like receptor signaling in the pathogenesis of intestinal inflammation. J. Leukoc. Biol. 83, 493–498 [DOI] [PubMed] [Google Scholar]
- 7. Nickoloff B. J., Xin H., Nestle F. O., Qin J. Z. (2007) The cytokine and chemokine network in psoriasis. Clin. Dermatol. 25, 568–573 [DOI] [PubMed] [Google Scholar]
- 8. Darveau R. P. (2010) Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 8, 481–490 [DOI] [PubMed] [Google Scholar]
- 9. Fukata M., Abreu M. T. (2009) Pathogen recognition receptors, cancer and inflammation in the gut. Curr. Opin. Pharmacol. 9, 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akira S., Uematsu S., Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 11. Kawai T., Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 [DOI] [PubMed] [Google Scholar]
- 12. Honda K., Taniguchi T. (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 [DOI] [PubMed] [Google Scholar]
- 13. Tamura T., Yanai H., Savitsky D., Taniguchi T. (2008) The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 [DOI] [PubMed] [Google Scholar]
- 14. Barnes B. J., Kellum M. J., Field A. E., Pitha P. M. (2002) Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell. Biol. 22, 5721–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casola A., Garofalo R. P., Haeberle H., Elliott T. F., Lin R., Jamaluddin M., Brasier A. R. (2001) Multiple cis regulatory elements control RANTES promoter activity in alveolar epithelial cells infected with respiratory syncytial virus. J. Virol. 75, 6428–6439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fitzgerald K. A., Rowe D. C., Barnes B. J., Caffrey D. R., Visintin A., Latz E., Monks B., Pitha P. M., Golenbock D. T. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin R., Heylbroeck C., Genin P., Pitha P. M., Hiscott J. (1999) Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell. Biol. 19, 959–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doyle S., Vaidya S., O'Connell R., Dadgostar H., Dempsey P., Wu T., Rao G., Sun R., Haberland M., Modlin R., Cheng G. (2002) IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263 [DOI] [PubMed] [Google Scholar]
- 19. Sato M., Suemori H., Hata N., Asagiri M., Ogasawara K., Nakao K., Nakaya T., Katsuki M., Noguchi S., Tanaka N., Taniguchi T. (2000) Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13, 539–548 [DOI] [PubMed] [Google Scholar]
- 20. Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777 [DOI] [PubMed] [Google Scholar]
- 21. Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., Udalova I. A. (2011) IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 12, 231–238 [DOI] [PubMed] [Google Scholar]
- 22. Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S., Honda K., Ohba Y., Mak T. W., Taniguchi T. (2005) Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249 [DOI] [PubMed] [Google Scholar]
- 23. Kato A., Favoreto S., Jr., Avila P. C., Schleimer R. P. (2007) TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 179, 1080–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsukura S., Kokubu F., Kurokawa M., Kawaguchi M., Ieki K., Kuga H., Odaka M., Suzuki S., Watanabe S., Takeuchi H., Kasama T., Adachi M. (2006) Synthetic double-stranded RNA induces multiple genes related to inflammation through Toll-like receptor 3 depending on NF-κB and/or IRF-3 in airway epithelial cells. Clin. Exp. Allergy 36, 1049–1062 [DOI] [PubMed] [Google Scholar]
- 25. Wang Q., Nagarkar D. R., Bowman E. R., Schneider D., Gosangi B., Lei J., Zhao Y., McHenry C. L., Burgens R. V., Miller D. J., Sajjan U., Hershenson M. B. (2009) Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 183, 6989–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kondo S., Schutte B. C., Richardson R. J., Bjork B. C., Knight A. S., Watanabe Y., Howard E., de Lima R. L., Daack-Hirsch S., Sander A., McDonald-McGinn D. M., Zackai E. H., Lammer E. J., Aylsworth A. S., Ardinger H. H., Lidral A. C., Pober B. R., Moreno L., Arcos-Burgos M., Valencia C., Houdayer C., Bahuau M., Moretti-Ferreira D., Richieri-Costa A., Dixon M. J., Murray J. C. (2002) Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bailey C. M., Khalkhali-Ellis Z., Kondo S., Margaryan N. V., Seftor R. E., Wheaton W. W., Amir S., Pins M. R., Schutte B. C., Hendrix M. J. (2005) Mammary serine protease inhibitor (Maspin) binds directly to interferon regulatory factor 6: identification of a novel serpin partnership. J. Biol. Chem. 280, 34210–34217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ingraham C. R., Kinoshita A., Kondo S., Yang B., Sajan S., Trout K. J., Malik M. I., Dunnwald M., Goudy S. L., Lovett M., Murray J. C., Schutte B. C. (2006) Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richardson R. J., Dixon J., Malhotra S., Hardman M. J., Knowles L., Boot-Handford R. P., Shore P., Whitmarsh A., Dixon M. J. (2006) Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 38, 1329–1334 [DOI] [PubMed] [Google Scholar]
- 30. Knight A. S., Schutte B. C., Jiang R., Dixon M. J. (2006) Developmental expression analysis of the mouse and chick orthologues of IRF6: the gene mutated in Van der Woude syndrome. Dev. Dyn. 235, 1441–1447 [DOI] [PubMed] [Google Scholar]
- 31. Bailey C. M., Abbott D. E., Margaryan N. V., Khalkhali-Ellis Z., Hendrix M. J. (2008) Interferon regulatory factor 6 promotes cell cycle arrest and is regulated by the proteasome in a cell cycle-dependent manner. Mol. Cell. Biol. 28, 2235–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Biggs L. C., Rhea L., Schutte B. C., Dunnwald M. (2012) Interferon regulatory factor 6 is necessary, but not sufficient, for keratinocyte differentiation. J. Invest. Dermatol. 132, 50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dickson M. A., Hahn W. C., Ino Y., Ronfard V., Wu J. Y., Weinberg R. A., Louis D. N., Li F. P., Rheinwald J. G. (2000) Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20, 1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Way K. J., Dinh H., Keene M. R., White K. E., Clanchy F. I., Lusby P., Roiniotis J., Cook A. D., Cassady A. I., Curtis D. J., Hamilton J. A. (2009) The generation and properties of human macrophage populations from hemopoietic stem cells. J. Leukoc. Biol. 85, 766–778 [DOI] [PubMed] [Google Scholar]
- 35. De Nardo D., Nguyen T., Hamilton J. A., Scholz G. M. (2009) Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerance in macrophages. Cell. Signal. 21, 246–252 [DOI] [PubMed] [Google Scholar]
- 36. Nguyen T., De Nardo D., Masendycz P., Hamilton J. A., Scholz G. M. (2009) Regulation of IRAK-1 activation by its C-terminal domain. Cell. Signal. 21, 719–726 [DOI] [PubMed] [Google Scholar]
- 37. Lin R., Génin P., Mamane Y., Hiscott J. (2000) Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of α/β interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 20, 6342–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garofalo R., Sabry M., Jamaluddin M., Yu R. K., Casola A., Ogra P. L., Brasier A. R. (1996) Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 70, 8773–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uematsu S., Sato S., Yamamoto M., Hirotani T., Kato H., Takeshita F., Matsuda M., Coban C., Ishii K. J., Kawai T., Takeuchi O., Akira S. (2005) Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR) 7- and TLR9-mediated interferon-α induction. J. Exp. Med. 201, 915–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schoenemeyer A., Barnes B. J., Mancl M. E., Latz E., Goutagny N., Pitha P. M., Fitzgerald K. A., Golenbock D. T. (2005) The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J. Biol. Chem. 280, 17005–17012 [DOI] [PubMed] [Google Scholar]
- 41. Saitoh T., Satoh T., Yamamoto N., Uematsu S., Takeuchi O., Kawai T., Akira S. (2011) Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 34, 352–363 [DOI] [PubMed] [Google Scholar]
- 42. Tun-Kyi A., Finn G., Greenwood A., Nowak M., Lee T. H., Asara J. M., Tsokos G. C., Fitzgerald K., Israel E., Li X., Exley M., Nicholson L. K., Lu K. P. (2011) Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat. Immunol. 12, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taniguchi T., Ogasawara K., Takaoka A., Tanaka N. (2001) IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655 [DOI] [PubMed] [Google Scholar]
- 44. De Nardo D., Masendycz P., Ho S., Cross M., Fleetwood A. J., Reynolds E. C., Hamilton J. A., Scholz G. M. (2005) A central role for the Hsp90.Cdc37 molecular chaperone module in interleukin-1 receptor-associated kinase-dependent signaling by toll-like receptors. J. Biol. Chem. 280, 9813–9822 [DOI] [PubMed] [Google Scholar]
- 45. Lin R., Heylbroeck C., Pitha P. M., Hiscott J. (1998) Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin R., Mamane Y., Hiscott J. (2000) Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275, 34320–34327 [DOI] [PubMed] [Google Scholar]
- 47. Marié I., Smith E., Prakash A., Levy D. E. (2000) Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol. Cell. Biol. 20, 8803–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharma S., tenOever B. R., Grandvaux N., Zhou G. P., Lin R., Hiscott J. (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 [DOI] [PubMed] [Google Scholar]
- 49. Barnes B. J., Field A. E., Pitha-Rowe P. M. (2003) Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J. Biol. Chem. 278, 16630–16641 [DOI] [PubMed] [Google Scholar]
- 50. Ullman K. S., Powers M. A., Forbes D. J. (1997) Nuclear export receptors: from importin to exportin. Cell 90, 967–970 [DOI] [PubMed] [Google Scholar]
- 51. Takeuchi O., Hoshino K., Akira S. (2000) Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165, 5392–5396 [DOI] [PubMed] [Google Scholar]
- 52. Mancuso G., Midiri A., Beninati C., Biondo C., Galbo R., Akira S., Henneke P., Golenbock D., Teti G. (2004) Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J. Immunol. 172, 6324–6329 [DOI] [PubMed] [Google Scholar]
- 53. Burns E., Eliyahu T., Uematsu S., Akira S., Nussbaum G. (2010) TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J. Immunol. 184, 1455–1462 [DOI] [PubMed] [Google Scholar]
- 54. Kobayashi Y. (2006) Neutrophil infiltration and chemokines. Crit. Rev. Immunol. 26, 307–316 [DOI] [PubMed] [Google Scholar]
- 55. Appay V., Rowland-Jones S. L. (2001) RANTES: a versatile and controversial chemokine. Trends Immunol. 22, 83–87 [DOI] [PubMed] [Google Scholar]
- 56. Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S. M., Maniatis T. (2003) IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 [DOI] [PubMed] [Google Scholar]
- 57. Panne D., McWhirter S. M., Maniatis T., Harrison S. C. (2007) Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282, 22816–22822 [DOI] [PubMed] [Google Scholar]
- 58. Hoshino K., Sugiyama T., Matsumoto M., Tanaka T., Saito M., Hemmi H., Ohara O., Akira S., Kaisho T. (2006) IκB kinase-α is critical for interferon-α production induced by Toll-like receptors 7 and 9. Nature 440, 949–953 [DOI] [PubMed] [Google Scholar]
- 59. Génin P., Algarté M., Roof P., Lin R., Hiscott J. (2000) Regulation of RANTES chemokine gene expression requires cooperativity between NF-κ B and IFN-regulatory factor transcription factors. J. Immunol. 164, 5352–5361 [DOI] [PubMed] [Google Scholar]
- 60. Krausgruber T., Saliba D., Ryzhakov G., Lanfrancotti A., Blazek K., Udalova I. A. (2010) IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 115, 4421–4430 [DOI] [PubMed] [Google Scholar]
- 61. Wathelet M. G., Lin C. H., Parekh B. S., Ronco L. V., Howley P. M., Maniatis T. (1998) Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol. Cell 1, 507–518 [DOI] [PubMed] [Google Scholar]