

Background: The insulin receptor (IR) is a transmembrane (TM) receptor tyrosine kinase that regulates nutrient homeostasis.
Results: Peptides derived from the IR-TM domain activate IR, independent of insulin binding.
Conclusion: IR can be activated through a pathway that bypasses its canonical ligand-binding domain.
Significance: Sites outside of canonical ligand-binding domains can be used as targets for the development of new therapeutic agents.
Keywords: Insulin, Insulin Receptor, Peptide Hormone, Receptor Tyrosine Kinase, Transmembrane Domain, Peptide Ligand, Receptor Activation
Abstract
Complementary surfaces are buried when peptide hormones, growth factors, or cytokines bind and activate cellular receptors. Although these extended surfaces provide high affinity and specificity to the interactions, they also present great challenges to the design of small molecules that might either mimic or antagonize the process. We show that the insulin receptor (IR) and downstream signals can be activated by targeting a site outside of its ligand-binding domain. A 24-residue peptide having the IR transmembrane (TM) domain sequence activates IR, but not related growth factor receptors, through specific interactions with the receptor TM domain. Like insulin-dependent activation, IR-TM requires that IR have a competent ATP-binding site and kinase activation loop. IR-TM also activates mutated receptors from patients with severe insulin resistance, which do not respond to insulin. These results show that IR can be activated through a pathway that bypasses its canonical ligand-binding domain.
Introduction
Distinct ligands typically activate cognate receptors with high affinity and specificity (1). This is certainly true for the receptor tyrosine kinases that regulate such critical cellular functions such as growth, survival, differentiation, metabolism, and inflammatory responses (2). The development of agonists and antagonists of these receptors represents a major goal in drug discovery. However, the design of potential pharmacophores is hampered by the fact that extended surfaces on both ligand and receptor are buried upon binding (3–5). This raises important questions about what natural ligands do, which features are sufficient for receptor activation, and whether receptor activation can be achieved using alternative methods.
Receptor oligomerization is believed to be a key step in the activation of a receptor (1), as it is believed to juxtapose cytoplasmic kinase domains and thereby facilitate their trans-phosphorylation, which stimulates catalysis and creates docking sites for downstream signaling proteins. Evidence supporting the necessity of oligomerization in receptor activation comes from studies using the following: (a) activating antibodies and lectins; (b) chimeric receptors containing the extracellular domain of one receptor linked to the intracellular domain of another; and (c) biochemically separated receptor monomers and oligomers (1). However, these studies do not distinguish whether ligands actually cause receptors to oligomerize or whether ligands stabilize preformed oligomers at the cell surface to shift equilibrium in that direction. It is also not clear whether oligomerization is sufficient for receptor activation or whether the binding of the ligand also induces additional conformational changes that facilitate phosphorylation in trans.
The insulin receptor (IR)5 is a heterotetrameric membrane protein that is composed of two α and two β subunits (6, 7). The α subunit, which is the insulin-binding subunit, is totally extracellular and is linked to the extracellular part of the β subunit as well as to the other α subunit through disulfide bridges (8). The β subunit is composed of an extracellular domain, a membrane-spanning transmembrane (TM) domain, and an intracellular kinase domain that can be activated by autophosphorylation. IR is very similar to other growth factor receptors except for the fact that its receptor protomers are covalently tethered. Thus, IR provides a special case where potential effects of ligand binding may be distinguished. Because insulin binding does not affect monomer/dimer equilibrium, the primary effect is presumably conformational. Lines of supporting evidence include the following: (a) studies using homodimeric chemical cross-linkers reveal differences in subunit contacts between ligand-occupied and -unoccupied IR (9, 10); (b) anti-receptor antibodies differentially recognize receptors depending on whether they are phosphorylated or insulin-bound (11–15); (c) the basal and the activated insulin receptor exhibit a differential response to sulfhydryl reagents (16); (d) changes in intrinsic fluorescence reveal real time conformational changes during discrete steps of receptor activation (insulin and ATP binding and autophosphorylation) (17); (e) analyses of x-ray crystallography structures of apo- versus insulin-bound IR ectodomains show that insulin binding induces conformational changes in both insulin and the IR ectodomain (18–20); and (f) comparisons between activation loops in nonphosphorylated versus phosphorylated IR kinase domains reveal similar conformational changes to those observed in other kinases (21, 22). This raises the possibility that the ligand-induced conformational change is a primary function in other signaling systems as well. Studies with cytokine receptors support this hypothesis. For example, fluorescence energy transfer experiments indicate that in the absence of their ligands the erythropoietin (EPO) and IL-2 receptors are oligomerized and that ligand binding alters the conformation of these receptors (23, 24). Moreover, crystal structures of extracellular EPO receptors reveal that they are dimeric, regardless of whether or not EPO is bound (5, 25). These results indicate that in addition to dimerization, which is clearly important, ligand binding stabilizes receptor dimers at the cell surface and alters the structure of the receptor (5, 26, 27).
Growth factor, insulin, and cytokine receptor protomers each span the membrane only once. Bargmann et al. (28) have shown that Val644 to Glu substitutions within the TM domain of HER2/erbB2/Neu activates this receptor tyrosine kinase in the absence of a ligand (29). We subsequently showed that chimeric IR containing the HER2/erbB2/Neu TM domain were similarly activated by the Val → Glu mutation and suggested that dimerization at the level of the TM domain may be sufficient to activate receptor tyrosine kinases (30, 31). Subsequent studies showed that related TM domain substitutions activate the EGF, PDGF, IGF-1, and FGF receptors, presumably by similar mechanisms (30, 32–34). Based on the hypothesis that TM domain homodimerization may cause the cytoplasmic domains to juxtapose, thereby activating the receptors, we reasoned that exogenous ligands that bind to receptor TM domains may also be able to modulate receptor function. We further reasoned that these exogenous ligands may be synthetic peptides that correspond to TM domain sequences and that may interact with TM domains.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
Peptide chains were assembled on Fmoc-amino acid Wang resin with the FastMoc protocol by using an Applied Biosystems 430A peptide synthesizer. Peptides were deprotected and cleaved from the resin using a mixture of TFA, thioanisole, phenol, ethanedithiol, and water (90:5:5:2.5:5) and purified by gel filtration and reverse phase-HPLC using a nitrile column. In certain cases, carboxyfluorescein was introduced using diisopropylcarbodiimide/1-hydroxybenzotriazole after removing the final Fmoc group. Peptide sequences were confirmed by mass spectrometry. Alternatively, the IR-TM peptide was synthesized and purified by the Tufts University Peptide Synthesis Facility.
Preparation of the Insulin Receptor
IR was purified from NIH3T3 cells expressing human IR (35). Briefly, the plasma membrane fraction was treated for 1 h in 30 mm HEPES buffer, pH 7.6, containing 2% Triton and 0.02% NaN3. Solubilized receptor collected from the supernatant following centrifugation at 105 × g for 60 min was loaded onto wheat germ agglutinin (WGA)-agarose overnight. After extensive washing with HTA (30 mm HEPES, pH 7.4, 0.1% Triton X-100, and 0.02% NaN3), the bound proteins were eluted with 0.3 m N-acetylglucosamine in 30 mm HEPES, pH 7.4, containing 0.1% Triton X-100. Column fractions containing insulin binding activity were pooled and stored at −80 °C.
Exogenous Kinase Activity
The exogenous kinase activity of the IR was examined using poly(Glu/Tyr) (4:1) as the substrate. The TM peptides were first dissolved in trifluoroethanol (10 mm) and then diluted in HTA (10 μm). The final concentration of trifluoroethanol in the reaction mixtures was 0.1%. WGA-purified IR was preincubated with the TM peptides for 2 h at 21 °C or overnight at 4 °C. Then the mixtures were further incubated with insulin for an additional hour at 21 °C and a mixture of ATP (50 μm), MgCl2 (10 mm), and MnCl2 (8 mm) for 5 min at 21 °C. To initiate substrate phosphorylation assays, [γ-32P]ATP (5 μCi/sample) and poly(Glu/Tyr) (4:1) (0.3 mg/ml final concentration) were added. Reactions were stopped after 5 min by adding 67 mm EDTA. Aliquots of the reaction mixtures were spotted onto 3 × 3-cm squares of Whatman No. 3 MM filter paper, which were washed extensively with 10% (w/v) ice-cold trichloroacetic acid (TCA) in 20 mm sodium pyrophosphate, dried, and counted.
Cellular Studies
Chinese hamster ovary (CHO) cells expressing either wild type (CHO-IRWT) or mutated human IR (CHO-IRPDGFR/TM, -IRK1006A, or -IRY3F) were grown to confluence in 6-well dishes containing DMEM supplemented with 10% fetal bovine serum (FBS) and 0.45 mg/ml G415. Prior to experiments, the cells were quiesced overnight in DMEM containing 0.2% BSA. To initiate experiments, the cells were incubated with the peptides for 30 min and then with insulin (10−10 m) for 5 min, all at 37 °C. The peptides were originally dissolved in DMSO (10 mm) and subsequently diluted with DMEM (the final concentration of DMSO in the culture medium was 0.5%). To terminate the assays, cells were washed with ice-cold PBS containing a protease and phosphatase inhibitors (1 mm PMSF, 1 μm leupeptin, 0.3 μm aprotinin, 1 μm pepstatin, 25 mm benzamidine HCl, 100 mm NaF, 2 mm sodium vanadate, 30 mm sodium pyrophosphate, 1 mm ammonium molybdate, 2 mm EGTA). Cells were subsequently lysed with 1% Triton X-100 in 30 mm HEPES buffer containing the same protease and phosphatase inhibitors. Activation of insulin signaling was determined by Western blot. Proteins were immunoprecipitated from cell lysates for 2 h at 4 °C using immobilized anti-IR and anti-IRS-1 antibodies (protein A-Sepharose beads, Amersham Biosciences) (36). After extensive washing, proteins were eluted with Laemmli sample buffer, separated by SDS-PAGE, and transferred to Immobilon PVDF membranes (Millipore). Blots were probed with the indicated antibodies, and proteins were detected by chemiluminescence (Pierce). The activations of the MAP kinase and AKT were determined in the total cell lysates by using phosphospecific MAP kinase (Promega) and AKT (Ser-473; Cell Signaling) antibodies.
Confocal Microscopy
CHO-IR cells were treated with 1 μm fluorescein-labeled IR-TM peptide as described above, washed extensively, and fixed with 4% formaldehyde in PBS. Cells were then incubated with IR antibody (4 μg/ml) and a rhodamine-labeled secondary antibody examined using a Zeiss confocal microscope.
Materials
CHO cells that express wild type or mutant IR (CHO-IRWT, CHO-IRPDGFR/TM, -IRK1006A, and -IRY3F) have been described previously (30, 37). NIH3T3 cells that express wild type or mutant IR were generously provided by Dr. Simeon Taylor (38). WGA-agarose was purchased from EY Labs. The anti-phosphotyrosine (4G10) and anti-IR antibodies that were used for confocal studies were from Millipore; the anti-activated MAPK antibody was from Promega, and the rhodamine-labeled donkey anti-rabbit IgG was from Jackson ImmunoResearch. All other reagents were from Sigma.
RESULTS
In Vitro Activation of IR by the IR-TM Peptide
Although the TM domains of insulin, growth factor, and cytokine receptors appear to adopt similar hydrophobic α-helical structures, the sequences of these membrane-spanning domains vary dramatically (Table 1). Even otherwise highly homologous receptors differ significantly within their TM domains. For example, insulin and IGF-1 receptors share 56% identity overall, but their TM domains are only 21% identical. We decided to test whether exogenous TM sequences altered receptor activity and, if so, whether this was sequence-specific.
TABLE 1.
TM peptide sequences
| Receptor | TM domain sequence | Identitya |
|---|---|---|
| % | ||
| hIR | KIIIGPLIFVFLFSVVIGSIYLFLRKR | 100 |
| hIGF-1R | LIIALPVAVLLIVGGLVIMLYVFHRKR | 21 |
| hPDGFR | KVVVISAILALVVLTIISLIILIMLWQKK | 12 |
| rNeu | RASPVTFIIATVVGVLLFLILVVVVGILIKRRR | 18 |
| rNeu(V→E) | RASPVTFIIATVEGVLLFLILVVVVGILIKRRR | 18 |
| hEGFR | KIPSIATGMVGALLLLLVVALGIGLFMRRR | 31 |
| hTrkA | KDETPFGVSVAVGLAVFACLFLSTLLLVLNK | 10 |
a The sequence identity of the different TM domains to that of the human (h) IR (hIR) was determined by using the Blast program.
Peptides corresponding to various TM domain sequences (Table 1) were incubated with solubilized IR, and the effects on tyrosine kinase activity were assessed. In the absence of insulin, the insulin receptor TM (IR-TM) peptide stimulated a 3–4-fold increase in the IR-catalyzed phosphorylation of the artificial substrate, poly(Glu/Tyr) (Fig. 1). Stimulation was concentration-dependent and showed a logarithmic dose-response profile (ED50 ∼1 μm). Under identical conditions, insulin stimulated a maximal 6-fold increase in substrate phosphorylation, with an ED50 value of ∼0.02 μm (Fig. 1). When submaximal insulin and IR-TM were combined, their effects were additive. However, IR-TM had no additional effects when added to maximal insulin (e.g. 10−7 m). This suggests that the two peptides might activate the IR by similar mechanisms.
FIGURE 1.
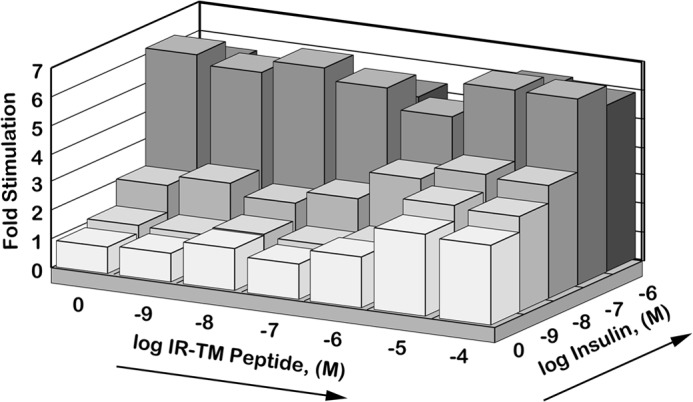
In vitro activation of the IR by the IR-TM peptide. The WGA-agarose-purified IR was incubated with various concentrations (x axis) of the IR-TM peptide for 2 h at 4 °C and then stimulated with various concentrations (z axis) of‘ insulin for an additional hour at room temperature. Its exogenous tyrosine kinase activity was measured using poly(Glu/Tyr) (4:1) as the substrate as described under “Experimental Procedures.”
To begin to assess a mechanism for IR-TM-mediated activation of IR, we asked whether IR-TM competed for or otherwise altered insulin binding. Binding of 125I-labeled insulin was not affected by IR-TM (data not shown), suggesting that IR-TM and insulin bind at distinct nonoverlapping sites. Because the IR-TM peptide stimulates IR kinase activity in the absence of insulin, activation is not through the modulation of insulin binding. Instead, the IR-TM peptide appears to be an insulin-mimetic as opposed to an insulin sensitizer.
Activation of IR in Cells
Next, we determined whether the IR-TM peptide could activate insulin signaling responses in cultured cells. We first examined the subcellular localization of IR-TM peptide by using confocal microscopy. FITC-labeled IR-TM exhibited a peripheral pattern of fluorescence in CHO-IR cells, consistent with incorporation of the peptide into the plasma membrane (Fig. 2A, left panel). As expected, immunofluorescence staining with a rhodamine-labeled antibody confirmed that IR was also located in the plasma membrane (Fig. 2A, middle panel). Overlays showed that IR and IR-TM were co-localized in the plasma membrane (Fig. 2A, right panel).
FIGURE 2.
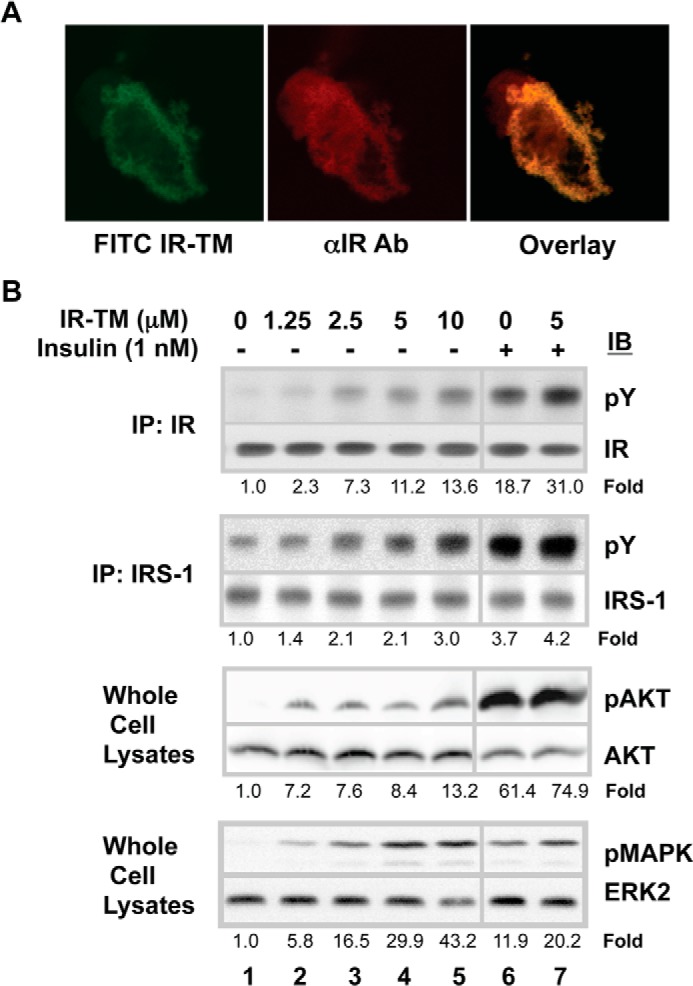
Localization of the IR-RM peptide in cells and its ability to activate the cellular IR. A, co-localization of IR-TM peptides with the IR in CHO-IR cells. CHO-IR cells were grown on the coverslip and then treated with the FITC-labeled IR-TM peptide for 30 min. After being washed with PBS, the cells were fixed with formaldehyde and incubated with an anti-IR antibody (αIR Ab) and then with a rhodamine-labeled donkey anti-rabbit IgG. Shown are the fluorescence patterns of the peptide alone (left) and the IR alone (middle) when the cells were visualized with confocal microscopy. The right panel shows the effect of overlaying the two images. B, activation of the IR, IRS-1, MAPK, and AKT in CHO-IR cells by the IR-TM peptide. CHO-IR cells were grown to confluence in 6-well plates, starved overnight with 2% BSA in DMEM, treated with various concentrations of IR-TM peptides dissolved in DMSO for 30 min at 37 °C, and then stimulated with 10−9 m insulin for 15 min. After washing the cells and lysing them, the IR and IRS-1 in the lysates were immunoprecipitated (IP) with specific antibodies and resolved by SDS-PAGE. The proteins were transferred onto PVDF membranes and subjected to Western blot analysis using an anti-phosphotyrosine antibody or antibodies specific for the IR or IRS-1. The proteins were visualized with chemiluminescence. Activated MAPK or ATK in total cell lysates was detected by an anti-phospho-specific MAPK or an anti-phospho-specific AKT antibody (Ser-473), respectively. Numbers represent the folds of specific phosphorylations (phosphorylation/amount of the protein). IB, immunoblot.
We next asked whether the peptide had related bioeffects in cells. Thirty min after addition of the IR-TM peptide, cells were lysed, and IR phosphorylation was determined by Western blotting (Fig. 2B). The IR-TM peptide stimulated a dose-dependent increase in IR tyrosine phosphorylation by 2.3-fold at a concentration as low as 1.25 μm and increased it to 13.6-fold at 10 μm (the limit of peptide solubility), compared with the control (Fig. 2B, lane 1). When CHO-IR cells were incubated with the IR-TM peptide prior to stimulation with submaximal insulin, the effects appeared to be additive (Fig. 2B, lane 6 versus lane 7). Tyrosine phosphorylation IRS-1 increased in parallel with IR phosphorylation (Fig. 2B), indicating that the IR-TM peptide activated IR in cells as does insulin. At 10 μm, the highest dose of the IR-TM peptide used, the level of IR or IRS-1 phosphorylation was roughly equivalent to that seen with a submaximal 10−9 m dose of insulin. Concomitant with its stimulation of proximal signaling, the IR-TM peptide stimulated AKT and MAPK, both of which are more distal end points downstream in the insulin action cascade (Fig. 2B).
Specificity of TM Domain Sequence
The specificity of IR-TM-mediated receptor activation was studied using two approaches. We first tested whether alternative peptide sequences activated IR. Peptide sequences corresponding to the TM domains of other receptor tyrosine kinases failed to activate IR in in vitro kinases assays (Fig. 3A). Sequences studied included those corresponding to the TM domains of the IGF-1, PDGF, HER2/erbB2/Neu, and TrkA receptors. Similar studies conducted with CHO-IR cells yielded identical results (Fig. 3B). IR-TM activated both substrate phosphorylation and IR autophosphorylation, whereas TM domain peptides from heterologous receptors did not.
FIGURE 3.
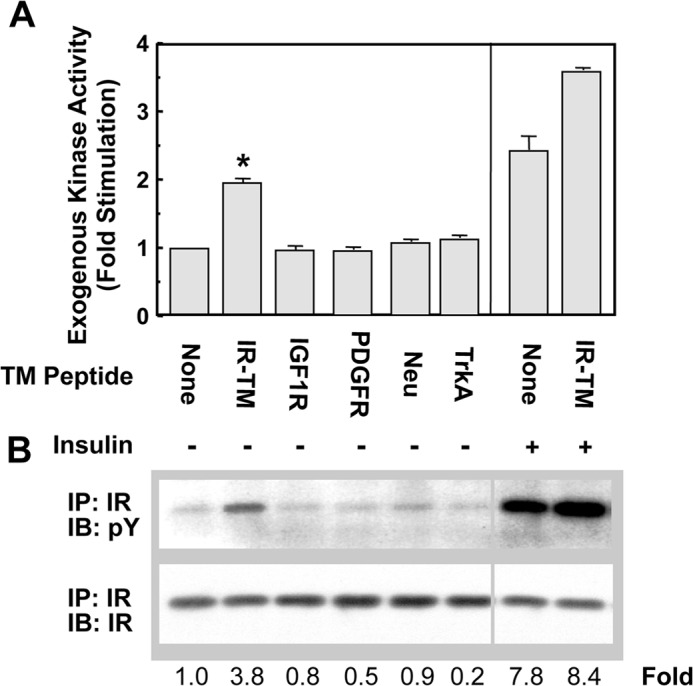
Ability of peptides corresponding to the TM sequences of other receptors to activate the IR in vitro and on cells. A, peptides corresponding to the TM domains of the IR, IGF-1, PDGF, erbB2, and TrkA receptors were incubated at 10 μm with the WGA-purified IR overnight at 4 °C. The tyrosine kinase activity of the IR was then measured as described in the legend of Fig. 1 in the presence or absence of 10−8 m insulin. *, p < 0.05. B, CHO-IR cells were grown as described previously and treated with the TM peptides of various receptors at 5 μm for 30 min at 37 °C. The cells were washed and lysed, and the phosphorylation status of the IR was analyzed as described in Fig. 2. Numbers represent the folds of specific phosphorylations (phosphorylation/amount of the protein). IP, immunoprecipitation; IB, immunoblot.
As a second approach to address specificity, we used a chimeric IR in which the TM domain had been substituted with the corresponding domain from a heterologous receptor. The sequence of IRPDGFR/TM is identical to WT IR, with the exception of the TM domain, which has the sequence of the PDGF receptor (30, 31). Although IR-TM consistently activates WT IR, IR-TM was unable to activate IRPDGFR/TM. In contrast, insulin activates IRPDGFR/TM normally (30, 31).
As additional controls, we compared the ability of either insulin or IR-TM to activate two kinase-defective receptors. The single amino acid substitution in the ATP-binding site of IRK1006A renders it incapable of catalyzing phosphoryl transfers, whereas the three tyrosines in the activation loop of IRY3F were substituted with phenylalanine (39, 40). Neither insulin nor IR-TM was capable of activating either receptor (Fig. 4). Our findings suggested that IR-TM might interact with IR through its TM domain.
FIGURE 4.
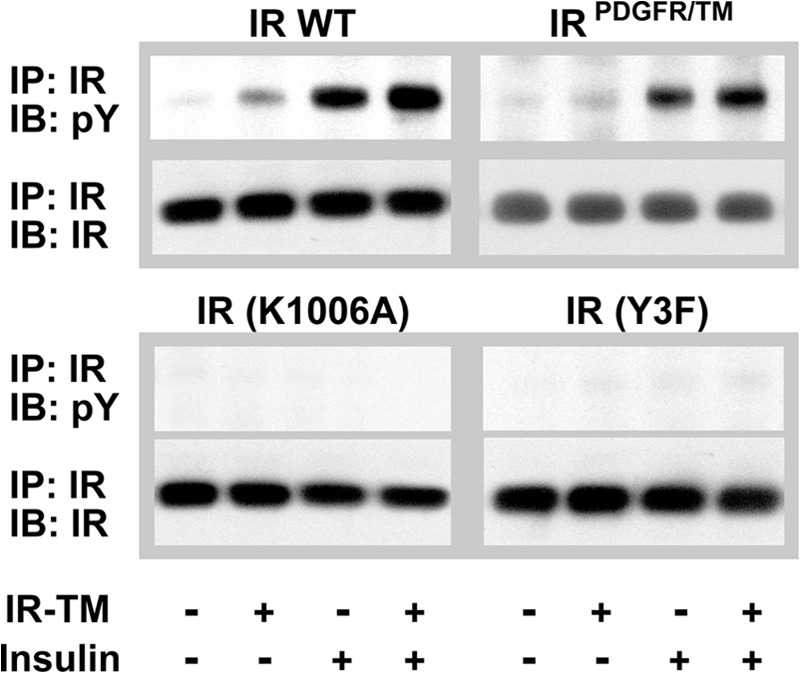
IR-TM fails to activate IRPDGFR/TM, IRK1006A, or IRY3F. CHO-IR cells expressing either the chimeric IR, the transmembrane domain of the PDGF receptor (IRPDGFR/TM), kinase-inactive IRs (IRK1006A and IRY3F), or wild type receptors were grown as described previously and treated with 5 μm IR-TM peptide for 30 min at 37 °C. After the cells were washed and lysed, the IR was immunoprecipitated (IP) and its phosphorylation state was determined by Western blotting analysis using antibodies specific for phosphotyrosine and the IR. IB, immunoblot.
Activation of “Unresponsive” IR
Numerous mutations have been identified in the IR of patients with extreme insulin resistance, including some that block insulin binding (41, 42). The S323L substitution within the insulin-binding site of IR abolishes the insulin binding without affecting surface expression or intrinsic kinase activity (38). We have found that IRS323L, expressed in NIH3T3 cells, is activated by IR-TM but not insulin (Fig. 5). In fact, IR-TM activates IRS323L as potently as it activates WT IR. These observations are consistent with the notion that IR-TM bypasses the insulin-binding site of IR and activates IR by interacting at an alternative site, possibly the TM domain.
FIGURE 5.
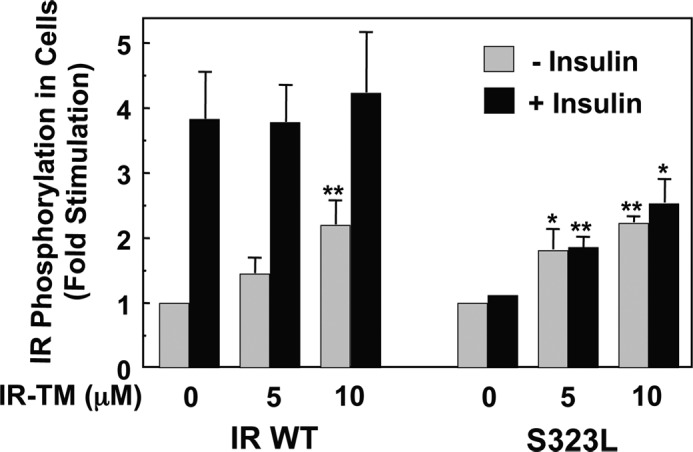
IR-TM peptide can activate a mutant IR that lacks insulin binding activity. NIH3T3 cells that express either the wild type receptor or an IR mutant that lacks insulin binding activity (S323L) were grown to confluence in 6-well plates as described previously, treated with 5 μm IR-TM peptide for 30 min at 37 °C, and then stimulated with 10−9 m insulin for 15 min. After the cells were washed and lysed, the IR was immunoprecipitated and its phosphorylation was assessed by Western blot analysis using antibodies against phosphotyrosine and the IR. The bands were quantitated with densitometry, and the fold of specific phosphorylation (phosphorylation of the IR/amount of the IR) was calculated. The results are shown with the average of three independent experiments. *, p < 0.05; **, p < 0.01.
DISCUSSION
We have found that a synthetic peptide corresponding to the TM domain sequence of IR possesses the ability to both activate purified IR and IR in cultured cells. The effects of submaximal doses of IR-TM and insulin are additive (Figs. 1 and 2). It appears that the IR-TM peptide influences the TM domain of the IR, as substituting the native TM domain abrogates IR-TM effects without altering insulin's ability to activate the receptor (Fig. 4). Moreover, as shown in Fig. 5, a mutation in the IR α subunits that abolishes insulin binding does not prevent the receptor from being activated by the IR-TM peptide. These data clearly demonstrate that the sites of interaction (signal initiation) are distinct between the two ligands, insulin and IR-TM peptide, but that they have indistinguishable net effects on the receptor kinase and downstream signaling. This suggests that insulin and IR-TM activate IR through similar mechanisms.
Conformational changes are key steps in the activation of the IR, including those caused by insulin binding to the IR ectodomain (18, 19, 43) and ATP binding to the intracellular IR kinase domain (21, 22). Although the nature of the conformational changes that are induced by IR-TM are unclear, studies on other membrane proteins provide some general insights into the structural changes that TM domains can undergo. For example, the dimerization of TM domains in glycophorin A has been well characterized by various biochemical and mutational studies (44, 45). The structure has also been resolved by an NMR technique (46). These studies show that the glycophorin A TM domain is constitutively dimerized as two parallel α-helices held at a constant distance by a network of hydrogen bonding between the amino acids facing each other on the inner dimer surface. Because of the high degree of hydrogen bonding between the two helices, the dimerized TM domains cannot even be dissociated during SDS-PAGE, a technique that has been used to analyze the effects of TM domain dimerization in the insulin and EPO receptors. When the TM domain of the IR was replaced with the TM domain of glycophorin A, the β subunits of the chimeric receptor remained dimerized in conditions that reduced the wild type receptor β subunits and rendered them entirely monomeric (47). This indicates that the chimeric receptor can exist as a heterotetrameric (functional dimeric) receptor even in the absence of disulfide bonds between the two α subunits (48). When the chimeric receptor was tested in vitro and in cells, it responded to insulin less well than wild type IR, which suggests that IR activation requires the separation of the two TM domains. This notion was supported by the structure of IR, as determined using cryoelectron microscopy. This analysis showed that the distance between the two TM domains was increased by insulin binding to the α subunits (49–51). By contrast, the TM of the EPO receptor appears to behave differently during the activation of this receptor. Comparison of the crystal structures of the liganded and unliganded EPO receptors suggests that the binding of the ligand initiates lateral (translational) movements that juxtapose the cytoplasmic kinase domains and promote trans-phosphorylation (Fig. 6A). Replacement of the EPO receptor TM domain with that of glycophorin A revealed that this dimerization event was independent of the ligand used and that this replacement of the TM domain did not impair the activation of the receptor by the EPO ligand (52). Thus, the TM domain replacement experiments with two different receptor types reveal completely different outcomes and suggest that the activation of some receptors requires that their TM domains be dimerized, although other receptors require the separation of their TM domains.
FIGURE 6.

Proposed mechanism for the activation of IR by the IR-TM peptide.
Based on our findings and those of others, we propose that the TM domains of IR in the nonactivated basal state are constitutively dimerized and that when insulin binds the α subunits, the TM domains dissociate from one another (Fig. 6B). This transition in turn brings the two β subunits of IR into closer proximity (53) and results in IR autophosphorylation and the activation of the kinase domains of the β subunits through a trans-phosphorylation mechanism (54). We further propose that the IR-TM peptide directly intercalates the interfaces of the two helices of the TM domains, thereby disrupting their interaction and resulting in the same dissociation of the TM domains that is induced by the binding of insulin to the receptor (Fig. 6C). This causes the β subunits to transit from their basal state to an activated state, which promotes tyrosine kinase domain activation in the absence of insulin binding. However, at present, we cannot exclude possible alternative mechanisms. For example, instead of the two β subunits of IR staying apart in the basal state (Fig. 6B), they may be interlocked with each other, an interaction that is then disrupted by insulin and IR-TM peptide binding to the IR; this in turn activates the IR kinases (43, 55). It is also possible that the protomers may rotate on ligand or IR-TM peptide binding such that the intracellular kinase domains are similarly aligned with little energetic expenditure. This is the mechanism proposed for the activation of the growth hormone (GH) receptor (Fig. 6D) (56, 57); comparison of the free and ligand-bound forms of the GH receptor in terms of the extracellular domain structures reveals that ligand binding causes one receptor chain to rotate toward the other receptor chain, causing the two C-terminal extracellular domains of the receptor to bind together (58). This rotation of extracellular domains has also been proposed to induce the rotation of the TM domain, which in turn causes the GH receptor to become fully activated. An elegant study by Waters and co-workers (58, 59) supports this notion. They showed that forcing the GH receptor TM domain to rotate by inserting Ala into the α-helix of the TM domain can activate the GH receptor in the absence of GH binding (58, 59). Interestingly, the NMR structure of the monomeric IR-TM domain in detergent micelles shows an α-helix structure with a kink that is induced by Pro-961 (60). This resembles the proposed model for the GH receptor TM domain (Fig. 6D) and suggests that IR may be activated by a mechanism similar to the one activating the GH receptor. However, the molecular mechanism by which IR is activated will not be fully understood until high resolution images of the holo-IR structure become available.
IR has been targeted for many years by researchers seeking small molecule insulin mimetics, which has had some success. Insulin mimetics identified to date include peptide dimers obtained by phage display screening that can bind the canonical insulin-binding sites (61, 62), a peptide from the MHC I antigen that binds to the extracellular domain of the IR and regulates the endocytosis of the receptor (63, 64), and small organic compounds that target the intracellular kinase domain of the receptor (65, 66). Our findings open up the possibility of targeting the IR-TM domain with novel therapeutic agents. However, identification of such agents would require considerable effort, including the screening of chemical libraries for compounds that activate the insulin holoreceptor. Such efforts would be complicated by the hydrophobic nature of the membrane. More importantly, our findings show that sites outside the canonical ligand-receptor-binding site can serve as potential targets in drug discovery research that aims to identify activators of receptor signaling pathways.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 DK80380 (to J. L.), R01 DK45943, R37 DK51729, and R01 DK73547 (to S. E. S.), T32 DK007260 (to G. R. R.), and P30 DK36836 (to Joslin Diabetes Research Center). This work was also supported by American Diabetes Association Grant RA 110BS97 (to J. L.) and the Helen and Morton Adler Chair (to S. E. S.).
- IR
- insulin receptor
- TM
- transmembrane
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- EPO
- erythropoietin
- WGA
- wheat germ agglutinin
- GH
- growth hormone.
REFERENCES
- 1. Ullrich A., Schlessinger J. (1990) Signal transduction by receptors with tyrosine kinase activity. Cell 61, 203–212 [DOI] [PubMed] [Google Scholar]
- 2. van der Geer P., Hunter T., Lindberg R. A. (1994) Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 10, 251–337 [DOI] [PubMed] [Google Scholar]
- 3. de Vos A. M., Ultsch M., Kossiakoff A. A. (1992) Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science 255, 306–312 [DOI] [PubMed] [Google Scholar]
- 4. Clackson T., Wells J. A. (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267, 383–386 [DOI] [PubMed] [Google Scholar]
- 5. Syed R. S., Reid S. W., Li C., Cheetham J. C., Aoki K. H., Liu B., Zhan H., Osslund T. D., Chirino A. J., Zhang J., Finer-Moore J., Elliott S., Sitney K., Katz B. A., Matthews D. J., Wendoloski J. J., Egrie J., Stroud R. M. (1998) Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature 395, 511–516 [DOI] [PubMed] [Google Scholar]
- 6. Ullrich A., Bell J. R., Chen E. Y., Herrera R., Petruzzelli L. M., Dull T. J., Gray A., Coussens L., Liao Y. C., Tsubokawa M. (1985) Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313, 756–761 [DOI] [PubMed] [Google Scholar]
- 7. Ebina Y., Ellis L., Jarnagin K., Edery M., Graf L., Clauser E., Ou J. H., Masiarz F., Kan Y. W., Goldfine I. D. (1985) The human insulin receptor cDNA: the structural basis for hormone activated transmembrane signalling. Cell 40, 747–758 [DOI] [PubMed] [Google Scholar]
- 8. Pilch P. F., Czech M. P. (1980) The subunit structure of the high affinity insulin receptor. Evidence for a disulfide-linked receptor complex in fat cell and liver plasma membranes. J. Biol. Chem. 255, 1722–1731 [PubMed] [Google Scholar]
- 9. Waugh S. M., DiBella E. E., Pilch P. F. (1989) Isolation of a proteolytically derived domain of the insulin receptor containing the major site of cross-linking/binding. Biochemistry 28, 3448–3455 [DOI] [PubMed] [Google Scholar]
- 10. Schenker E., Kohanski R. A. (1988) Conformational states of the insulin receptor. Biochem. Biophys. Res. Commun. 157, 140–145 [DOI] [PubMed] [Google Scholar]
- 11. Perlman R., Bottaro D. P., White M. F., Kahn C. R. (1989) Conformational changes in the α- and β-subunits of the insulin receptor identified by anti-peptide antibodies. J. Biol. Chem. 264, 8946–8950 [PubMed] [Google Scholar]
- 12. Herrera R., Rosen O. M. (1986) Autophosphorylation of the insulin receptor in vitro. Designation of phosphorylation sites and correlation with receptor kinase activation. J. Biol. Chem. 261, 11980–11985 [PubMed] [Google Scholar]
- 13. Baron V., Gautier N., Komoriya A., Hainaut P., Scimeca J. C., Mervic M., Lavielle S., Dolais-Kitabgi J., Van Obberghen E. (1990) Insulin binding to its receptor induces a conformational change in the receptor C terminus. Biochemistry 29, 4634–4641 [DOI] [PubMed] [Google Scholar]
- 14. Maddux B. A., Goldfine I. D. (1991) Evidence that insulin plus ATP may induce a conformational change in the β subunit of the insulin receptor without inducing receptor autophosphorylation. J. Biol. Chem. 266, 6731–6736 [PubMed] [Google Scholar]
- 15. Baron V., Kaliman P., Gautier N., Van Obberghen E. (1992) The insulin receptor activation process involves localized conformational changes. J. Biol. Chem. 267, 23290–23294 [PubMed] [Google Scholar]
- 16. Clark S., Konstantopoulos N. (1993) Sulphydryl agents modulate insulin- and epidermal growth factor (EGF)-receptor kinase via reaction with intracellular receptor domains: differential effects on basal versus activated receptors. Biochem. J. 292, 217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee J., Pilch P. F., Shoelson S. E., Scarlata S. F. (1997) Conformational change of the insulin receptor upon insulin binding and activation as monitored by fluorescence spectroscopy. Biochemistry 36, 2701–2708 [DOI] [PubMed] [Google Scholar]
- 18. McKern N. M., Lawrence M. C., Streltsov V. A., Lou M. Z., Adams T. E., Lovrecz G. O., Elleman T. C., Richards K. M., Bentley J. D., Pilling P. A., Hoyne P. A., Cartledge K. A., Pham T. M., Lewis J. L., Sankovich S. E., Stoichevska V., Da Silva E., Robinson C. P., Frenkel M. J., Sparrow L. G., Fernley R. T., Epa V. C., Ward C. W. (2006) Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 443, 218–221 [DOI] [PubMed] [Google Scholar]
- 19. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., Ward C. W., Lawrence M. C. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith B. J., Huang K., Kong G., Chan S. J., Nakagawa S., Menting J. G., Hu S. Q., Whittaker J., Steiner D. F., Katsoyannis P. G., Ward C. W., Weiss M. A., Lawrence M. C. (2010) Structural resolution of a tandem hormone-binding element in the insulin receptor and its implications for design of peptide agonists. Proc. Natl. Acad. Sci. U.S.A. 107, 6771–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hubbard S. R., Wei L., Ellis L., Hendrickson W. A. (1994) Crystal structure of the tryosine kinase domain of the human insulin receptor. Nature 372, 746–754 [DOI] [PubMed] [Google Scholar]
- 22. Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Damjanovich S., Bene L., Matkó J., Alileche A., Goldman C. K., Sharrow S., Waldmann T. A. (1997) Preassembly of interleukin 2 (IL-2) receptor subunits on resting kit 225 K6 T cells and their modulation by IL-2, IL-7, and IL-15: a fluorescence resonance energy transfer study. Proc. Natl. Acad. Sci. U.S.A. 94, 13134–13139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Remy I., Wilson I. A., Michnick S. W. (1999) Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993 [DOI] [PubMed] [Google Scholar]
- 25. Livnah O., Stura E. A., Middleton S. A., Johnson D. L., Jolliffe L. K., Wilson I. A. (1999) Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283, 987–990 [DOI] [PubMed] [Google Scholar]
- 26. Livnah O., Stura E. A., Johnson D. L., Middleton S. A., Mulcahy L. S., Wrighton N. C., Dower W. J., Jolliffe L. K., Wilson I. A. (1996) Functional mimicry of a protein hormone by a peptide agonist: the EPO receptor complex at 2.8 A (see comments). Science 273, 464–471 [DOI] [PubMed] [Google Scholar]
- 27. Livnah O., Johnson D. L., Stura E. A., Farrell F. X., Barbone F. P., You Y., Liu K. D., Goldsmith M. A., He W., Krause C. D., Pestka S., Jolliffe L. K., Wilson I. A. (1998) An antagonist peptide-EPO receptor complex suggests that receptor dimerization is not sufficient for activation. Nat. Struct. Biol. 5, 993–1004 [DOI] [PubMed] [Google Scholar]
- 28. Bargmann C. I., Hung M. C., Weinberg R. A. (1986) Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45, 649–657 [DOI] [PubMed] [Google Scholar]
- 29. Weiner D. B., Liu J., Cohen J. A., Williams W. V., Greene M. I. (1989) A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature 339, 230–231 [DOI] [PubMed] [Google Scholar]
- 30. Yamada K., Goncalves E., Kahn C. R., Shoelson S. E. (1992) Substitution of the insulin receptor transmembrane domain with the c-neu/erbB2 transmembrane domain constitutively activates the insulin receptor kinase in vitro. J. Biol. Chem. 267, 12452–12461 [PubMed] [Google Scholar]
- 31. Cheatham B., Shoelson S. E., Yamada K., Goncalves E., Kahn C. R. (1993) Substitution of the erbB-2 oncoprotein transmembrane domain activates the insulin receptor and modulates the action of insulin and insulin-receptor substrate 1. Proc. Natl. Acad. Sci. U.S.A. 90, 7336–7340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takahashi K., Yonezawa K., Nishimoto I. (1995) Insulin-like growth factor I receptor activated by a transmembrane mutation. J. Biol. Chem. 270, 19041–19045 [DOI] [PubMed] [Google Scholar]
- 33. Webster M. K., Donoghue D. J. (1996) Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 15, 520–527 [PMC free article] [PubMed] [Google Scholar]
- 34. Petti L. M., Irusta P. M., DiMaio D. (1998) Oncogenic activation of the PDGF β receptor by the transmembrane domain of p185neu*. Oncogene 16, 843–851 [DOI] [PubMed] [Google Scholar]
- 35. Lee J., O'Hare T., Pilch P. F., Shoelson S. E. (1993) Insulin receptor autophosphorylation occurs asymmetrically. J. Biol. Chem. 268, 4092–4098 [PubMed] [Google Scholar]
- 36. Werner E. D., Lee J., Hansen L., Yuan M., Shoelson S. E. (2004) Insulin resistance due to phosphorylation of IRS-1 at serine 302. J. Biol. Chem. 279, 35298–35305 [DOI] [PubMed] [Google Scholar]
- 37. Wilden P. A., Siddle K., Haring E., Backer J. M., White M. F., Kahn C. R. (1992) The role of insulin receptor kinase domain autophosphorylation in receptor-mediated activities. Analysis with insulin and anti-receptor antibodies. J. Biol. Chem. 267, 13719–13727 [PubMed] [Google Scholar]
- 38. Taouis M., Levy-Toledano R., Roach P., Taylor S. I., Gorden P. (1994) Rescue and activation of a binding-deficient insulin receptor. Evidence for intermolecular transphosphorylation. J. Biol. Chem. 269, 27762–27766 [PubMed] [Google Scholar]
- 39. White M. F., Shoelson S. E., Keutmann H., Kahn C. R. (1988) A cascade of tyrosine autophosphorylation in the β-subunit activates the phosphotransferase of the insulin receptor. J. Biol. Chem. 263, 2969–2980 [PubMed] [Google Scholar]
- 40. Wilden P. A., Kahn C. R., Siddle K., White M. F. (1992) Insulin receptor kinase domain autophosphorylation regulates receptor enzymatic function. J. Biol. Chem. 267, 16660–16668 [PubMed] [Google Scholar]
- 41. Taylor S. I., Cama A., Accili D., Barbetti F., Quon M. J., de la Luz Sierra M., Suzuki Y., Koller E., Levy-Toledano R., Wertheimer E. (1992) Mutations in the insulin receptor gene. Endocr. Rev. 13, 566–595 [DOI] [PubMed] [Google Scholar]
- 42. Accili D. (1995) Molecular defects of the insulin receptor gene. Diabetes Metab. Rev. 11, 47–62 [DOI] [PubMed] [Google Scholar]
- 43. Ward C. W., Menting J. G., Lawrence M. C. (2013) The insulin receptor changes conformation in unforeseen ways on ligand binding: sharpening the picture of insulin receptor activation. BioEssays 35, 945–954 [DOI] [PubMed] [Google Scholar]
- 44. Lemmon M. A., Flanagan J. M., Treutlein H. R., Zhang J., Engelman D. M. (1992) Sequence specificity in the dimerization of transmembrane α-helices. Biochemistry 31, 12719–12725 [DOI] [PubMed] [Google Scholar]
- 45. Treutlein H. R., Lemmon M. A., Engelman D. M., Brünger A. T. (1992) The glycophorin A transmembrane domain dimer: sequence-specific propensity for a right-handed supercoil of helices. Biochemistry 31, 12726–12732 [DOI] [PubMed] [Google Scholar]
- 46. MacKenzie K. R., Prestegard J. H., Engelman D. M. (1997) A transmembrane helix dimer: structure and implications. Science 276, 131–133 [DOI] [PubMed] [Google Scholar]
- 47. Gardin A., Auzan C., Clauser E., Malherbe T., Aunis D., Crémel G., Hubert P. (1999) Substitution of the insulin receptor transmembrane domain with that of glycophorin A inhibits insulin action. FASEB J. 13, 1347–1357 [DOI] [PubMed] [Google Scholar]
- 48. Böni-Schnetzler M., Kaligian A., DelVecchio R., Pilch P. F. (1988) Ligand-dependent intersubunit association within the insulin receptor complex activates its intrinsic kinase activity. J. Biol. Chem. 263, 6822–6828 [PubMed] [Google Scholar]
- 49. Luo R. Z., Beniac D. R., Fernandes A., Yip C. C., Ottensmeyer F. P. (1999) Quaternary structure of the insulin-insulin receptor complex. Science 285, 1077–1080 [DOI] [PubMed] [Google Scholar]
- 50. Ottensmeyer F. P., Beniac D. R., Luo R. Z., Yip C. C. (2000) Mechanism of transmembrane signaling: insulin binding and the insulin receptor. Biochemistry 39, 12103–12112 [DOI] [PubMed] [Google Scholar]
- 51. Yip C. C., Ottensmeyer P. (2003) Three-dimensional structural interactions of insulin and its receptor. J. Biol. Chem. 278, 27329–27332 [DOI] [PubMed] [Google Scholar]
- 52. Constantinescu S. N., Keren T., Socolovsky M., Nam H., Henis Y. I., Lodish H. F. (2001) Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc. Natl. Acad. Sci. U.S.A. 98, 4379–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Waugh S. M., Pilch P. F. (1989) Insulin binding changes the interface region between α subunits of the insulin receptor. Biochemistry 28, 2722–2727 [DOI] [PubMed] [Google Scholar]
- 54. Frattali A. L., Treadway J. L., Pessin J. E. (1992) Transmembrane signalling by the human insulin receptor kinase: relationship between intramolecular β subunit trans- and cis-autophosphorylation and substrate kinase activation. J. Biol. Chem. 267, 19521–19528 [PubMed] [Google Scholar]
- 55. Shoelson S. E., Boni-Schnetzler M., Pilch P. F., Kahn C. R. (1991) Autophosphorylation within insulin receptor β-subunits can occur as an intramolecular process. Biochemistry 30, 7740–7746 [DOI] [PubMed] [Google Scholar]
- 56. Brooks A. J., Dai W., O'Mara M. L., Abankwa D., Chhabra Y., Pelekanos R. A., Gardon O., Tunny K. A., Blucher K. M., Morton C. J., Parker M. W., Sierecki E., Gambin Y., Gomez G. A., Alexandrov K., Wilson I. A., Doxastakis M., Mark A. E., Waters M. J. (2014) Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344, 1249783. [DOI] [PubMed] [Google Scholar]
- 57. Wells J. A., Kossiakoff A. A. (2014) Cell biology. New tricks for an old dimer. Science 344, 703–704 [DOI] [PubMed] [Google Scholar]
- 58. Brown R. J., Adams J. J., Pelekanos R. A., Wan Y., McKinstry W. J., Palethorpe K., Seeber R. M., Monks T. A., Eidne K. A., Parker M. W., Waters M. J. (2005) Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 12, 814–821 [DOI] [PubMed] [Google Scholar]
- 59. Waters M. J., Hoang H. N., Fairlie D. P., Pelekanos R. A., Brown R. J. (2006) New insights into growth hormone action. J. Mol. Endocrinol. 36, 1–7 [DOI] [PubMed] [Google Scholar]
- 60. Li Q., Wong Y. L., Kang C. (2014) Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim. Biophys. Acta 1838, 1313–1321 [DOI] [PubMed] [Google Scholar]
- 61. Pillutla R. C., Hsiao K. C., Beasley J. R., Brandt J., Østergaard S., Hansen P. H., Spetzler J. C., Danielsen G. M., Andersen A. S., Brissette R. E., Lennick M., Fletcher P. W., Blume A. J., Schäffer L., Goldstein N. I. (2002) Peptides identify the critical hotspots involved in the biological activation of the insulin receptor. J. Biol. Chem. 277, 22590–22594 [DOI] [PubMed] [Google Scholar]
- 62. Schäffer L., Brissette R. E., Spetzler J. C., Pillutla R. C., Østergaard S., Lennick M., Brandt J., Fletcher P. W., Danielsen G. M., Hsiao K. C., Andersen A. S., Dedova O., Ribel U., Hoeg-Jensen T., Hansen P. H., Blume A. J., Markussen J., Goldstein N. I. (2003) Assembly of high-affinity insulin receptor agonists and antagonists from peptide building blocks. Proc. Natl. Acad. Sci. U.S.A. 100, 4435–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stagsted J., Reaven G. M., Hansen T., Goldstein A., Olsson L. (1990) Regulation of insulin receptor functions by a peptide derived from a major histocompatibility complex class I antigen. Cell 62, 297–307 [DOI] [PubMed] [Google Scholar]
- 64. Stagsted J., Ziebe S., Satoh S., Holman G. D., Cushman S. W., Olsson L. (1993) Insulinomimetic effect on glucose transport by epidermal growth factor when combined with a major histocompatibility complex class I-derived peptide. J. Biol. Chem. 268, 1770–1774 [PubMed] [Google Scholar]
- 65. Zhang B., Salituro G., Szalkowski D., Li Z., Zhang Y., Royo I., Vilella D., Díez M. T., Pelaez F., Ruby C., Kendall R. L., Mao X., Griffin P., Calaycay J., Zierath J. R., Heck J. V., Smith R. G., Moller D. E. (1999) Discovery of a small molecule insulin mimetic with antidiabetic activity in mice. Science 284, 974–977 [DOI] [PubMed] [Google Scholar]
- 66. Air E. L., Strowski M. Z., Benoit S. C., Conarello S. L., Salituro G. M., Guan X. M., Liu K., Woods S. C., Zhang B. B. (2002) Small molecule insulin mimetics reduce food intake and body weight and prevent development of obesity. Nat. Med. 8, 179–183 [DOI] [PubMed] [Google Scholar]