

Abstract
There is a large body of evidence that many protein–ligand cocrystal structures contain poorly refined ligand geometries. These errors result in bound structures that have nonideal bond lengths and angles, are strained, contain improbable conformations, and have bad protein–ligand contacts. Many of these problems can be greatly reduced with better refinement models.
Keywords: Protein−ligand cocrystal, structure-induced fit, structure-based design, structure refinement, bound ligand strain
The ability to accurately determine the 3D structure of protein–ligand complexes using X-ray crystallography has provided an important tool for drug discovery. The number of publicly available structures in the RCSB PDB (www.pdb.org) has grown to almost 100,000, and of course this does not include the many thousands of proprietary structures that have been determined. During this growth there have been numerous studies that raise concerns about the fidelity of many of these structures with regard to the bound ligand.1−4 They all find that many bound ligands in the PDB incorporate a surprising amount of internal strain. Further, inspection of these structures shows a litany of distorted rings, bad contacts, and unusual conformations/configurations. In short, the prevailing literature suggests that current refinement procedures often do a poor job of correctly refining the bound ligand.
Evidence of a Problem
Just to give a few examples: 1xqd contains three planar oxygens as part of a phosphate group; 1pme features a planar sulfur in the sulfoxide; 1tnk, a 1.8 Å resolution structure, contains a nonplanar tetrahedral aromatic carbon as part of a substituted aniline; and 4g93 contains an olefin that is twisted nearly 90° out of the plane. While it is surprising that such egregious chemical structures could find their way into the literature much less the PDB, we might dismiss them as anomalies. However, the truth is any systematic evaluation of the bound ligands in the PDB will uncover countless, less dramatic, albeit still serious structural errors. While the tone of some early work seems to be more in the direction of attempting to explain this phenomenon,1 there has gradually been a widespread realization that induced fit cannot explain the large number of strained and distorted ligands.
Studies of bound ligand strain commonly entail assembling a selection of cocrystal structures from the PDB, extracting the bound ligand, and then optimizing the ligand outside the confines of the protein active site. While this sounds simple, there are many important computational details that can have a significant effect on the results. For example, selection of the bound and free reference states, inclusion of solvation (or other medium effects), and the model employed (e.g., force field or quantum). The most common measures of bound ligand strain are usually referred to as local or global strain as defined in eqs 1 and 2, respectively.
| 1 |
| 2 |
The first definition (eq 1) is problematic in that it demands extensive conformational analysis in order to obtain the global minimum (a result that can never be proved conclusively). However, it is also true that this represents a lower boundary on the error since finding a better global minimum only makes the strain energy larger. For our purposes, the details are not that consequential given that the trends are consistent regardless of definition. A 2004 study, by Perola and Charifson,5 of 150 cocrystal structures found that almost half the bound ligands had strain energies (in this case global) in excess of 5 kcal/mol, and approximately 10% of the structures had ligand strains in excess of 9 kcal/mol. A more recent study by Liebeschuetz et al.3 is notable for assembling three separate data sets based on the year the structure was determined. The three sets encompassed all years before 2000, 2006, and 2009. They also employed a more categorical evaluation of the quality of the structures: OK, strained, and questionable. The results? Approximately 70% of recent structures have errors that might be corrected with better restraints, and at least 25% have errors that could lead to misleading interpretation of key binding interactions. Significantly, little evidence was found that ligand geometries have improved in any systematic way since 2006. Two studies using quantum calculations to evaluate ligand strain provide more evidence that this problem is pervasive.4,6
Common Issues
How can so many bound-ligand structures have problems? There are many reasons. First it is sometimes forgotten, but all structures, unless done at very high resolution (i.e., less than 1.0 Å), are fitted models.2 They cannot be assigned using the experimental density information alone. This means that the underlying theoretical model that is used as a constraint on refinement is important. Typical X-ray refinement protocols use force fields (e.g., Engh–Huber)7 that employ united atom representations, neglect electrostatics, and have not been parametrized for small molecule drugs. In most refinement paradigms this means that the crystallographer is responsible for determining appropriate structural constraints for the ligand, a task that is not simple. In addition, the ligand is very small relative to the protein and has limited statistical weight in the overall fitting function. This means that large errors in the ligand have only a small effect on the overall goodness of fit metric (typically Rfree). These technical issues are often compounded by the fact that many, if not most, crystallographers do not come from a chemistry background. So the myriad ad hoc decisions required of the crystallographer such as proper molecular connectivity; ideal bond lengths, angles, and dihedrals; and the best conformation for functional groups and rings are often thrust on scientists with limited chemistry experience. It must also be remembered that in addition to getting all the molecular data right for the ligand there is still the complex issue of determining the correct binding orientation in the protein. Getting everything “correct” can be demanding and time-consuming. Unfortunately, it is often the case that speed and productivity are given higher priorities. Adding these factors all together, it is no wonder so many structures have problems.
Typical structural problems for protein-bound ligands include the following:
-
(1)
Errors in the ligand structure, such as missing atoms, incorrect bond orders, or other connectivity issues.
-
(2)
Incorrect bond distances, angles, or dihedral angles due to problems with geometric constraints and ideal values.
-
(3)
Bad steric clashes between the protein and ligand.
-
(4)
Conformational errors such as cis- or twisted amides, distorted rings (e.g., boat or twist), nonplanar aromatic groups, or planar structures that should not be planar (e.g., sulfones and sulfoxides).
-
(5)
Incorrect orientations with respect to the protein active site. In some cases the proper pose may be obvious from the experimental data, but in others it is not. There may also be problems with protonation states and charges. These can be difficult to get right.
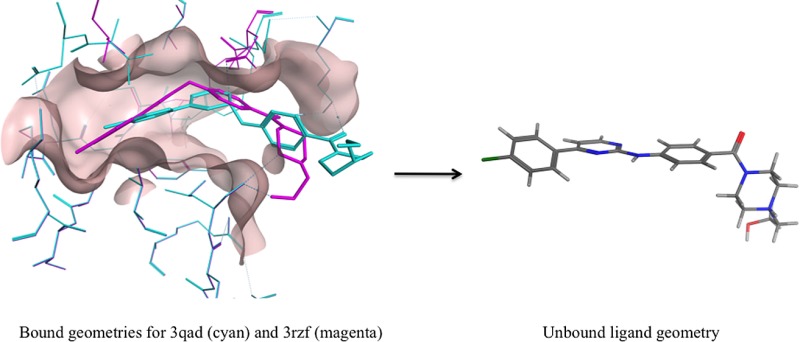
An example that highlights some of these issues is the cocrystal structure for κB kinaseβ (3qad). This is a low-resolution structure, so the structural constraints are particularly important in obtaining a reasonable model. It was pointed out in previous work by Liebeschuetz et al.3 that the original structure deposited in the PDB (3qad) suffered from a serious error in the amino-pyrimidine moiety that lead to a pyramidal, not planar, structure (Figure 1a). The ligand also contained a piperazine in an unfavorable boat conformation. Subsequently the structure was refined again with the correct planar C (sp2) in the aminopyrimidine and a chair conformation for the piperazine (3rzf). However, even this structure was highly strained (Figure 1b) and contained many bad contacts between ligand and protein. For comparison the ligand structure in 3rzf was minimized outside the protein, resulting in the very different structure shown in Figure 1c.
Figure 1.
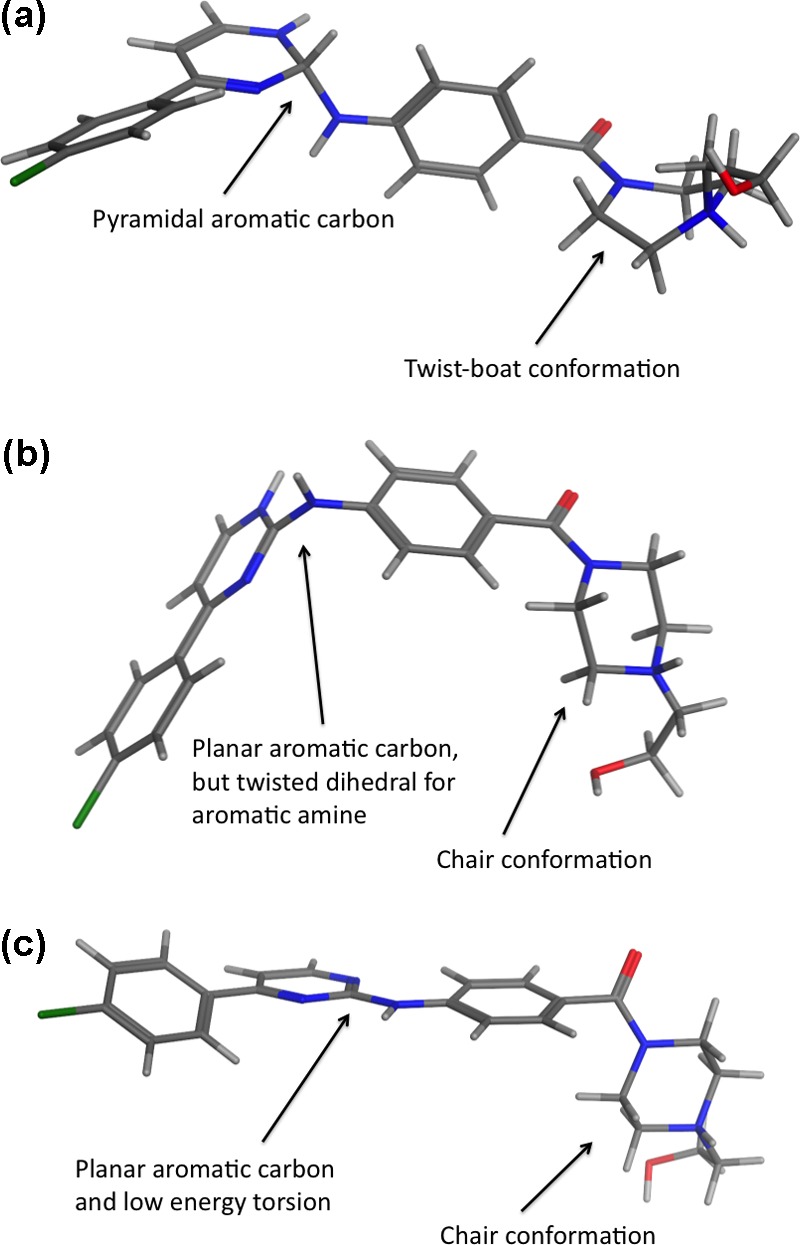
Comparison of ligand structures in (a) 3qad, (b) the revised structure 3rzf, and (c) the 3rzf ligand after minimization outside the protein using the MMFF force field in MOE (Chemical Computing Group).
Better Models Are Available
There are now a variety of new computational tools that can address at least some of the problems discussed above.6,8−10 In the case of steric clashes, Bell et al. have shown that all atom refinement dramatically reduces these clashes. For a collection of 94 moderate resolution (i.e., 1.5–2.8 Å) structures, the number of clashes/structure dropped from 28 to 6, and the number of severe clashes/structure was reduced from 3.7 to 0.2, relative to the original PDB structures. With regard to bound ligand strain, a recent QM refinement of 50 cocrystal structures led to significant reductions in bound-ligand strain for all but one structure.6 In both studies there are many specific examples cited that show large deviations in bond lengths, angles, and dihedrals that can be corrected using better reference models. In many cases the difference between the original and rerefined structures are certainly large enough to adversely affect ligand design. It has become common practice to apply a variety of protein preparation tools to structures before modeling in order to deal with some of these issues on a post hoc basis, but this is obviously not optimal. It would be far better to deal with these issues during structure determination.
The newer models are not terribly obscure and can at least help with some of the issues outlined above. Indeed several are commercially available and are reasonably straightforward to implement, either as stand alone tools8,10 or as a plugin to PHENIX.6 Given the overwhelming evidence that current refinement protocols are too often failing for bound ligands, one wonders why adoption of these tools has not been more rapid and general.
Summary
There is ample evidence in the literature of widespread problems with ligand geometries in protein–ligand cocrystal structures. This is an issue that should be of general concern given the tendency of medicinal chemists to regard these structures uncritically. The ligand and associated active site are particularly important for drug discovery. While all of the problems outlined above are difficult to eliminate, there are newer refinement models available that can improve the quality of bound ligand structures considerably. Moreover, stricter inspection of final structures with regard to the ligand might improve the situation. Part of the problem is due to the emphasis on automation and “numbers of structures” in the industry. In some cases production comes at the expense of quality. There is a great need for improved refinement protocols that are more robust, and analysis tools for assessing bound-ligand structure quality. The standard should certainly be higher for acceptance of cocrystal structures published in the PDB.
Acknowledgments
Thanks to Bruce Maryanoff, Kennie Merz, Dagmar Ringe, and Joe Vacca for their valuable comments and input. Chemical Computing Group kindly provided MOE.
Glossary
Abbreviations Used
- RCSB
Research Collaboratory for Structural Bioinformatics
- PDB
protein data bank
- 3D
three-dimensional
- QM
quantum mechanical
- MOE
Molecular Operating Environment
- MMFF
Merck Molecular Force Field
Views expressed in this editorial are those of the author and not necessarily the views of the ACS.
The authors declare the following competing financial interest(s): CHR has advised and collaborated with QuantumBio.
References
- Nicklaus M. C.; Wang S.; Driscoll J. S.; Milne G. W. Conformational changes of small molecules binding to proteins. Bioorg. Med. Chem. 1995, 3, 411–28. [DOI] [PubMed] [Google Scholar]
- Davis A. M.; Teague S. J.; Kleywegt G. J. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew. Chem., Int. Ed. 2003, 42, 2718–2736. [DOI] [PubMed] [Google Scholar]
- Liebeschuetz J.; Hennemann J.; Olsson T.; Groom C. R. The good, the bad and the twisted: a survey of ligand geometry in protein crystal structures. J. Comput.-Aided Mol. Des. 2012, 26, 169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitzmann M.; Weidlich I. E.; Filippov I. V.; Liao C.; Peach M. L.; Ihlenfeldt W. D.; Karki R. G.; Borodina Y. V.; Cachau R. E.; Nicklaus M. C. PDB ligand conformational energies calculated quantum-mechanically. J. Chem. Inf. Model. 2012, 52, 739–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E.; Charifson P. S. Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47, 2499–510. [DOI] [PubMed] [Google Scholar]
- Borbulevych O. Y.; Plumley J. A.; Martin R. M.; Merz J.; Kenneth M.; Westerhoff L. M. Accurate macromolecular crystallographic refinement: incorporation of the linear scaling, semiempirical quantum-mechanics program DivCon into the PHENIX refinement package. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, D70, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh R. A.; Huber R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr., Sect. A: Found. Crystallogr. 1991, 47, 392–400. [Google Scholar]
- Bell J. A.; Ho K. L.; Farid R. Significant reduction in errors associated with nonbonded contacts in protein crystal structures: automated all-atom refinement with PrimeX. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 935–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn T. D.; Schnieders M. J.; Mustyakimov M.; Wu C.; Langan P.; Pande V. S.; Brunger A. T. Reintroducing electrostatics into macromolecular crystallographic refinement: application to neutron crystallography and DNA hydration. Structure 2011, 19, 523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodek S.; Skillman A. G.; Nicholls A. Automated ligand placement and refinement with a combined force field and shape potential. Acta Crystallogr., Sect. D: Biol. Crystallogr.. 2006, 62, 741–9. [DOI] [PubMed] [Google Scholar]