

Abstract
Oral infection of certain inbred mouse strains with the protozoan Toxoplasma gondii triggers inflammatory pathology resembling lesions seen during human inflammatory bowel disease, in particular Crohn's disease (CD). Damage triggered by the parasite is largely localized to the distal portion of the small intestine, and as such is one of only a few models for ileal inflammation. This is important because ileal involvement is a characteristic of CD in over two-thirds of patients. The disease induced by Toxoplasma is mediated by Th1 cells and the cytokines tumor necrosis factor-α and interferon-γ. Inflammation is dependent upon IL-23, also identified by genome-wide association studies as a risk factor in CD. Development of lesions is concomitant with emergence of E. coli that display enhanced adhesion to the intestinal epithelium and subepithelial translocation. Furthermore, depletion of gut flora renders mice resistant to Toxoplasma-triggered ileitis. Recent findings suggest complex CCR2-dependent interactions between lamina propria T cells and intraepithelial lymphocytes in fueling proinflammatory pathology in the intestine. The advantage of the Toxoplasma model is that disease develops rapidly (within 7–10 days of infection) and can be induced in immunodeficient mice by adoptive transfer of mucosal T cells from infected donors. We propose that Toxoplasma acts as a trigger setting into motion a series of events culminating in loss of tolerance in the intestine and emergence of pathogenic T cell effectors. The Toxoplasma trigger model is providing new leaps in our understanding of immunity in the intestine.
Keywords: infection, inflammation, mucosal immunity, protozoan
Inflammatory bowel diseases (IBD) in humans are immune-mediated disorders typically identified as Crohn's disease (CD) and ulcerative colitis (UC) depending on the disease phenotype and histological characteristics.1–3 UC manifests as severe inflammation of the colon and rectum that is generally thought to be mediated by cytokines of the Th2 family, in particular IL-13. In patients with CD, lesions are found in the ileum and colon. The inflammatory cytokine milieu during CD is predominantly Th1. More recently, IL-23/Th17 has emerged as an important inflammatory axis during CD.4 The incidence of both CD and UC are increasing in developed countries. While these conditions are rarely fatal, they are debilitating diseases to live with and in many cases they may progress to colorectal cancer.5 In economic terms, the cost of treating IBD has been estimated to be approximately 6 billion dollars per year in the United States alone.6
IBD is characterized as chronic relapsing intestinal inflammation associated with a compromised epithelial barrier, inappropriate immune responses to commensal intestinal microbes and overgrowth of pathogenic enteroadhesive E. coli and other microbes such as Bacteroides spp. A general model for disease pathogenesis involves an underlying genetic susceptibility on which an ill-defined trigger event is superimposed, followed by intestinal dysbiosis leading to fulminant pathology.7 Treatment approaches involve alleviation of symptoms with drugs and immune modulators, but some patients are less responsive than others.8 Therefore, an important research goal is to identify new tools and targets to treat the disease, whether they are aimed at restoring the balance of pathogenic versus commensal microflora, or whether they are targeted at host immunity and epithelial healing mechanisms.9–11 Using multiple disease models we can hope to understand immune function and dysfunction in the intestine, we can map out the cellular and molecular pathways leading from health to disease, and we can ultimately identify new targets to treat the disease.
A number of mouse models have been used to gain insight into IBD.12,13 Most involve Th1/Th17 responses that also characterize CD. A smaller number of models encompass Th2 responses that mimic UC. Generally, models can be classified as those that involve inappropriate effector responses and those that include defects in regulatory cells.14 An example of the former is the tumor necrosis factor (TNF)ΔARE mouse that overexpresses TNF-α.15 An example of the latter is the severe combined immunodeficiency-transfer model, in which transfer of splenic CD45RBhi T cells from wild-type mice induces colitis in severe combined immunodeficiency mice unless cells are co-transferred with regulatory T cells.16 IBD in mice can be induced genetically, chemically and by certain microbial infections. Among the microorganisms that trigger IBD-like lesions is the protozoan parasite Toxoplasma gondii.
Toxoplasma causes inflammation in the small intestine of certain inbred mice that resembles CD, in that it is characterized by Th1-induced lesions in the ileum. Lesion development is dependent upon orally acquired infection insofar as intraperitoneal injection of parasites does not elicit intestinal lesions. Mouse models for ileitis are scarce. The SAMP1/Yit mouse is a newly characterized strain that spontaneously develops CD-like lesions, and the TNF ΔARE strain that is genetically engineered to overexpress TNF-α also develops pathology resembling CD.17 Mice deficient for IL-10 also develop progressive lesions with resemblance to CD in many ways. An advantage of the Toxoplasma-trigger model is that disease onset is rapid and displays 100% penetrance. Unlike other CD models, T. gondii infection in this model leads to acute lethality, although (as described below) recent modifications to this model allow for induction of disease in the absence of lethality by adoptive cell transfer from infected to non-infected immunodeficient recipients. In this Review we discuss the Toxoplasma trigger model in the context of IBD, drawing attention to several recent findings that are yielding new insight into mechanisms of inflammatory disease in the small intestine.
T. Gondii—an Opportunistic TH1 Microbe
Toxoplasma is an extremely successful intracellular pathogen that infects 10–50% of the human population worldwide. The parasite is transmitted by carnivorism or predation, and also by ingestion of oocysts shed by members of the cat family.18 Infection is characterized by an acute phase, in which parasites cross the intestinal epithelium and widely disseminate as tachyzoites that undergo rapid cycles of invasion, replication, escape by host cell lysis, and re-invasion. Although most often asymptomatic, acute inection can sometimes lead to ocular involvement and other manifestations of infection.19 This is followed by chronic infection, characterized by emergence of long-lived cysts containing quiescent bradyzoites in tissues of the skeletal muscle and central nervous system. This stage of infection, also called the latent phase, is usually asymptomatic. However, during immunodeficiency such as in AIDS progression or immunosuppression following organ transplant, the parasite may undergo reactivation consisting of emergence from cysts as tissue-destructive tachyzoites.20,21 Without chemotherapy this can be lethal. Toxoplasma may also cross the placenta to cause serious damage to the fetus during primary infection. Furthermore, sequelae of infection often emerge later in life following in utero infection.22
Infection with T. gondii normally triggers robust protective cell-mediated immunity in which interferon (IFN) -γ has a central role. Early on, a model was established where the parasite triggers IL-12 production that acts with TNF-α to induce IFN-γ production by NK cells.23–26 Although macrophages and neutrophils produce IL-12 in response to Toxoplasma,24,27 studies in cell-specific knockout mice indicate that dendritic cells are likely the major IL-12 source in vivo.28 In mice, production of IL-12 and resistance to infection is highly dependent upon MyD88, implicating Toll-like receptors (TLR) in immune recognition.29–32 In this regard, there is evidence that TLR2, 4, 9 and 11 are involved in recognition, although knockout of no single TLR results in the high susceptibility observed following infection of Myd88−/− mice.33–37 Parasite ligands for TLR2 and 4 have been identified as glycosylphosphoinositol lipid moieties on the tachyzoite surface, and profilin in the parasite cytoplasm is recognized by TLR11, presumably following phagocytosis of dead or antibody-coated parasites.33,38,39 At present, direct recognition of Toxoplasma ligands by TLR9 is lacking. As described below, it is possible that recognition of gut bacterial DNA underlies the effects seen during T. gondii infection.
IL-12 production and NK cell activation is followed by emergence of antigen-specific Th1 cells as well as CD8+ T lymphocyte effectors. Through production of IFN-γ, CD4+ and CD8+ cells mediate protection during both acute and chronic stages of infection.26,40–42 Perforin-dependent cytolytic T cell activity also contributes to control of long-term infection, although this appears to be secondary to production of IFN-γ43,44 The latter cytokine provides protection in mice through STAT1-dependent induction of mediators such as nitric oxide and the IFN-γ-dependent immunity-related p47 GTPase (IRG) proteins.45–49 What provides protection in humans is less clear insofar as the IRG family is absent and nitric oxide-dependent killing of T. gondii has never been shown in our species.50,51 However, there is evidence for IFN-γ-independent killing in human macrophages that involves CD40 signaling and autophagy.52,53
Protective Immunity During the Mucosal Response to Toxoplasma
As an orally acquired infection, the host immune system first encounters Toxoplasma in the intestinal mucosa. The mucosal immune system consists of a single layer of epithelial cells that separates the underlying lamina propria (LP) from trillions of bacteria contained within the lumen of the gut.54 Interspersed in the epithelium are a heterogenous population of T cells collectively called intraepithelial lymphocytes (IEL) that contribute to homeostasis and defense in the intestine, but that can also cause damage under certain conditions.55 Most IEL express CD8αβ or CD8αα, and cells expressing the CD8α homodimer are composed of approximately equal numbers of αβ T-cell receptor (TCR) and γδTCR-expressing cells. The LP compartment is made up of myofibroblasts, T cells, B cells, macrophages and dendritic cells. Organized lymphoid tissues called Peyer's patches line the intestine. The Peyer's patches and LP are drained by lymphatics into mesenteric lymph nodes.
Following ingestion of oocysts or bradyzoites, the cyst wall is digested in the stomach, sporozoites or bradyzoites cross the small intestinal epithelium, enter the LP and differentiate into tachyzoites within ∼24 h.18 After 24–48 h, parasites begin to disseminate to extra-intestinal sites in a process that most likely involves utilizing macrophages and dendritic cells as carriers.56–58 Inflammatory macrophages are important microbicidal effectors at this stage of infection. Recruitment of these cells is dependent upon expression of chemokine receptor CCR2, resulting in killing of parasites.59 This is most likely dependent upon IFN-γ-induced IRG-mediated destruction of the parasitophorous vacuole.60
Initiation of immunity in the intestinal mucosa involves recognition of pathogen-associated molecular patterns such as profilin and glycosylphosphoinositol moieties.61 In the mouse model, activation of mucosal dendritic cells through TLRs such as TLR11 and possibly TLR2 and 4 results in antigen presentation and activation of T cells in regional lymphoid tissues. However, a recent study suggests that lack of these TLR only minimally affects generation of IFN-γ-secreting T cells.62 Instead, it was suggested that translocating bacteria have a central role in initiating immunity to the parasite, essentially acting as an adjuvant to kick start antigen-specific immunity to Toxoplasma. Evidence for this model is that mice depleted of gut flora display impaired generation of cytokine-secreting T cells after T. gondii infection, and this defect is corrected by oral administration of bacterial lipopolysaccharide.62,63
A collection of studies indicate the importance of three-way interactions between CD4+ LP T cells, CD8αβ IEL, and enterocytes in maintaining gut homeostasis and providing protection during Toxoplasma infection. CD8αβ IEL from infected mice produce IFN-γ and display cytotoxic activity against infected enterocytes and macrophages in vitro.64 Following adoptive transfer, these cells traffic back to the intestinal mucosa dependent upon chemokine receptor CCR5 and provide protective immunity to challenge infection in the gut.65–67 The mechanism of protection is not clear, but is known to require IFN-γ expression in recipient animals but not in donor cells.68
IEL populations also display immunoregulatory activity in the gut during T. gondii infection. LP CD4+ T cells synergize with intestinal epithelial cells resulting in increased proinflammatory cytokine and chemokine responses.69 It has been shown that an important function of IEL is to downmodulate this response resulting in protection against parasite-mediated intestinal pathology. This activity is dependent upon production of transforming growth factor-β by CD8αβ IEL that downregulates IFN-γ expression by LP CD4+ T cells.65,70 Other studies indicate an important role for γδTCR+ IEL in maintaining the integrity of the epithelium during Toxoplasma infection.71 Thus, under normal conditions IEL are important cells for both host defense against Toxoplasma and in protection against inflammatory pathology. Yet despite this rigorous response, T. gondii escapes the intestinal mucosa, establishes systemic infection and ultimately encysts forming latent infection.
Immunopathology in the Intestine During Toxoplasma Infection
Cells, cytokines and other soluble mediators
Infection of C57BL/6 mice with a relatively low dose of T. gondii (typically 20 cysts of Type II strain parasites) elicits mucosal immune defense and animals survive into chronic infection. However, when the infectious dose is raised to 100 cysts, infection is lethal in the C57BL/6 mouse strain.72 Death occurs around 7–10 days after infection, and is associated with massive necrosis of the villi and mucosal cells in the ileum of the small intestine (Figure 1). In addition to the case of C57BL/6 mice, there is evidence that this type of pathology occurs during T. gondii infection in other host species.73 Lesions in mice bare many interesting resemblances to IBD in humans. Necrosis is dependent upon CD4+ T lymphocytes, IFN-γ and nitric oxide, as determined by antibody depletion and gene knockout studies.72,74,75 Furthermore, the original study showed that induction of damage was dependent upon αβTCR+ and not γδTCR+ cells.72 It has also been found that CCR5-dependent NK cell recruitment contributes to intestinal pathology, most likely because these cells serve as an early source of IFN-γ during infection.76 Toxoplasma-triggered necrosis can be prevented by depletion of TNF-α, an important observation insofar as neutralization of this cytokine is used as a strategy to induce and maintain remission in CD.8,75
Figure 1.
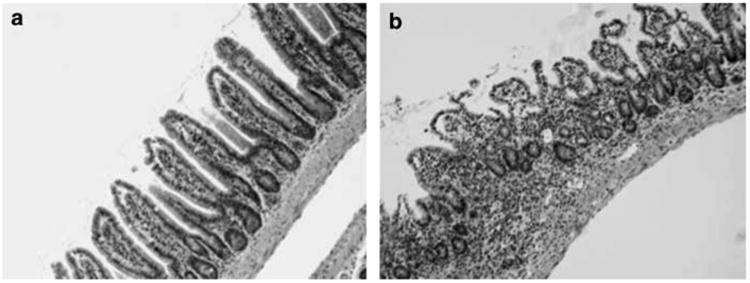
Toxoplasma-induced immunopathology in the C57BL/6 ileum. (a), Non-infected mouse, showing normal architecture of the intestinal mucosa. (b) Typical ileal lesions occurring 8 days following oral infection of C57BL/6 mice (100 cysts; ME49 parasite strain). Damage is characterized histopathologically by transmural inflammation, sloughing of epithelial tips, fusion of villi and increased necrosis.
Activation of CD4+ T cells by IL-12p40 and to a lesser extent IL-18 was found to be required for parasite-triggered necrosis.77 The p40 chain of IL-12 is also shared by IL-23, and recent studies indicate that IL-23 mediates intestinal immunopathology triggered by T. gondii as well as in other IBD models.78,79 Importantly, polymorphisms in the IL-23 receptor gene are known to influence IBD, and the IL-23p19 chain has been shown to be upregulated in CD colon biopsy samples.80,81
During Toxoplasma infection, IL-23-dependent upregulation of matrixmetalloprotease-2 induces intestinal lesions, most likely by mediating damage to the epithelial border.78 Increased expression of both matrixmetalloprotease-2 and matrixmetalloprotease-9 has been observed during IBD in both humans and experimental animals.82,83 In the Toxoplasma study, IL-23-induced T-cell IL-22 production was found to contribute to colitis during T. gondii infection.78 This is interesting because IL-22 has a protective role in other models of IBD, namely the CD45RBhi transfer model and during dextran sulfate sodium-induced colitis in mice.84,85 Yet, as with the case of T. gondii-induced colitis, several studies have linked IL-22 upregulation to CD86,87
Although IL-23 is strongly associated with Th17 responses, the above Toxoplasma study found no evidence for a role of IL-17 in parasite-induced ileitis using IL-17A−/− mice.78 However, two other recent studies reported that IL17RA−/− animals are resistant to development of intestinal lesions during T. gondii infection.88,89 Therefore, at this point it is unclear whether IL-17 is involved in promoting damage in the intestine during Toxoplasma infection. Disparities may stem from differences in strains of mice or parasites used in the studies.
The cytokine IL-10 has a central immunoregulatory role in preventing cytokine overproduction during Toxoplasma infection. Mice lacking expression of IL-10 are hypersusceptible to parasite-induced immunopathology after both oral and intraperitoneal infection.90,91 Immunoregulatory Foxp3+ Treg cells are known to have a role in maintained tolerance in the intestine, and it was found during T. gondii infection that there is both a collapse in this population and conversion to a T-bet/IFN-γ-positive phenotype in the remaining cells associated with lethal pathology.92,93 This was shown to result from limited amounts of IL-2 in an overwhelmingly Th1-dominated cytokine environment. It is possible that collapse in the Treg population results in loss of the source of IL-10 that would normally prevent inflammatory pathology during Toxoplasma infection. Alternatively, Th1 cells or NK cells are additional possible sources of IL-10 during oral infection whose possible loss during oral infection might contribute to intestinal immunopathology.94,95
Role of intestinal flora and TLR signaling in Toxoplasma-triggered ileitis
It is now recognized that intestinal flora aggravate IBD, including CD.3 Inflamed lesions contain accumulations of gram-negative Escherichia coli and Bacteroides spp., and there is evidence for increased adherence and translocation in microlesions in the intestine, as well as systemic immune responses to bacterial antigens.96–99 This has led to the view that CD may result from alterations in the endogenous flora, including emergence of enteroadhesive bacteria, loss of tolerance and inappropriate immune responses to gut flora. These imbalances are collectively called dysbiosis. Several models of experimental colitis support the view that dysbiosis is an important component of IBD pathogenesis.100,101 However, it is not clear whether abnormalities in the mucosal immune system lead to dysbiosis, or whether dysbiosis causes abnormal immunity.
During acute ileitis triggered by T. gondii there is substantial damage to the epithelium, and subepithelial bacterial translocation is readily apparent.63,102 Indeed, in LP preparations from infected mice, macrophages and neutrophils with intracellular bacteria as well as tachyzoites are easily seen (Figure 2). Adherence to epithelial cells and translocation of gut flora are associated with increased bacterial load during Toxoplasma infection. In addition, there is a decrease in species diversity characterized by dominance of gram-negative E. coli and Bacteroides/Prevotella spp and increased intestinal concentrations of bacterial TLR ligands such as lipopolysaccharide, lipopeptides and flagellin.103 Depletion of flora with antibiotics renders mice resistant to Toxoplasma-induced ileitis, and reconstitution with E. coli and Bacteroides/Prevotella restores sensitivity to parasite-induced disease. In parallel, depletion of gut flora results in decreased cytokine responses during T. gondii infection.62 Collectively, these studies make it clear that gut flora have an integral role in ileitis caused by Toxoplasma in the gut.
Figure 2.
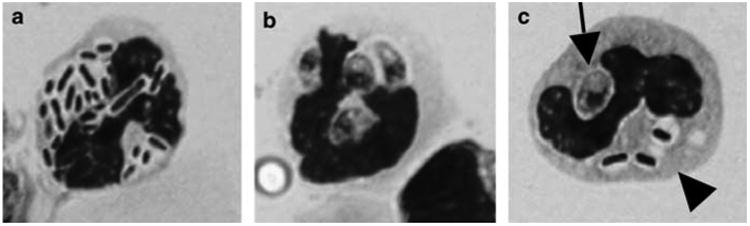
Bacteria and Toxoplasma are present in the lamina propria. C57BL/6 strain mice were orally infected and the lamina propria compartment was isolated 8 days later. Neutrophil-like cells containing intracellular bacteria (a) and Toxoplasma tachyzoites (b) are readily identified. Panel c shows a cell containing an intracellular parasite (arrow) and intracellular bacteria (arrowhead).
The TLR system has an important role in recognition of endogenous bacteria and immunity in the gut. Under some conditions, signaling through TLR helps to maintain homeostasis, as mice lacking MyD88 (the major adaptor of TLR signaling) are more sensitive to dextran sulfate sodium-induced colitis.104–106 However, Myd88−/− mice do not develop intestinal inflammation after Toxoplasma infection suggesting TLR recognition promotes inflammation in this circumstance.62 Consistent with the observation that LPS-expressing bacteria overgrow in the inflamed ileum of infected C57BL/6 mice, animals lacking TLR4, the major LPS receptor, do not develop ileitis after peroral T. gondii infection.107 In addition, ileitis is ameliorated by administration of the LPS scavenger molecule polymyxin B. Nonetheless, an independent study reported that lack of functional TLR4 resulted in increased susceptibility to intestinal damage during Toxoplasma infection.108 The difference between the studies may stem from the fact that Heimesaat et al.107 used TLR4 knockout on a C57BL/6 background whereas the Furuta et al.108 compared C3H/HeN (LPS responder) to C3H/HJ (LPS non-responder) mouse strains.
It has also been found that mice lacking TLR9 display decreased intestinal pathology during peroral T. gondii infection.35,62,109 This seems likely due to recognition of bacterial nucleic acid triggered by Toxoplasma infection. A role for Toxoplasma profilin-TLR1 1 interaction as an early signal for pathogenesis is indicated by the finding that Tlr11−/− mice are resistant to ileitis following peroral infection.62 Taking these findings collectively, it appears that emergence of intestinal inflammation during T. gondii infection involves TLR-dependent responses to both parasite and endogenous gut flora. We propose that proinflammatory parasite-TLR interactions combined with infection-mediated bacterial translocation results in abnormal TLR activation in response to prokaryotic ligands. In turn, this culminates in intestinal dysbiosis and massive subepithelial invasion of gut flora (summarized in Figure 3).
Figure 3.
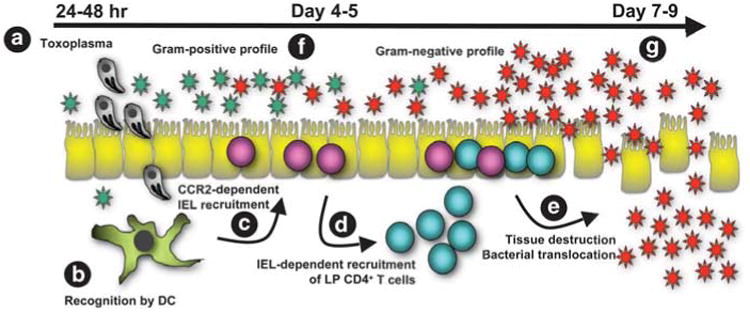
Model for emergence of intestinal pathology during Toxoplasma infection. (a) At 24–48hr after cyst ingestion invasive parasites cross the small intestinal epithelium to initiate infection in cells such as macrophages and dendritic cells. (b) Dendritic cells recognize and initiate immunity to Toxoplasma. They also respond to intestinal bacteria, possibly as a result of translocation occurring as parasites cross the epithelial barrier. (c) Early inflammation results in recruitment of CD8+ T cells into the intraepithelial lymphocyte compartment, detectable 4–5 days after infection. (d) In turn, IEL are involved in recruiting lamina propria CD4+ T cells into the intraepithelial compartment. (e) This results in damage to the intestine and bacterial translocation with fulminant Th1-type immunopathology that peaks around 7–9 days post-infection. In ways that are not clear, concurrent with emergence of disease, the intestinal microbiota increases in number and shifts from a predominantly gram-positive population (f) to a gram-negative population with increased adherence characteristics (g).
Interactions between IEL and LP T lymphocytes fuel Inflammation
The chemokine receptor CCR2 is required for Toxoplasma-induced ileitis.102,110 This receptor and its ligand are also found to be upregulated during CD, and increased numbers of CCR2-positive LP CD4+ T cells are also seen in human disease.111,112 We found that a subpopulation of IEL express this receptor, and CD8α+TCRαβ+ IEL from infected mice induced pathology following transfer into infected Ccr2−/− hosts.102 There is growing evidence that CD8+ cells contribute to and even initiate the disease in IBD113 In a hapten-induced colitis model, CD8+ T cells were identified as the earliest initiators of inflammation.114 Along similar lines, CD8+ T cells specific for influenza hemagglutinin A triggered colitis in transgenic mice expressing this molecule in the intestinal epithelial compartment.115 In CD patients, IEL have been found to possess enhanced cytotoxic activity and increased IFN-γ production compared with cells from normal individuals.116,117 Therefore, although CD8+ IEL have important roles in homeostasis and protection against infection, under some inflammatory conditions these cells display pathogenic activity. For the case of Toxoplasma infection, down-modulatory transforming growth factor-β-producing IEL activity may be replaced by CCR2-dependent recruitment of lesion-inducing CD8 T cells into the intestinal epithelium.
Previous studies found that CD4+ T cells are required for Toxoplasma-induced ileitis.72 In this regard, we found that an LP-derived CCR2+CD4+ T cell population emerged in the IEL compartment concomitant with lesion onset.118 By adoptive transfer of LP and IEL alone or together into non-infected Rag1−/− mice, we found that CD8α+ IEL act to recruit LP CD4+ T cells to cause proinflammatory pathology. In addition, the chemokine and cytokine environment in the intestine was remarkably similar in this co-transfer model to that found in Toxoplasma-infected C57BL/6 mice. The observation that lesions are induced in the absence of Toxoplasma provides more evidence that the role of the parasite is to act as an initiator for dysfunctional immunity that damages tissue rather than a view that the parasite directly destroys the intestinal mucosa through replication in the tissues (Figure 3). Subsequently, we found that endogenous microflora in recipient mice is required to induce damage, suggesting that IEL, LP CD4+ T cells, or both populations may recognize bacterial antigens.118 Together, these findings suggest that IEL and LP T cells act together in mediating damage in the intestinal mucosa during Toxoplasma-initiated inflammation.
Toxoplasma as a Trigger for IBD
How exactly does T. gondii induce onset of inflammation? Resistance to parasite-induced ileitis in Tlr11−/− mice suggests the obvious possibility that profilin-TLR11-signaled IL-12 production sets into motion an uncontrolled proinflammatory cytokine cascade.62 It has also been reported that parasites engineered to lack expression of SAG-1, a major tachyzoite surface protein, lose their ability to trigger ileitis, suggesting that this molecule might also be involved in lesion pathogenesis.119 Conversely, a rhoptry kinase called ROP16 seems to act as an ‘off’ switch to protect against intestinal damage.120 Toxoplasma-induced disease requires the presence of gut flora.63 Accordingly, it is possible that the trigger for inflammation is provided by localized epithelial barrier disruption during early invasion and replication of the parasites, resulting in bacterial translocation and activation of innate immunity (Figure 3).
It is known that Toxoplasma suppresses TLR signaling in macrophages and dendritic cells, and possibly emergence of inflammation results from this activity.121 Macrophages from CD patients have been reported to be defective in cytokine secretion, and it has also been found that neutrophil recruitment is impaired.122–124 One proposal is that pathogenesis involves defective macrophage sentinel responses to occasional bacterial ingress, resulting in failure to recruit neutrophils that would otherwise control infection.124 Loss of early control would then result in an overwhelming inflammatory reaction to translocated bacteria. Possibly, Toxoplasma achieves the same effect through its immunosuppressive effects on macrophages and dendritic cells.
Could Toxoplasma be a trigger for IBD in humans? It has long been speculated that an infectious agent may underlie onset of IBD, although no single pathogen has ever been identified.7 Intracellular bacteria have been suggested as an IBD trigger, and there is strong evidence that Johne's disease in cattle, which resembles CD, is caused by Mycobacterium avium paratuberculosis.7 A role for Toxoplasma in human IBD seems unlikely, based on the fact that intestinal disease is rarely associated with acute infection. Yet, one recent study found a higher rate of Toxoplasma seropositivity among CD-afflicted individuals compared with normal or UC patients.125 Conceivably, Toxoplasma might actually serve as an etiological agent of human disease with the appropriate environmental conditions and genetic predisposition, although we emphasize that this is highly speculative.
Conclusions:Toxoplasma-Induced Inflammation as a Model for Human IBD
The overall pathogenesis of IBD during Toxoplasma infection in mice parallels what has been posited for human disease.3,7 The individual must possess an underlying genetic susceptibility (examples in humans are Nod2 or IL23R susceptibility alleles, in mice the C57BL/6 background) on which a trigger event is superimposed. In the mouse model this is Toxoplasma infection, in humans a trigger has not been identified. This results in dysbiosis in the intestine leading in turn to fulminant IBD. An advantage of the Toxoplasma trigger model is that disease onset is rapid (on the order of 7–10 days) and extremely reproducible. As pointed out by others,126 the model is unlike human IBD in that it is not a chronic relapsing inflammation. This suggests that the major value of peroral Toxoplasma infection is as a tool to understand IBD pathogenesis.
Histopathologically, Toxoplasma-triggered intestinal inflammation resembles CD in that it preferentially occurs in the ileum, a site that is involved in ∼two-thirds of CD patients. Like CD lesions, those induced by T. gondii are discontinuous and transmural damage is often seen. Unlike CD, parasite-induced inflammation does not observably involve granuloma formation at sites of inflammation. Emergence of intestinal lesions during Toxoplasma infection, as in CD, is associated with increased numbers of gram-negative bacteria that display epithelial adhesiveness and translocation into the mucosa.
In immunological terms there are many interesting parallels between CD and Toxoplasma-triggered ileitis. Both are associated with upregulation of CCR2 and its ligands, and recruitment of CCR2+CD4+ T cells is found in both cases. It is also notable that IL-23 signaling is implicated in both cases, although whether Th17 responses are involved in Toxoplasma-triggered disease is presently unclear. Although CCR5 seems to contribute to parasite-induced ileitis, there is no evidence for a role of this chemokine receptor in CD, based upon analysis of Ccr5 gene polymorphisms in CD patients and normal controls.127,128 Regardless, Th1 cytokines are a prominent characteristic of both Toxoplasma-triggered disease and CD, and blocking TNF-α action can ameliorate the disease in both cases. The many parallels between CD and Toxoplasma-induced ileitis argue that the T. gondii-trigger system is a highly useful addition to the collection of IBD mouse models.
Future Directions
Toxoplasma is normally the cause of benign infection, but as discussed here the parasite causes IBD in C57BL/6 strain mice. The recent identification of cells such as IEL and immune receptors such as CCR2 in the Toxoplasma model offers potential new strategies for therapy in human IBD. Other findings are that novel interactions between LP T cells and IEL mediate intestinal damage, but precisely how this occurs is not known. Important matters to address in the future are the antigen specificity and MHC restriction of these cell types. The major effector molecules of damage and necrosis in the intestine also await identification. Also, the function of intestinal dendritic cells as proinflammatory lesions emerge is unexplored. The tractability of the Toxoplasma trigger model suggests that these issues are amenable to resolution, and working towards this goal can provide us with insight into the intestinal mucosa in health and disease. By translating these findings from the bench to the bedside, the Toxoplasma trigger model ultimately may prove valuable in identifying new cells, receptors and soluble mediators as targets to treat IBD in humans.
Acknowledgments
Our work is supported by US Public Health Service grant AI083526.
References
- 1.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 4.Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut. 2009;58:1152–1167. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes JM, Campbell BJ. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trends Mol Med. 2002;8:10–16. doi: 10.1016/s1471-4914(01)02194-3. [DOI] [PubMed] [Google Scholar]
- 6.Kappelman MD, Rifas-Shiman SL, Porter CQ, Ollendorf DA, Sandler RS, Galanko JA, et al. Direct health care costs of Crohn's disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135:1907–1913. doi: 10.1053/j.gastro.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen R, Thomson JM, El-Omar EM, Hold GL. The role of infection in the aetiology of inflammatory bowel disease. J Gastroenterol. 2010;45:266–276. doi: 10.1007/s00535-009-0191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blonski W, Buchner AM, Lichtenstein GR. Inflammatory bowel disease therapy: current state-of-the-art. Curr Opin Gastroenterol. 2011;27:346–357. doi: 10.1097/MOG.0b013e328347aef3. [DOI] [PubMed] [Google Scholar]
- 9.Korzenik JR, Podolsky DK. Evolving knowledge and therapy of inflammatory bowel disease. Nat Rev Drug Discov. 2006;5:197–209. doi: 10.1038/nrd1986. [DOI] [PubMed] [Google Scholar]
- 10.Podolsky DK. Beyond tumor necrosis factor: next-generation biologic therapy for inflammatory bowel disease. Dig Dis. 2009;27:366–369. doi: 10.1159/000228575. [DOI] [PubMed] [Google Scholar]
- 11.Reiff C, Kelly D. Inflammatory bowel disease, gut bacteria and probiotic therapy. Int J Med Microbiol. 2010;300:25–33. doi: 10.1016/j.ijmm.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. 2010;8:564–577. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 13.Strober W. Why study animal models of IBD? Inflamm Bowel Dis. 2008;14(Suppl 2):S129–S131. doi: 10.1002/ibd.20667. [DOI] [PubMed] [Google Scholar]
- 14.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 15.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 16.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–3943. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 17.Pizarro TT, Arseneau KO, Bamias G, Cominelli F. Mouse models for the study of Crohn's disease. Trends Mol Med. 2003;9:218–222. doi: 10.1016/s1471-4914(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 18.Dubey JP. The history and life-cycle of Toxoplasma gondii. In: Weiss LM, Kim K, editors. Toxoplasma Gondii The Model Apicomplexan: Perspective and Methods. Academic Press; San Diego: 2007. pp. 1–17. [Google Scholar]
- 19.Latkany P. Ocular disease due to Toxoplasma gondii. In: Weiss LM, Kim K, editors. Toxoplasma Gondii, the Model Apicomplexan: Perspectives and Methods. Academic Press; Amsterdam: 2007. pp. 101–131. [Google Scholar]
- 20.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 21.Peterson E, Liesenfeld O. Clinical disease and diagnostics. In: Weiss LM, Kim K, editors. Toxoplasma Gondii The Model Apicomplexan: Perspectives and Methods. Academic Press; Amsterdam: 2007. pp. 81–100. [Google Scholar]
- 22.Montoya JG, Remington JS. Management of Toxoplasma gondii infection during pregnancy. Clin Infect Dis. 2008;47:554–566. doi: 10.1086/590149. [DOI] [PubMed] [Google Scholar]
- 23.Sher A, Oswald IO, Hieny S, Gazzinelli RT. Toxoplasma gondii induces a T-independent IFN-g response in NK cells which requires both adherent accessory cells and TNF-a. J Immunol. 1993;150:3982–3989. [PubMed] [Google Scholar]
- 24.Gazzinelli RT, Hieny S, Wynn T, Wolf S, Sher A. IL-12 is required for the T-cell independent induction of IFN-g by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc Natl Acad Sci USA. 1993;90:6115–6119. doi: 10.1073/pnas.90.13.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, et al. Parasite-induced IL-12 stimulates early IFN-g synthesis and resistance during acute infection with Toxoplasma gondii. J Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- 26.Denkers EY, Gazzinelli RT. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bliss SK, Marshall AJ, Zhang Y, Denkers EY. Human polymorphonuclear leukocytes produce IL-12, TNF-a, and the chemokines macrophage-inflammatory protein-1a and -1b in response to Toxoplasma gondii antigens. J Immunol. 1999;162:7369–7375. [PubMed] [Google Scholar]
- 28.Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, et al. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J Immunol. 2006;177:31–35. doi: 10.4049/jimmunol.177.1.31. [DOI] [PubMed] [Google Scholar]
- 29.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 30.Sukhumavasi W, Egan CE, Warren AL, Taylor GA, Fox BA, Bzik DJ, et al. TLR adaptor MyD88 is essential for pathogen control during oral Toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol. 2008;181:3464–3473. doi: 10.4049/jimmunol.181.5.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Micro. 2005;6:837–848. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- 32.Hou B, Benson A, Kuzmich L, DeFranco AL, Yarovinsky F. Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proc Natl Acad Sci U S A. 2011;108:278–283. doi: 10.1073/pnas.1011549108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol. 2007;179:1129–1137. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- 34.Del Rio L, Butcher BA, Bennouna S, Hieny S, Sher A, Denkers EY. Toxoplasma gondii triggers MyD88-dependent and CCL2(MCP-1) responses using distinct parasite molecules and host receptors. J Immunol. 2004;172:6954–6960. doi: 10.4049/jimmunol.172.11.6954. [DOI] [PubMed] [Google Scholar]
- 35.Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, et al. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol. 2006;176:7589–7597. doi: 10.4049/jimmunol.176.12.7589. [DOI] [PubMed] [Google Scholar]
- 36.Mun HS, Aosai F, Norose K, Chen M, Piao LX, Takeuchi O, et al. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int Parasitol. 2003;15:1081–1087. doi: 10.1093/intimm/dxg108. [DOI] [PubMed] [Google Scholar]
- 37.Yarovinsky F, Zhang D, Anderson JF, Bannenberg GL, Serhan CN, Hayden MS, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 38.Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, et al. Toxoplasma profilin is essential for host cell invasion and TLR dependent induction of interleukin-12. Cell Host Microbe. 2008;14:77–87. doi: 10.1016/j.chom.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12 dependent host resistance to Toxoplasma Gondii. J Biol Chem. 2011;286:3307–3314. doi: 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J Immunol. 1992;149:175–180. [PubMed] [Google Scholar]
- 41.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-g: the major mediator of resistance against Toxoplasma gondii. Science. 1988;240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki Y, Remington JS. Dual regulation of resistance against Toxoplasma gondii infection by Lyt-2+ and Lyt1+, L3T4+ T cells in mice. J Immunol. 1988;140:3943–3946. [PubMed] [Google Scholar]
- 43.Denkers EY, Yap G, Scharton-Kersten T, Charest H, Butcher B, Caspar P, et al. Perforin-mediated cytolysis plays a limited role in host resistance to Toxoplasma gondii. J Immunol. 1997;159:1903–1908. [PubMed] [Google Scholar]
- 44.Wang X, Kang H, Kikuchi T, Suzuki Y. Gamma interferon production, but not perforin-mediated cytolytic activity, of T cells is required for prevention of toxoplasmic encephalitis in BALB/c mice genetically resistant to the disease. Infect Immun. 2004;72:4432–4438. doi: 10.1128/IAI.72.8.4432-4438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gavrilescu LC, Butcher BA, Del Rio L, Taylor GA, Denkers EY. STAT1 is essential for antimicrobial function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect Immun. 2004;72:1257–1264. doi: 10.1128/IAI.72.3.1257-1264.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lieberman LA, Banica M, Reiner SL, Hunter CA. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. J Immunol. 2004;172:457–463. doi: 10.4049/jimmunol.172.1.457. [DOI] [PubMed] [Google Scholar]
- 47.Scharton-Kersten T, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med. 1997;185:1–13. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 2009;5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Howard JC, Hunn JP, Steinfeldt T. The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr Opin Microbiol. 2011;14:414–421. doi: 10.1016/j.mib.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, et al. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacMicking J, Xie Q, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 52.Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andrade RM, Wessendarp M, Subauste CS. CD154 activates macrophage antimicrobial activity in the absence of IFN-g through a TNF-a-dependent mechanism. J Immunol. 2003;171:6750–6756. doi: 10.4049/jimmunol.171.12.6750. [DOI] [PubMed] [Google Scholar]
- 54.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3:331–341. doi: 10.1038/nri1057. [DOI] [PubMed] [Google Scholar]
- 55.Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol. 2011;11:445–456. doi: 10.1038/nri3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli A, Denkers EY. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol. 2008;181:8445–8491. doi: 10.4049/jimmunol.181.12.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. CD11c and CD11b expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. 2006;107:309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol. 2006;8:1611–1623. doi: 10.1111/j.1462-5822.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 59.Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Tox-oplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fentress SJ, Behnke MS, Dunay IR, Mashayekhi M, Rommereim LM, Fox BA, et al. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe. 2010;8:484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Egan CE, Sukhumavasi W, Butcher BA, Denkers EY. Functional aspects of toll-like receptor/MyD88 signalling during protozoan infection: focus on Toxoplasma gondii. Clin Exp Immunol. 2009;156:17–24. doi: 10.1111/j.1365-2249.2009.03876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benson A, Pifer R, Behrendt CL, Hooper LV, Yarovinsky F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe. 2009;6:187–196. doi: 10.1016/j.chom.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, et al. Gram-negative bacteria aggravate murine small intestinal Th1-Type immunopathology following oral infection with Toxoplasma gondii. J Immunol. 2006;177:8785–8795. doi: 10.4049/jimmunol.177.12.8785. [DOI] [PubMed] [Google Scholar]
- 64.Chardes T, Buzoni-Gatel D, Lepage A, Bernard F, Bout D. Toxoplasma gondii oral infection induces specific cytotoxic CD8a/b+ Thy-1+ gut iintraepithelial lymphocytes, lytic for parasite-infected enterocytes. J Immunol. 1994;153:4596–4603. [PubMed] [Google Scholar]
- 65.Buzoni-Gatel D, Debbabi H, Mennechet FJD, Martin V, LePage AC, Schwartzman JD, et al. Murine ileitis after intracellular parasite infection is controlled by TGF-b-producing intraepithelial lymphocytes. Gastroenterol. 2001;120:914–924. doi: 10.1053/gast.2001.22432a. [DOI] [PubMed] [Google Scholar]
- 66.Buzoni-Gatel D, Debbabi H, Moretto M, Dimier-Poisson IH, Lepage AC, Bout DT, et al. Intraepithelial lymphocytes traffic to the intestine and enhance resistance to Tox-oplasma gondii oral infection. J Immunol. 1999;162:5846–5852. [PubMed] [Google Scholar]
- 67.Luangsay S, Kasper LH, Rachinel N, Minns LA, Mennechet FJD, Vandewalle A, et al. CCR5 mediates specific migration of Toxoplasma gondii -primed CD8+ lymphocytes to inflammatory intestinal epithelial cells. Gastroenterol. 2003;125:491–500. doi: 10.1016/s0016-5085(03)00903-x. [DOI] [PubMed] [Google Scholar]
- 68.Lepage AC, Buzoni-Gatel D, Bout DT, Kasper LH. Gut-derived intraepithelial lymphocytes induce long term immunity against Toxoplasma gondii. J Immunol. 1998;161:4902–4908. [PubMed] [Google Scholar]
- 69.Mennechet FJD, Kasper LH, Rachinel N, Li W, Vandewalle A, Buzoni-Gatel D. Lamina propria CD4+ T lymphocytes synergize with murine intestinal epithelial cells to enhance proinflammatory response against an intracellular pathogen. J Immunol. 2002;168:2988–2996. doi: 10.4049/jimmunol.168.6.2988. [DOI] [PubMed] [Google Scholar]
- 70.Mennechet FJ, Kasper LH, Rachinel N, Minns LA, Luangsay S, Vandewalle A, et al. Intestinal intraepithelial lymphocytes prevent pathogen-driven inflammation and regulate the Smad/T-bet pathway of lamina propria CD4+ T cells. Eur J Immunol. 2004;34:1059–1067. doi: 10.1002/eji.200324416. [DOI] [PubMed] [Google Scholar]
- 71.Dalton JE, Cruickshank SM, Egan CE, Mears R, Newton DJ, Andrew EM, et al. Intraepithelial gammadelta+ lymphocytes maintain the integrity of intestinal epithelial tight junctions in response to infection. Gastroenterology. 2006;131:818–829. doi: 10.1053/j.gastro.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 72.Liesenfeld O, Kosek J, Remington JS, Suzuki Y. Association of CD4+ T cell-dependent, IFN-g-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med. 1996;184:597–607. doi: 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schreiner M, Liesenfeld O. Small intestinal inflammation following oral infection with Toxoplasma gondii does not occur exclusively in C57BL/6 mice: review of 70 reports from the literature. Mem Inst Oswaldo Cruz. 2009;104:221–233. doi: 10.1590/s0074-02762009000200015. [DOI] [PubMed] [Google Scholar]
- 74.Khan IA, Schwartzman JD, Matsuura T, Kasper LH. A dichotomous role for nitric oxide during acute Toxoplasma gondii infection in mice. Proc Natl Acad Sci USA. 1997;94:13955–13960. doi: 10.1073/pnas.94.25.13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liesenfeld O, Kang H, Park D, Nguyen TA, Parkhe CV, Watanabe H, et al. TNF-a, nitric oxide and IFN-g are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 1999;21:365–376. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- 76.Khan IA, Thomas SY, Moretto MM, Lee FS, Islam SA, Combe C, et al. CCR5 is essential for NK cell trafficking and host survival following Toxoplasma gondii infection. PLoS Pathog. 2006;2:e49. doi: 10.1371/journal.ppat.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vossenkamper A, Struck D, Alvarado-Esquivel C, Went T, Takeda K, Akira S, et al. Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur J Immunol. 2004;34:3197–3207. doi: 10.1002/eji.200424993. [DOI] [PubMed] [Google Scholar]
- 78.Munoz M, Heimesaat MM, Danker K, Struck D, Lohmann U, Plickert R, et al. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J Exp Med. 2009;206:3047–3059. doi: 10.1084/jem.20090900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, et al. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 82.Baugh MD, Evans GS, Hollander AP, Davies DR, Perry MJ, Lobo AJ, et al. Expression of matrix metalloproteases in inflammatory bowel disease. Ann N Y Acad Sci. 1998;859:249–253. doi: 10.1111/j.1749-6632.1998.tb11139.x. [DOI] [PubMed] [Google Scholar]
- 83.Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 84.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 86.Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;290:G827–G838. doi: 10.1152/ajpgi.00513.2005. [DOI] [PubMed] [Google Scholar]
- 87.Schmechel S, Konrad A, Diegelmann J, Glas J, Wetzke M, Paschos E, et al. Linking genetic susceptibility to Crohn's disease with Th17 cell function: IL-22 serum levels are increased in Crohn's disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis. 2008;14:204–212. doi: 10.1002/ibd.20315. [DOI] [PubMed] [Google Scholar]
- 88.Guiton R, Vasseur V, Charron S, Arias MT, Van Langendonck N, Buzoni-Gatel D, et al. Interleukin 17 receptor signaling is deleterious during Toxoplasma gondii infection in susceptible BL6 mice. J Infect Dis. 2010;202:427–435. doi: 10.1086/653738. [DOI] [PubMed] [Google Scholar]
- 89.Kelly MN, Kolls JK, Happel K, Schwartzman JD, Schwarzenberger P, Combe C, et al. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect Immun. 2005;73:617–621. doi: 10.1128/IAI.73.1.617-621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent upon CD4+ Tcells and accompanied by overproduction of IL-12, IFN-g, and TNF-a. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 91.Suzuki Y, Sher A, Yap G, Park D, Ellis Neyer L, Liesenfeld O, et al. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J Immunol. 2000;164:5375–5382. doi: 10.4049/jimmunol.164.10.5375. [DOI] [PubMed] [Google Scholar]
- 92.Murai M, Krause P, Cheroutre H, Kronenberg M. Regulatory T-cell stability and plasticity in mucosal and systemic immune systems. Mucosal Immunol. 2010;3:443–449. doi: 10.1038/mi.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3(+) Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, et al. Conventional T-bet+Foxp3- Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, et al. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe. 2009;6:503–512. doi: 10.1016/j.chom.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 97.Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005;43:3380–3389. doi: 10.1128/JCM.43.7.3380-3389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tabaqchali S, O'Donoghue DP, Bettelheim KA. Escherichia coli antibodies in patients with inflammatory bowel disease. Gut. 1978;19:108–113. doi: 10.1136/gut.19.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hans W, Scholmerich J, Gross V, Falk W. The role of the resident intestinal flora in acute and chronic dextran sulfate sodium-induced colitis in mice. Eur J Gastroenterol Hepatol. 2000;12:267–273. doi: 10.1097/00042737-200012030-00002. [DOI] [PubMed] [Google Scholar]
- 101.Madsen KL, Doyle JS, Tavernini MM, Jewell LD, Rennie RP, Fedorak RN. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology. 2000;118:1094–1105. doi: 10.1016/s0016-5085(00)70362-3. [DOI] [PubMed] [Google Scholar]
- 102.Egan CE, Craven MD, Leng J, Mack M, Simpson KW, Denkers EY. CCR2-dependent intraepithelial lymphocytes mediate inflammatory gut pathology during Toxoplasma gondii infection. Mucosal Immunol. 2009;2:527–535. doi: 10.1038/mi.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Erridge C, Duncan SH, Bereswill S, Heimesaat MM. The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PLoS One. 2010;5:e9125. doi: 10.1371/journal.pone.0009125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Araki A, Kanai T, Ishikura T, Makita S, Uraushihara K, Iiyama R, et al. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J Gastroenterol. 2005;40:16–23. doi: 10.1007/s00535-004-1492-9. [DOI] [PubMed] [Google Scholar]
- 105.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial transloca-tion in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–G1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 106.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 107.Heimesaat MM, Fischer A, Jahn HK, Niebergall J, Freudenberg M, Blaut M, et al. Exacerbation of murine ileitis by toll-like receptor 4 meditated sensing of lipopoly-saccharide from commensal escherichia coli. Gut. 2007;56:941–948. doi: 10.1136/gut.2006.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Furuta T, Kikuchi T, Akira S, Watanabe N, Yoshikawa Y. Roles of the small intestine for induction of toll-like receptor 4-mediated innate resistance in naturally acquired murine toxoplasmosis. Int Immunol. 2006;18:1655–1662. doi: 10.1093/intimm/dxl099. [DOI] [PubMed] [Google Scholar]
- 109.Foureau DM, Mielcarz DW, Menard LC, Schulthess J, Werts C, Vasseur V, et al. TLR9-dependent induction of intestinal alpha-defensins by Toxoplasma gondii. J Immunol. 2010;184:7022–7029. doi: 10.4049/jimmunol.0901642. [DOI] [PubMed] [Google Scholar]
- 110.Benevides L, Milanezi CM, Yamauchi LM, Benjamim CF, Silva JS, Silva NM. CCR2 receptor is essential to activate microbicidal mechanisms to control Toxoplasma gondii infection in the central nervous system. Am J Pathol. 2008;173:741–751. doi: 10.2353/ajpath.2008.080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Connor SJ, Paraskevopoulos N, Newman R, Cuan N, Hampartzoumian T, Lloyd AR, et al. CCR2 expressing CD4+ T lymphocytes are preferentially recruited to the ileum in Crohn's disease. Gut. 2004;53:1287–1294. doi: 10.1136/gut.2003.028225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhong W, Kolls JK, Chen H, McAllister F, Oliver PD, Zhang Z. Chemokines orchestrate leukocyte trafficking in inflammatory bowel disease. Front Biosci. 2008;13:1654–1664. doi: 10.2741/2789. [DOI] [PubMed] [Google Scholar]
- 113.Cheroutre H. In IBD eight can come before four. Gastroenterology. 2006;131:667–670. doi: 10.1053/j.gastro.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 114.Nancey S, Holvoet S, Graber I, Joubert G, Philippe D, Martin S, et al. CD8+ cytotoxic T cells induce relapsing colitis in normal mice. Gastroenterology. 2006;131:485–496. doi: 10.1053/j.gastro.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 115.Westendorf AM, Fleissner D, Deppenmeier S, Gruber AD, Bruder D, Hansen W, et al. Autoimmune-mediated intestinal inflammation-impact and regulation of antigen-specific CD8+ T cells. Gastroenterology. 2006;131:510–524. doi: 10.1053/j.gastro.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 116.Nussler NC, Stange B, Hoffman RA, Schraut WH, Bauer AJ, Neuhaus P. Enhanced cytolytic activity of intestinal intraepithelial lymphocytes in patients with Crohn's disease. Langenbecks Arch Surg. 2000;385:218–224. doi: 10.1007/s004230050268. [DOI] [PubMed] [Google Scholar]
- 117.Watanabe M, Hayashi A, Hosoda Y, Ohara M, Iwao Y, Ishii H, et al. Preferential activation of CD4+V beta 5.2/5.3+ intestinal intraepithelial lymphocytes in the inflamed lesions of Crohn's disease. Clin Immunol Immunopathol. 1996;78:130–139. doi: 10.1006/clin.1996.0022. [DOI] [PubMed] [Google Scholar]
- 118.Egan CE, Maurer KJ, Cohen SB, Mack M, Simpson KW, Denkers EY. Synergy between intraepithelial lymphocytes and lamina propria T cells drives intestinal inflammation during infection. Mucosal Immunol. 2011;4:658–670. doi: 10.1038/mi.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rachinel N, Buzoni-Gatel D, Dutta C, Mennechet FJD, Laungsay S, Minns LA, et al. The induction of acute ileitis by a single microbial antigen of Toxoplasma gondii. J Immunol. 2004;173:2725–2735. doi: 10.4049/jimmunol.173.4.2725. [DOI] [PubMed] [Google Scholar]
- 120.Jensen KD, Wang Y, Wojno ED, Shastri AJ, Hu K, Cornel L, et al. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe. 2011;9:472–483. doi: 10.1016/j.chom.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Leng J, Butcher BA, Denkers EY. Dysregulation of macrophage signal transduction by Toxoplasma gondii: past progress and recent advances. Parasite Immunol. 2009;31:717–728. doi: 10.1111/j.1365-3024.2009.01122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marks DJ, Harbord MW, MacAllister R, Rahman FZ, Young J, Al-Lazikani B, et al. Defective acute inflammation in Crohn's disease: a clinical investigation. Lancet. 2006;367:668–678. doi: 10.1016/S0140-6736(06)68265-2. [DOI] [PubMed] [Google Scholar]
- 123.Segal AW, Loewi G. Neutrophil dysfunction in Crohn's disease. Lancet. 1976;2:219–221. doi: 10.1016/s0140-6736(76)91024-2. [DOI] [PubMed] [Google Scholar]
- 124.Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med. 2009;206:1883–1897. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lidar M, Langevitz P, Barzilai O, Ram M, Porat-Katz BS, Bizzaro N, et al. Infectious serologies and autoantibodies in inflammatory bowel disease: insinuations at a true pathogenic role. Ann N Y Acad Sci. 2009;1173:640–648. doi: 10.1111/j.1749-6632.2009.04673.x. [DOI] [PubMed] [Google Scholar]
- 126.Liesenfeld O. Oral infection of C57BL/6 mice with Toxoplasma gondii: a new model of inflammatory bowel disease? J Infect Dis. 2002;185:S96–S101. doi: 10.1086/338006. [DOI] [PubMed] [Google Scholar]
- 127.Craggs A, Welfare M, Donaldson PT, Mansfield JC. The CC chemokine receptor 5 delta32 mutation is not associated with inflammatory bowel disease (IBD) in NE England. Genes Immun. 2001;2:114–116. doi: 10.1038/sj.gene.6363735. [DOI] [PubMed] [Google Scholar]
- 128.Rector A, Vermeire S, Thoelen I, Keyaerts E, Struyf F, Vlietinck R, et al. Analysis of the CC chemokine receptor 5 (CCR5) delta-32 polymorphism in inflammatory bowel disease. Hum Genet. 2001;108:190–193. doi: 10.1007/s004390100462. [DOI] [PubMed] [Google Scholar]