

Background: The deubiquitinating enzyme USP33 controls several important cellular functions.
Results: HERC2 and p97 are critical for USP33 degradation.
Conclusion: HERC2 and p97 constitute a novel pathway to degrade USP33.
Significance: The HERC2/p97 degradative pathway may control USP33-dependent processes.
Keywords: Deubiquitination, E3 Ubiquitin Ligase, Proteasome, Protein Degradation, Ubiquitination, HERC2, USP33, p97
Abstract
Because the deubiquitinating enzyme USP33 is involved in several important cellular processes (β-adrenergic receptor recycling, centrosome amplification, RalB signaling, and cancer cell migration), its levels must be carefully regulated. Using quantitative mass spectrometry, we found that the intracellular level of USP33 is highly sensitive to the activity of p97. Knockdown or chemical inhibition of p97 causes robust accumulation of USP33 due to inhibition of its degradation. The p97 adaptor complex involved in this function is the Ufd1-Npl4 heterodimer. Furthermore, we identified HERC2, a HECT domain-containing E3 ligase, as being responsible for polyubiquitination of USP33. Inhibition of p97 causes accumulation of polyubiquitinated USP33, suggesting that p97 is required for postubiquitination processing. Thus, our study has identified several key molecules that control USP33 degradation within the ubiquitin-proteasome system.
Introduction
USP332 is a deubiquitinating enzyme (DUB), a class of proteins that regulate ubiquitin-dependent processes. By removing monoubiquitin or disassembling polyubiquitin chains from substrate proteins, DUBs counteract or regulate the activities of E3 ubiquitin ligases (1, 2). USP33 has been implicated in several important cellular functions. These functions include centrosome amplification (3), RalB-dependent signaling (4), and recycling of membrane receptors (5). In addition, USP33 is involved in SLIT-dependent axon guidance and cell migration events (6, 7), as well as stabilization of type 2 iodothyronine deiodinase which is important for supplying active thyroid hormones for brain development (8). Many of these pathways are sensitive to USP33 levels, as both knockdown and overexpression of USP33 result in specific cellular defects. Consequently, there appears to be a need to tightly regulate the levels of USP33. USP33 is a short lived DUB that is constantly turned over by the ubiquitin-proteasome system (9).
A previous study identified USP33 as an interacting protein of the von Hippel-Lindau protein (pVHL), an E3 ligase. pVHL-mediated polyubiquitination was proposed to target USP33 for degradation via the ubiquitin-proteasome system (9). However, this study utilized in vitro assays or cellular assays with exogenously expressed USP33, and the role of pVHL in regulating endogenous USP33 has not been validated. It therefore remains possible that additional pathways are involved in USP33 degradation.
In this study, we have identified and characterized a number of proteins critical for regulating the intracellular levels of USP33 using quantitative mass spectrometry, RNA interference, and biochemical approaches. These proteins consist of p97 (also known as VCP; CDC48 in yeast), its adaptor complex Ufd1-Npl4, and the E3 ubiquitin ligase HERC2. Together, p97, Ufd-Npl4, and HERC2 constitute a novel pathway to target USP33 for degradation via the ubiquitin-proteasome system.
EXPERIMENTAL PROCEDURES
Cell Culture and Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)
HeLa, 293T, NIH-3T3, 786-O, RCC4, and 293 Flp-In T-Rex cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. 293 Flp-In T-Rex HERC2-ShB cells expressing doxycycline-inducible and shRNA-resistant HERC2, or catalytically inactive (C4762S) HERC2 were generated by the Flp-In T-Rex system according to the manufacturer's instructions (Invitrogen). 786-O and RCC4 cells stably infected with control vector or HA-pVHL were kind gifts from William G. Kaelin Jr. and were described previously (10). For SILAC, HeLa cells were cultured in DMEM supplemented with 10% dialyzed fetal bovine serum (light labeling). For heavy labeling, Arg6 (U-13C6) and Lys8 (U-13C6, U-15N2) (Cambridge Isotopes) were supplemented at the same concentration as in the standard DMEM formulation.
Sample Preparation for Mass Spectrometry Analysis
For SILAC, a membrane fraction enriched with mitochondria was prepared from a 1:1 mixture of heavy and light SILAC-labeled HeLa p97-V1 cells (treated in the absence or presence of 1 μg/ml doxycycline for 2 days). This fraction was prepared as described previously (11). Isoelectric focusing of peptides was performed as described previously (11).
Mass Spectrometry Analysis
Mass spectrometry experiments were performed on an EASY-nLC connected to a hybrid LTQ-Orbitrap classic (Thermo Scientific) equipped with a nanoelectrospray ion source (Proxeon Biosystems) as described previously (12). Peptides were separated on a 15-cm reversed-phase analytical column (75-μm internal diameter) in-house packed with 3-μm C18 beads (ReproSil-Pur C18-AQ medium; Dr. Maisch GmbH) with a 160-min gradient from 2 to 30% acetonitrile in 0.2% formic acid at a flow rate of 350 nl/min. The mass spectrometer was operated in data-dependent mode to switch automatically between full scan MS and tandem MS acquisition. Survey full scan mass spectra were acquired in the Orbitrap (300–1700 m/z), after accumulation of 500,000 ions, with a resolution of 60,000 at 400 m/z. The top 10 most intense ions from the survey scan were isolated and, after the accumulation of 5,000 ions, fragmented in the linear ion trap by collision-induced dissociation (collisional energy 35% and isolation width 2 Da). Precursor ion charge state screening was enabled and all singly charged and unassigned charge states were rejected. The dynamic exclusion list was set with a maximum retention time of 90 s, a relative mass window of 10 ppm. Early expiration was enabled.
Data Analysis
Raw data files were analyzed by MaxQuant (v 1.4.1.2) (13, 14) and searched against the UniProt human database (15) (148,298 sequences), a contaminant database (247 sequences), and an equally sized decoy database. The latter consisted of reversed sequences with tryptic digestion and was searched with a maximum of two missed cleavages, fixed carboxyamidomethyl modifications of cysteine, variable oxidation modifications of methionine, and variable protein N terminus acetylations, with 1% false discovery rate thresholds for both peptides and proteins as estimated by the target decoy approach (16). At least two different peptide sequences were required for protein identification, and two different ratio measurements were required for protein quantitation. Protein SILAC ratio p values were calculated as described previously using a hierarchical bootstrap statistical model (17).
DNA Plasmids
N-terminally 3×FLAG-tagged HERC2 fragments (F1–F6) were PCR-amplified from HeLa cDNA library and cloned into pcDNA3.1+. N-terminally 3×FLAG-tagged full-length HERC2 (WT), and C4762S constructs were cloned into pcDNA5 FRT/TO and were generated by a combination of fusion PCR and multiple cloning steps. N-terminal 3×FLAG-tagged Npl4 and C-terminally single FLAG-tagged Ufd1 were PCR-amplified and cloned into pcDNA3.1(+). All DNA transfection experiments were performed using Lipofectamine 2000 (Invitrogen). For RNAi-resistant USP33, silent mutations that render the construct refractory to the USP33-ShA construct were introduced into the coding sequence of USP33 via PCR and cloned into a modified version of pRetroX Tet-on Advanced, where replacement of the rtTA segment results in constitutive expression of USP33 under the cytomegalovirus (CMV) promoter. Expression of this construct was mediated via retroviral transduction.
Antibodies
All antibodies used in this study are commercially available. The following monoclonal antibodies were used: USP33 (clone 5B5; Sigma), actin (clone C4; Millipore), FLAG and FLAG-peroxidase (clone M2; Sigma). The rabbit polyclonal antibodies used were: USP33 (Millipore), USP33 (Bethyl Laboratories), USP20 (Bethyl Laboratories), p97 (Cell Signaling), Oxa1 (Proteintech Group), HERC2 (Bethyl Laboratories), actin (Sigma), Npl4 (Bethyl Laboratories), UfD1 (Bethyl Laboratories), pVHL (Thermo Scientific).
RNA Interference
Cell lines (HeLa, 293T, NIH-3T3) with doxycycline-inducible expression of short hairpin RNA (shRNA) against p97 were established using the TRIPZ lentiviral shRNA system (Thermo Scientific). The targeted sequences for p97-V1 and p97-V4 are 5′-AACAGCCATTCTCAAACAGAA-3′ and 5′-GAATAGAGTTGTTCGGAATAA-3′, respectively. For UBXD7-V1, the targeted sequence is 5′-AAGCAACGAAGCTGTGAAGAAT-3′. In all experiments, 1 μg/ml doxycycline was used to induce shRNA expression, and a nontargeting shRNA was used as control (Thermo Scientific). For rescue experiments, p97 containing silent mutations that render the construct refractory to the p97-V1 shRNA was expressed via retroviral transduction in HeLa p97-V1 cells. The shRNA-resistant p97 construct was cloned into pRetroX-Tet-On Advanced (Clontech) to replace the rtTA-Advanced segment, resulting in constitutive expression of p97 under the CMV promoter. Transduced cells were selected by 1 mg/ml G418. For shRNA-mediated knockdown of USP33, HERC2, and Npl4, the indicated cell lines were transduced with retrovirus expressing shRNA from the human H1 promoter. The targeted sequences were: USP33-ShA (5′-GGACCAAATCTTTGGGCATGT-3′), HERC2-ShA (5′-GCCAATGTGATTGGTCCAATC-3′), HERC2-ShB (5′-GCACGCACATTGGAGATATAC-3′), HERC2-ShC (5′-GCAGTTCCTTTCTTAGCTTCG-3′), HERC2-ShD (5′-GGAAGAAAGTCATCGCCATCG-3′), Npl4-ShA (5′-GCTGAAGTGGCTGCGATTTAT-3″), Npl4-ShC (5′-GCGGAAGGTTGGCTGGATATT-3′).
For small interfering RNA (siRNA) experiments, dicer-substrate RNA (DsiRNA) duplexes were used (IDT). The target sequences were: Npl4 siRNA1 (5′-CGGTTTACATCAATAGAAACAAGAC-3′), Ufd1 siRNA1 (5′-CAAACTGACCAATAAGAATTCGGAC-3′), nontargeting control (5′-CGTTAATCGCGTATAATACGCGTAT-3′). Transfections of siRNA were performed using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's protocol.
Co-immunoprecipitation
Freshly harvested cells were washed once in ice-cold phosphate-buffered saline (PBS) followed by lysis in immunoprecipitation (IP) buffer (50 mm HEPES-KOH, pH 7.0, 250 mm NaCl, 5 mm EDTA, 10% glycerol, 0.1% Igepal CA-630) supplemented with 1× Halt protease inhibitors mixture (Pierce) and 10 μm MG132. Cell lysis was carried out at 4 °C with constant rotation for 1 h, and insoluble materials were removed by centrifugation at 13,000 × g for 15 min. Protein concentrations of the cleared lysates were quantified, and 1–5 mg of total protein was used for co-immunoprecipitation with the indicated antibodies for 2 h at 4 °C. For control experiments, an equal amount of purified immunoglobulin G (IgG) from the host species was used. Protein A/G-agarose (Pierce) was subsequently used to capture the immuno-complexes (1.5 h at 4 °C), followed by four washes with IP buffer. Bound proteins were eluted by boiling in SDS-PAGE sample loading buffer and analyzed by SDS-PAGE and immunoblotting. For immunoprecipitation of FLAG-tagged proteins, anti-FLAG M2 affinity gel (Sigma) was used.
Affinity Purification of Polyubiquitinated Proteins
Freshly harvested cells were washed once in ice-cold PBS, followed by lysis in IP buffer containing 0.5% Igepal CA-630, 10 mm N-ethylmaleimide, 10 μm MG132, 50 μm PR-619 (LifeSensors Inc.), and 1× Halt protease inhibitors mixture. Cleared lysates were prepared as described for the co-immunoprecipitation experiments, and ubiquitinated proteins were affinity-purified from ∼2 mg of total protein using Ubiquilin 1 Tandem UBA (TUBE2) agarose (Boston Biochem) at 4 °C, with constant rotation for 2 h. Bound proteins were washed four times using the same buffer and eluted by boiling in SDS-PAGE sample loading buffer.
RESULTS
p97 Is Required for the Degradation of USP33
p97 is an abundant, ubiquitously expressed member of the AAA (ATPase associated with diverse cellular activities) protein family and is involved in a broad range of cellular activities (18–20). With help from distinct cofactor proteins and using energy derived from ATP hydrolysis, p97 can structurally remodel or unfold ubiquitinated client proteins (21, 22). This ability enables p97 to function as a “segregase,” whereby it mechanically extracts client proteins from their native environment. Examples include dissociation of protein complexes or extraction of proteins from membrane surfaces. In doing so, p97 can facilitate membrane trafficking, protein complex remodeling, and proteasomal protein degradation (19, 20, 23).
To identify p97-regulated proteins, quantitative mass spectrometry was used to measure protein levels in a mitochondria-enriched fraction after depletion of p97. We used a HeLa cell line (HeLa p97-V1) in which efficient silencing of p97 is achieved via doxycycline-inducible expression of an shRNA construct against p97 (Fig. 1A). After 2 days of p97 depletion, the cells reproducibly showed a ∼3.3-fold increase in the levels of USP33 (Fig. 1B). Other DUBs in the mass spectrometry dataset were unaffected. Although we initially identified USP33 from a mitochondria-enriched fraction, other studies have indicated localization of USP33 to nonmitochondrial sites, such as the endoplasmic reticulum, Golgi, and centrosome (3, 8, 24). We also have not found evidence for a mitochondrial localization, and therefore our identification of USP33 in the mitochondrial fraction likely resulted from the high sensitivity of mass spectrometry coupled with low level contamination of this fraction by other cellular components. Nevertheless, these data suggest that USP33 accumulates upon depletion of p97.
FIGURE 1.

p97 inhibition reduces the degradation of USP33. A, efficient doxycycline (dox)-induced knockdown of p97 in HeLa p97-V1 cells. HeLa cells were transduced with retrovirus encoding doxycycline-induced shRNA against p97 and treated with the presence or absence of 1 μg/ml doxycycline for the indicated times. Total cell lysates were isolated and analyzed by immunoblotting. B, SILAC data comparing p97-depleted cells with control cells. SILAC-labeled HeLa p97-V1 cells were treated in the presence (heavy-labeled) or absence (light-labeled) of 1 μg/ml doxycycline for 2 days to knock down p97. A mitochondria-enriched membrane fraction was analyzed by mass spectrometry. The SILAC (heavy/light) ratios combined from two independent biological samples are presented. For each indicated protein, at least 10 independent peptide measurements were obtained. Error bars indicate the S.E. Asterisks indicate p value <0.0001. C, analysis of USP33 levels after p97 knockdown. HeLa p97-V1 cells were cultured in the absence or presence of 1 μg/ml doxycycline (dox) for the indicated time, and total cell lysates were analyzed by immunoblotting against USP33, p97, and the loading control Oxa1. D, same as in C, except the murine fibroblast NIH-3T3 and human embryonic kidney 293T cells were used, respectively. E, same as C, except that an independent shRNA (p97-V4) against p97 was used. For control, a nontargeting shRNA was used. F, rescue of p97 knockdown cells. HeLa p97-V1 cells expressing empty vector or an shRNA-resistant version of p97 (p97R) were treated with 1 μg/ml doxycycline to induce knockdown of endogenous p97. Total cell lysates were isolated and analyzed by immunoblotting. G, effects of p97 knockdown on exogenously expressed USP33. p97-V1 cells in which the endogenous expression of USP33 was constitutively silenced (USP33-ShA) were transduced with a retrovirus encoding a shRNA-resistant version of untagged USP33 (exo USP33) where indicated. Cells were treated with or without 1 μg/ml doxycycline to knock down p97 for the indicated time, and the levels of USP33 were analyzed by immunoblotting. H, analysis of USP33 degradation upon chemical inhibition of p97. HeLa cells were treated with 100 μg/ml cycloheximide (CHX) and 10 μm NMS-873 where indicated, and total cell lysates were analyzed by immunoblotting. I, accumulation of ubiquitinated USP33 upon inhibition of p97. HeLa cells were treated with control (dimethyl sulfoxide) or 10 μm NMS-873 for 12 h, and total cellular ubiquitinated proteins were affinity-purified with TUBE2-agarose. Ubiquitinated USP33 (Ub-USP33) was detected by immunoblotting using anti-USP33 antibody.
To validate the mass spectrometry results, we cultured HeLa p97-V1 cells in the presence of doxycycline for 3 days and analyzed the steady-state levels of endogenous USP33 by immunoblotting total cell lysates. We found that the levels of USP33 increased up to 8-fold upon depletion of p97 (Fig. 1C). The effect of p97 knockdown on USP33 was not limited to HeLa cells, as similar results were obtained from the human cell line HEK293 and the murine cell line NIH-3T3 (Fig. 1D). An independent shRNA targeted against a different region of the p97 transcript caused similar accumulation, whereas a nontargeting shRNA did not (Fig. 1E). Further arguing against an off-target effect, expression of shRNA-resistant p97 completely suppressed the accumulation of USP33 caused by knockdown of endogenous p97 (Fig. 1F).
p97 facilitates the degradation of a number of proteins via the ubiquitin-proteasome pathway (25–32). To test whether p97 regulates USP33 posttranslationally, we exogenously expressed an RNAi-resistant version of USP33 in a modified p97-V1 cell line where the endogenous expression of USP33 is constitutively silenced. Upon knockdown of p97 via doxycycline treatment, we found that the exogenously expressed USP33 also accumulated (Fig. 1G), thus demonstrating that USP33 is regulated by p97 at a posttranslational level. Using cycloheximide chase experiments, we compared the degradation rate of endogenous USP33 with and without p97 activity. For these experiments, shRNA against p97 is not ideal, because long term knockdown of p97 results in higher starting levels of USP33. We therefore used the recently described p97 inhibitor NMS-873 (33, 34), which causes acute inhibition of p97 activity and circumvents potential secondary effects that may result from long term p97 depletion. Upon addition of cycloheximide to inhibit new protein synthesis, USP33 degradation was monitored. Addition of NMS-873 to the cycloheximide chase experiment did not impact the starting levels of USP33 but caused complete inhibition of USP33 degradation over the course of 9 h (Fig. 1H). In control cells, ∼70% of USP33 was degraded by 9 h. We therefore conclude that the effect of p97 knockdown on USP33 is at the level of protein degradation.
Because USP33 is degraded by the ubiquitin-proteasome system (9), we tested whether inhibition of p97 results in changes in the polyubiquitination levels of endogenous USP33. A 12-h treatment with NMS-873 caused a modest increase in the steady-state levels of USP33 (Fig. 1I). Upon affinity purification of polyubiquitinated proteins, we found a substantial increase in the level of high molecular weight USP33 species in the NMS-873 treated cells (Fig. 1I). These data support the notion that p97 is important for targeting polyubiquitinated USP33 for proteasomal degradation.
The p97 Adaptor Complex Ufd1-Npl4 Is Important for USP33 Degradation
p97 can interact with a multitude of protein cofactors and binding partners. These cofactor proteins are generally thought to act as adaptors to direct p97 to its different substrates, thereby enabling the functional diversity of p97 (22, 35–37). To elucidate which p97 cofactor regulates USP33, we used RNAi to knockdown Npl4, a subunit of the Ufd1-Npl4 heterodimer that has been previously implicated in p97-dependent proteolysis in the cytoplasm, as well as during endoplasmic reticulum-associated degradation and outer mitochondrial membrane protein degradation (25, 27, 29, 30). Two shRNA constructs against Npl4 were expressed independently via retroviral transduction, and we analyzed the total cell extracts isolated 48 h after infection by immunoblotting. Both shRNA constructs efficiently silenced endogenous Npl4 and caused obvious accumulation of USP33 (Fig. 2A). Likewise, when the expression of Ufd1 was silenced by siRNA, we also found a similar accumulation of USP33 (Fig. 2B). These effects were specific for the Ufd1-Npl4 complex, because knockdown of UBXD7, another p97 cofactor, had no effect on the steady-state levels of USP33 (Fig. 2C).
FIGURE 2.

The p97 adaptor Ufd1-Npl4 regulates USP33 levels. A, accumulation of USP33 upon knockdown of Npl4. HeLa cells were transduced with retrovirus encoding control (nontargeting) shRNA or shRNA against Npl4. 48 h after infection, total cell lysates were isolated and analyzed by immunoblotting. B, accumulation of USP33 upon knockdown of Ufd1. HeLa cells were transfected with control (nontargeting) siRNA or siRNA against Ufd1. Cell lysates were isolated at the indicated time after transfection and analyzed by immunoblotting. C, USP33 levels upon UBXD7 knockdown. HeLa cells transduced with doxycyline (dox)-inducible shRNA against UBXD7 were treated with 1 μg/ml doxycycline where indicated for 72 h, and total cell lysates were analyzed by immunoblotting. D, co-immunoprecipitation (IP) of USP33 with Npl4. HeLa cells were transiently transfected with pcDNA FLAG-Npl4 or empty vector (control). 40 h after transfection, total cell lysates were prepared, and co-immunoprecipitation was performed using anti-FLAG antibodies conjugated to agarose resin. E, co-immunoprecipitation of USP33 with Ufd1. Results are same as D, except cells were transfected with FLAG-Ufd1. F, accumulation of ubiquitinated USP33 upon knockdown of Npl4. HeLa cells were transfected with control (nontargeting) siRNA or Npl4 siRNA1. 48 h after transfection, total ubiquitinated proteins were affinity-purified using TUBE2-agarose. Ubiquitinated USP33 was detected by immunoblotting with antibodies specific to USP33. G, same as in F, except Ufd1 was silenced by transfection of siRNA against Ufd1.
The effects of silencing Ufd1 or Npl4 on USP33 likely represent a direct involvement of the Ufd1-Npl4 heterodimer in regulating USP33, as both FLAG-Npl4 (Fig. 2D) and FLAG-Ufd1 (Fig. 2E) readily co-immunoprecipitated endogenous USP33. Furthermore, knockdown of Npl4 (Fig. 2F) or Ufd1 (Fig. 2G) caused accumulation of polyubiquitinated USP33. Collectively, these data strongly suggest that the p97 adaptor Ufd1-Npl4 regulates the degradation of USP33.
The HERC2 Ubiquitin E3 Ligase Targets USP33 for p97-dependent Degradation
To identify the E3 ligase important for targeting USP33 for degradation, we initially focused our attention on pVHL, a component of a cullin E3 ligase that had previously been implicated in mediating the proteasomal degradation of USP33 (9). However, we did not observe any significant changes to the steady-state levels of USP33 upon ectopic expression of pVHL in two independent renal carcinoma cell lines, 786-O and RCC4, which are naturally deficient of pVHL (Fig. 3A). Furthermore, knockdown of p97 in 786-O cells still caused dramatic accumulation of USP33 (Fig. 3B). This result indicates that pVHL is dispensable for p97-dependent degradation of USP33; however, pVHL may play a role in regulating USP333 under other cellular conditions.
FIGURE 3.
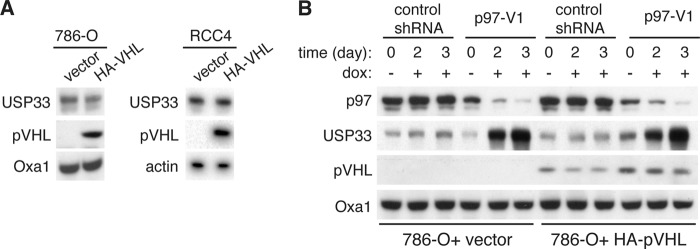
pVHL is dispensable for p97-dependent degradation of USP33. A, steady-state levels of USP33 upon expression of pVHL. Total cell lysates were isolated from 786-O or RCC4 (both naturally deficient of pVHL expression) cells stably expressing control vector or HA-tagged pVHL. Protein levels were analyzed by immunoblotting. B, accumulation of USP33 upon p97 knockdown in 786-O cells. 786-O cells expressing control vector or HA-pVHL were transduced with lentivirus encoding a doxycycline (dox)-inducible shRNA against p97 (p97-V1). Cells were treated with or without 1 μg/ml doxycycline for the indicated time, and total cell lysates were isolated and analyzed by immunoblotting.
We therefore searched for other candidate E3 ligases that may regulate USP33. HERC2 is a giant 550-kDa HECT domain-containing E3 ligase that has roles in orchestrating the DNA damage response (38), proteasomal degradation of proteins (39, 40), and regulation of centrosome morphology (41). In a proteomics study, a high confidence physical interaction was identified between HERC2 and the deubiquitinating enzyme USP20, and a potential interaction between HERC2 and USP33 was suggested (42). USP20 is a homologue of USP33, and they share ∼60% sequence identity (43). To test whether HERC2 can interact with USP33, we performed co-immunoprecipitation experiments with an anti-HERC2 antibody. We found that endogenous HERC2 could indeed interact with USP33, as well as with USP20 (Fig. 4A). To further characterize the interaction between these proteins, we generated a series of partially overlapping FLAG-tagged constructs (F1–F6) that span the entire protein sequence of HERC2 and tested their interaction with endogenous USP33 (Fig. 4B). USP33 bound selectively to the F4 and F5 fragments, which encompass a region upstream of the C-terminal HECT E3 ligase domain of HERC2. Both the F4 and F5 fragments have an RCC1-like domain, which consists of a seven-blade β-propeller fold that has been implicated in mediating protein-protein interactions, lipid binding, and nucleotide exchange (44). Within the F4 fragment there is a zinc finger domain and a DOC (destruction of cyclin B) domain, both known to mediate protein-protein interactions (45–48). DOC domains are found exclusively in subclasses of E3 ligases. In the case of the APC10 subunit of the APC ubiquitin ligase involved in mitotic progression, the DOC domain has been shown to enhance binding of the ligase to its substrate, thus promoting substrate polyubiquitination (45, 47, 49). Of note, USP20 bound exclusively to the F4 fragment.
FIGURE 4.

HERC2 is an E3 ligase important for the degradation of USP33. A, co-immunoprecipitation (IP) of endogenous USP33 and USP20 with endogenous HERC2. Cell lysates were prepared from HeLa cells and immunoprecipitation experiments were performed using control or anti-HERC2 antibody. Co-immunoprecipitated proteins were detected by immunoblotting. Asterisk, nonspecific band detected by the anti-USP20 antibody. B, characterization of HERC2-USP33 interaction. FLAG-tagged HERC2 fragments were transiently transfected into HeLa cells, and anti-FLAG antibody was used to immunoprecipitate the HERC2 fragments. Co-immunoprecipitated endogenous proteins were detected by immunoblotting. DOC, doxycyline. C, accumulation of USP33 upon knockdown of HERC2. Control (nontargeting) shRNA or four independent shRNAs targeting different regions of the HERC2 transcript were expressed by retroviral transduction in HeLa cells. Cell lysates were isolated and analyzed by immunoblotting. D, analysis of USP33 degradation upon HERC2 knockdown. HeLa cells transduced with retrovirus encoding a control (nontargeting) shRNA or an shRNA against HERC2 (HERC2-ShB) were treated with 100 μg/ml cycloheximide (CHX) for the indicated time. Total cell lysates were isolated and analyzed by immunoblotting. E, rescue of HERC2 knockdown cells. 293 Flp-In T-Rex HERC2 knockdown cells (HERC2-ShB) expressed control vector, FLAG-tagged (RNAi-resistant) wild-type (WTR), or catalytically inactive C4762S (CSR) HERC2. To induce expression, cells were treated with 0.1 μg/ml doxycycline (dox) for 40 h, and total cell lysates were analyzed by immunoblotting.
To determine whether HERC2 is important for regulating the degradation of USP33, we generated four HeLa cell lines each expressing a distinct retroviral shRNA construct against HERC2. These shRNA constructs silenced HERC2 expression with varying efficiencies and caused corresponding increases in USP33 levels (Fig. 4C). Using a cycloheximide chase experiment, we compared the degradation rate of USP33 between HeLa cells expressing HERC2-ShB versus a nontargeting shRNA. HERC2 knockdown cells contained much higher levels of endogenous USP33, and no significant reduction of USP33 was detected within a 9-h experiment. In contrast, ∼75% of USP33 was degraded in control cells by 9 h (Fig. 4D).
A previous study suggested that the catalytic activity of HERC2 is not strictly required for all of its functions (38). To determine whether the E3 ligase activity is required for regulating USP33, we introduced shRNA-resistant wild-type or catalytically inactive (C4762S) FLAG-tagged HERC2 into the genome of HERC2-knockdown 293 Flp-In T-Rex cells. The integration of these constructs was achieved via Flp recombinase-mediated recombination into a single FRT site within the genome of the parental 293 Flp-In T-Rex cells, and the expression of the FLAG-tagged HERC2 constructs is doxycycline inducible. Consistent with our data in HeLa cells, knockdown of HERC2 caused significant accumulation of USP33. Overexpression of wild-type HERC2, but not the C4762S mutant, suppressed this accumulation (Fig. 4E). In fact, cells expressing the C4762S mutant accumulated an even higher level of USP33, suggesting a dominant negative effect. These data suggest that HERC2 functions as a canonical E3 ligase to target USP33 for proteasomal degradation.
Next, we sought to determine whether HERC2 and p97 function within the same pathway to regulate the degradation of USP33. Because loss of p97 activity resulted in accumulation of polyubiquitinated USP33 (Fig. 1I), we asked whether these ubiquitination modifications are dependent on HERC2. To address this issue, we modified the inducible p97 knockdown cell line to express HERC2 shRNA or a nontargeting shRNA. Upon affinity purification of total ubiquitinated proteins and immunoblotting for USP33, we found that the accumulation of ubiquitinated USP33 caused by p97 knockdown was lost in HERC2-depleted cells (Fig. 5A). Similar results were obtained when p97 activity was inhibited by NMS-873 (Fig. 5B). Strikingly, despite a substantially higher level of total cellular USP33 in HERC2 knockdown cells, the amount of ubiquitinated USP33 in HERC2 knockdown cells was lower than that in control cells that were treated with NMS-873. Together, these data demonstrate that HERC2 and p97 participate in the same pathway to mediate degradation of USP33 and that HERC2 functions as an E3 ligase upstream of p97.
FIGURE 5.

HERC2 is required for accumulation of ubiquitinated USP33 upon p97 inhibition. A, analysis of ubiquitinated USP33 in p97 knockdown cells lacking HERC2. HeLa p97-V1 cells were transduced with retrovirus expressing either a control (nontargeting) shRNA or an shRNA against HERC2 (HERC-ShB). Cells were treated with 1 μg/ml doxycycline (dox) for 48 h where indicated, and total cell lysates were isolated. Total ubiquitinated proteins were affinity-purified using TUBE2-resin, and ubiquitinated USP33 (Ub-USP33) was detected by immunoblotting. B, analysis of ubiquitinated USP33 upon NMS-873 treatment in cells depleted for HERC2. Control (expressing nontargeting shRNA) or stable knockdown cell lines of HERC2 (HERC2-ShB) or USP33 (USP33-ShA) were treated with 10 μm NMS-873 for 12 h. Total cell lysates were isolated, and TUBE2-resin was used to affinity purify total ubiquitinated proteins. Ubiquitinated USP33 (Ub-USP33) was detected by immunoblotting.
To understand the role of p97 in the HERC2-dependent degradation of USP33, we analyzed the interaction between endogenous HERC2 and USP33 in response to inhibition of p97. NMS-873 treatment had no effect on the amount of USP20 that co-immunoprecipitated with HERC2. However, the amount of co-immunoprecipitated USP33 was substantially increased (Fig. 6A). Likewise, when we performed a reciprocal co-immunoprecipitation experiment using an anti-USP33 antibody, the levels of co-immunoprecipitated HERC2 were also selectively increased (Fig. 6B). These experiments suggest that p97 may regulate the interaction of USP33 with HERC2.
FIGURE 6.
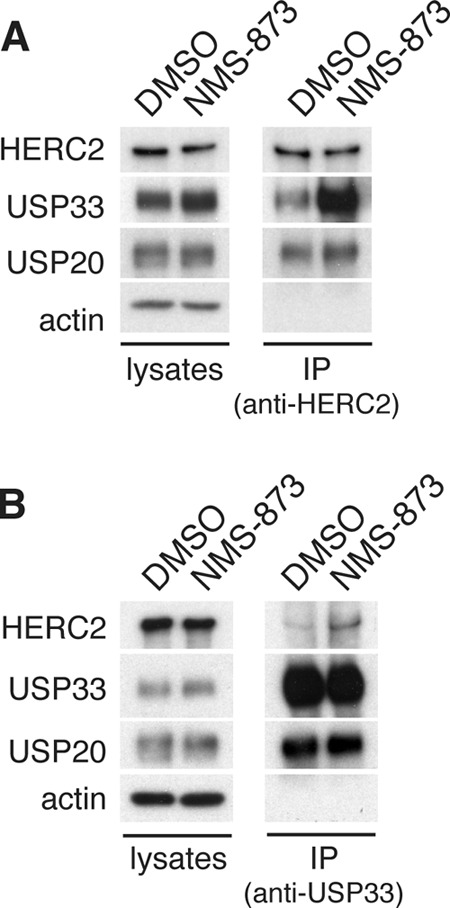
p97 regulates the interaction between HERC2 and USP33. A, co-immunoprecipitation of HERC2 and USP33 upon p97 inhibition. HeLa cells were treated with control (DMSO) or 10 μm NMS-873 for 12 h, and total lysates were harvested for immunoprecipitation (IP) using an anti-HERC2 antibody. Co-immunoprecipitated proteins were detected by immunoblotting. B, similar to A, except anti-USP33 antibodies were used to immunoprecipitate USP33.
DISCUSSION
In this study, we identified a pathway of USP33 degradation within the ubiquitin-proteasome system. This pathway consists of three regulators: the E3 ligase HERC2, the AAA ATPase p97, and its heterodimeric cofactor Ufd1-Npl4. Our data support a model in which HERC2 polyubiquitinates USP33 to target it for proteasomal degradation. Subsequently, p97 and its adaptor Ufd1-Npl4 are required for efficient proteolysis of the polyubiquitinated USP33 (Fig. 7).
FIGURE 7.
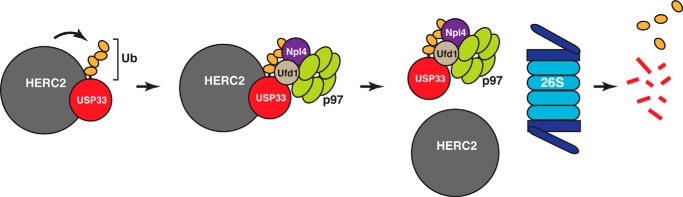
Model of the HERC2-p97-dependent degradation of USP33. HERC2 physically interacts with USP33, leading to USP33 polyubiquitination. p97 and its adaptor complex Ufd1-Npl4 are necessary for the postubiquitination processing of USP33. They facilitate the dissociation of polyubiquitinated USP33 from HERC2, a step that may be important for efficient proteolysis of USP33 by the 26S proteasome.
HERC2 is a HECT domain-containing E3 ligase that has been shown to participate in diverse cellular pathways including the DNA damage response, centrosome morphogenesis, and ubiquitin-proteasome-dependent protein degradation (38–41). Because depletion of HERC2 causes dramatic accumulation of intracellular USP33, decreased USP33 polyubiquitination, and inhibition of degradation, HERC2 appears to be the predominant E3 ligase that targets USP33 for proteasomal degradation. At present, our data do not formally eliminate the possibility that USP33 might be regulated by yet another E3 ligase that is in turn activated by HERC2. However, given that endogenous HERC2 and USP33 physically interact with each other in co-immunoprecipitation experiments, it seems likely that USP33 is a bona fide HERC2 substrate.
USP33 has been implicated in a number of disparate cellular processes. Although the dramatic increase in USP33 levels as a result of HERC2 or p97 depletion highlights the importance of these proteins in regulating USP33, we currently do not know whether HERC2 and p97 act upstream of all processes that involve USP33.
Further work will be necessary to clarify the mechanism of p97 in facilitating USP33 degradation. Because loss of p97 activity causes increased association between HERC2 and USP33, the most straightforward interpretation is that p97 functions as a segregase to dissociate USP33 from HERC2 so that USP33 can be efficiently recognized and degraded by the proteasome (Fig. 7). More speculatively, it is also possible that p97 regulates the activity of HERC2 by controlling its interaction with USP33. During endoplasmic reticulum-associated degradation, deubiquitinating enzymes have been demonstrated to facilitate substrate discrimination by E3 ligases (50). Analogously, binding of USP33 to HERC2 may allow the deubiquitinating activity of USP33 to counteract or balance the E3 ligase activity of HERC2, thereby controlling substrate polyubiquitination.
The molecules we have identified as USP33 regulators are involved in pathological conditions in humans and mice. Mutations in p97 are causal for inclusion body myopathy with early onset Paget's disease and frontotemporal dementia (IBMPFD) (51), whereas mutations in HERC2 lead to the rjs (runty jerky sterile) phenotype in mice (52, 53) and neurodevelopmental delay with Angelman-like features in humans (54). It will be interesting to test whether defects in USP33 degradation play a role in the pathogenesis of these conditions.
Acknowledgments
We are grateful to Tomohiko Ohta for HERC2 constructs that helped us reconstruct full-length HERC2; William G. Kaelin Jr. for the 786-O and RCC4 cell lines; Tsui-Fen Chou (Harbor-UCLA Medical Center) for helpful discussions and reagents during the early stage of this work; Robert Graham (currently at University of Manchester) from the Proteosome Exploration Laboratory (PEL) for technical help with the mass spectrometry experiments; and Michael Walters and Lev G. Lis (University of Minnesota) for NMS-873. The PEL is supported by the Gordon and Betty Moore Foundation through Grant GBMF775 and the Beckman Institute.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM062967. This work was also supported by the Howard Hughes Medical Institute.
- USP
- ubiquitin-specific protease
- DUB
- deubiquitinating enzyme
- HECT
- homologous to the E6-AP C terminus
- IP
- immunoprecipitation
- pVHL
- von Hippel-Lindau protein
- SILAC
- stable isotope labeling by amino acids in cell culture
- DOC
- destruction of cyclin B.
REFERENCES
- 1. Reyes-Turcu F. E., Ventii K. H., Wilkinson K. D. (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Komander D., Clague M. J., Urbé S. (2009) Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 3. Li J., D'Angiolella V., Seeley E. S., Kim S., Kobayashi T., Fu W., Campos E. I., Pagano M., Dynlacht B. D. (2013) USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 495, 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simicek M., Lievens S., Laga M., Guzenko D., Aushev V. N., Kalev P., Baietti M. F., Strelkov S. V., Gevaert K., Tavernier J., Sablina A. A. (2013) The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol. 15, 1220–1230 [DOI] [PubMed] [Google Scholar]
- 5. Berthouze M., Venkataramanan V., Li Y., Shenoy S. K. (2009) The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 28, 1684–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yuasa-Kawada J., Kinoshita-Kawada M., Wu G., Rao Y., Wu J. Y. (2009) Midline crossing and Slit responsiveness of commissural axons require USP33. Nat. Neurosci. 12, 1087–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yuasa-Kawada J., Kinoshita-Kawada M., Rao Y., Wu J. Y. (2009) Deubiquitinating enzyme USP33/VDU1 is required for Slit signaling in inhibiting breast cancer cell migration. Proc. Natl. Acad. Sci. U.S.A. 106, 14530–14535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Curcio-Morelli C., Zavacki A. M., Christofollete M., Gereben B., de Freitas B. C., Harney J. W., Li Z., Wu G., Bianco A. C. (2003) Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J. Clin. Invest. 112, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z., Na X., Wang D., Schoen S. R., Messing E. M., Wu G. (2002) Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 277, 4656–4662 [DOI] [PubMed] [Google Scholar]
- 10. Li L., Zhang L., Zhang X., Yan Q., Minamishima Y. A., Olumi A. F., Mao M., Bartz S., Kaelin W. G., Jr. (2007) Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol. Cell. Biol. 27, 5381–5392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan N. C., Salazar A. M., Pham A. H., Sweredoski M. J., Kolawa N. J., Graham R. L., Hess S., Chan D. C. (2011) Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20, 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalli A., Smith G. T., Sweredoski M. J., Hess S. (2013) Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: focus on LTQ-Orbitrap mass analyzers. J. Proteome Res. 12, 3071–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 14. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 15. UniProt Consortium. (2013) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 41, D43–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elias J. E., Gygi S. P. (2010) Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 604, 55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pierce N. W., Lee J. E., Liu X., Sweredoski M. J., Graham R. L., Larimore E. A., Rome M., Zheng N., Clurman B. E., Hess S., Shan S. O., Deshaies R. J. (2013) Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 153, 206–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyer H., Bug M., Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 19. Dantuma N. P., Hoppe T. (2012) Growing sphere of influence: Cdc48/p97 orchestrates ubiquitin-dependent extraction from chromatin. Trends Cell Biol. 22, 483–491 [DOI] [PubMed] [Google Scholar]
- 20. Baek G. H., Cheng H., Choe V., Bao X., Shao J., Luo S., Rao H. (2013) Cdc48: a Swiss army knife of cell biology. J Amino Acids 2013:183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stolz A., Hilt W., Buchberger A., Wolf D. H. (2011) Cdc48: a power machine in protein degradation. Trends Biochem. Sci. 36, 515–523 [DOI] [PubMed] [Google Scholar]
- 22. Schuberth C., Buchberger A. (2008) UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell. Mol. Life Sci. 65, 2360–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bug M., Meyer H. (2012) Expanding into new markets: CP/p97 in endocytosis and autophagy. J. Struct. Biol. 179, 78–82 [DOI] [PubMed] [Google Scholar]
- 24. Thorne C., Eccles R. L., Coulson J. M., Urbé S., Clague M. J. (2011) Isoform-specific localization of the deubiquitinase USP33 to the Golgi apparatus. Traffic 12, 1563–1574 [DOI] [PubMed] [Google Scholar]
- 25. Heo J. M., Livnat-Levanon N., Taylor E. B., Jones K. T., Dephoure N., Ring J., Xie J., Brodsky J. L., Madeo F., Gygi S. P., Ashrafi K., Glickman M. H., Rutter J. (2010) A stress-responsive system for mitochondrial protein degradation. Mol. Cell 40, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu S., Peng G., Wang Y., Fang S., Karbowski M. (2011) The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 22, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barbin L., Eisele F., Santt O., Wolf D. H. (2010) The Cdc48-Ufd1-Npl4 complex is central in ubiquitin-proteasome triggered catabolite degradation of fructose-1,6-bisphosphatase. Biochem. Biophys. Res. Commun. 394, 335–341 [DOI] [PubMed] [Google Scholar]
- 28. Ghislain M., Dohmen R. J., Levy F., Varshavsky A. (1996) Cdc48p interacts with Ufd3p, a WD repeat protein required for ubiquitin-mediated proteolysis in Saccharomyces cerevisiae. EMBO J. 15, 4884–4899 [PMC free article] [PubMed] [Google Scholar]
- 29. Neuber O., Jarosch E., Volkwein C., Walter J., Sommer T. (2005) Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 7, 993–998 [DOI] [PubMed] [Google Scholar]
- 30. Schuberth C., Buchberger A. (2005) Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 7, 999–1006 [DOI] [PubMed] [Google Scholar]
- 31. Alberts S. M., Sonntag C., Schäfer A., Wolf D. H. (2009) Ubx4 modulates cdc48 activity and influences degradation of misfolded proteins of the endoplasmic reticulum. J. Biol. Chem. 284, 16082–16089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soetandyo N., Ye Y. (2010) The p97 ATPase dislocates MHC class I heavy chain in US2-expressing cells via a Ufd1-Npl4-independent mechanism. J. Biol. Chem. 285, 32352–32359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Polucci P., Magnaghi P., Angiolini M., Asa D., Avanzi N., Badari A., Bertrand J., Casale E., Cauteruccio S., Cirla A., Cozzi L., Galvani A., Jackson P. K., Liu Y., Magnuson S., Malgesini B., Nuvoloni S., Orrenius C., Sirtori F. R., Riceputi L., Rizzi S., Trucchi B., O'Brien T., Isacchi A., Donati D., D'Alessio R. (2013) Alkylsulfanyl-1,2,4-triazoles, a new class of allosteric valosine containing protein inhibitors: synthesis and structure-activity relationships. J. Med. Chem. 56, 437–450 [DOI] [PubMed] [Google Scholar]
- 34. Magnaghi P., D'Alessio R., Valsasina B., Avanzi N., Rizzi S., Asa D., Gasparri F., Cozzi L., Cucchi U., Orrenius C., Polucci P., Ballinari D., Perrera C., Leone A., Cervi G., Casale E., Xiao Y., Wong C., Anderson D. J., Galvani A., Donati D., O'Brien T., Jackson P. K., Isacchi A. (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 9, 548–556 [DOI] [PubMed] [Google Scholar]
- 35. Yeung H. O., Kloppsteck P., Niwa H., Isaacson R. L., Matthews S., Zhang X., Freemont P. S. (2008) Insights into adaptor binding to the AAA protein p97. Biochem. Soc. Trans. 36, 62–67 [DOI] [PubMed] [Google Scholar]
- 36. Dreveny I., Pye V. E., Beuron F., Briggs L. C., Isaacson R. L., Matthews S. J., McKeown C., Yuan X., Zhang X., Freemont P. S. (2004) p97 and close encounters of every kind: a brief review. Biochem. Soc. Trans. 32, 715–720 [DOI] [PubMed] [Google Scholar]
- 37. Kloppsteck P., Ewens C. A., Förster A., Zhang X., Freemont P. S. (2012) Regulation of p97 in the ubiquitin-proteasome system by the UBX protein family. Biochim. Biophys. Acta 1823, 125–129 [DOI] [PubMed] [Google Scholar]
- 38. Bekker-Jensen S., Rendtlew Danielsen J., Fugger K., Gromova I., Nerstedt A., Lukas C., Bartek J., Lukas J., Mailand N. (2010) HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 12, 80–86 [DOI] [PubMed] [Google Scholar]
- 39. Wu W., Sato K., Koike A., Nishikawa H., Koizumi H., Venkitaraman A. R., Ohta T. (2010) HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 70, 6384–6392 [DOI] [PubMed] [Google Scholar]
- 40. Lee T. H., Park J. M., Leem S. H., Kang T. H. (2014) Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 33, 19–25 [DOI] [PubMed] [Google Scholar]
- 41. Al-Hakim A. K., Bashkurov M., Gingras A. C., Durocher D., Pelletier L. (2012) Interaction proteomics identify NEURL4 and the HECT E3 ligase HERC2 as novel modulators of centrosome architecture. Mol. Cell. Proteomics 10.1074/mcp.M111.014233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sowa M. E., Bennett E. J., Gygi S. P., Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Z., Wang D., Na X., Schoen S. R., Messing E. M., Wu G. (2002) Identification of a deubiquitinating enzyme subfamily as substrates of the von Hippel-Lindau tumor suppressor. Biochem. Biophys. Res. Commun. 294, 700–709 [DOI] [PubMed] [Google Scholar]
- 44. Hadjebi O., Casas-Terradellas E., Garcia-Gonzalo F. R., Rosa J. L. (2008) The RCC1 superfamily: from genes, to function, to disease. Biochim. Biophys. Acta 1783, 1467–1479 [DOI] [PubMed] [Google Scholar]
- 45. Garcia-Gonzalo F. R., Rosa J. L. (2005) The HERC proteins: functional and evolutionary insights. Cell. Mol. Life Sci. 62, 1826–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ponting C. P., Blake D. J., Davies K. E., Kendrick-Jones J., Winder S. J. (1996) ZZ and TAZ: new putative zinc fingers in dystrophin and other proteins. Trends Biochem. Sci. 21, 11–13 [PubMed] [Google Scholar]
- 47. Passmore L. A., McCormack E. A., Au S. W., Paul A., Willison K. R., Harper J. W., Barford D. (2003) Doc1 mediates the activity of the anaphase-promoting complex by contributing to substrate recognition. EMBO J. 22, 786–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wendt K. S., Vodermaier H. C., Jacob U., Gieffers C., Gmachl M., Peters J. M., Huber R., Sondermann P. (2001) Crystal structure of the APC10/DOC1 subunit of the human anaphase-promoting complex. Nat. Struct. Biol. 8, 784–788 [DOI] [PubMed] [Google Scholar]
- 49. Carroll C. W., Morgan D. O. (2002) The Doc1 subunit is a processivity factor for the anaphase-promoting complex. Nat. Cell Biol. 4, 880–887 [DOI] [PubMed] [Google Scholar]
- 50. Zhang Z. R., Bonifacino J. S., Hegde R. S. (2013) Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 154, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., Kimonis V. E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 [DOI] [PubMed] [Google Scholar]
- 52. Ji Y., Walkowicz M. J., Buiting K., Johnson D. K., Tarvin R. E., Rinchik E. M., Horsthemke B., Stubbs L., Nicholls R. D. (1999) The ancestral gene for transcribed, low-copy repeats in the Prader-Willi/Angelman region encodes a large protein implicated in protein trafficking, which is deficient in mice with neuromuscular and spermiogenic abnormalities. Hum. Mol. Genet. 8, 533–542 [DOI] [PubMed] [Google Scholar]
- 53. Lehman A. L., Nakatsu Y., Ching A., Bronson R. T., Oakey R. J., Keiper-Hrynko N., Finger J. N., Durham-Pierre D., Horton D. B., Newton J. M., Lyon M. F., Brilliant M. H. (1998) A very large protein with diverse functional motifs is deficient in rjs (runty, jerky, sterile) mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9436–9441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harlalka G. V., Baple E. L., Cross H., Kühnle S., Cubillos-Rojas M., Matentzoglu K., Patton M. A., Wagner K., Coblentz R., Ford D. L., Mackay D. J., Chioza B. A., Scheffner M., Rosa J. L., Crosby A. H. (2013) Mutation of HERC2 causes developmental delay with Angelman-like features. J. Med. Genet. 50, 65–73 [DOI] [PubMed] [Google Scholar]