

Abstract
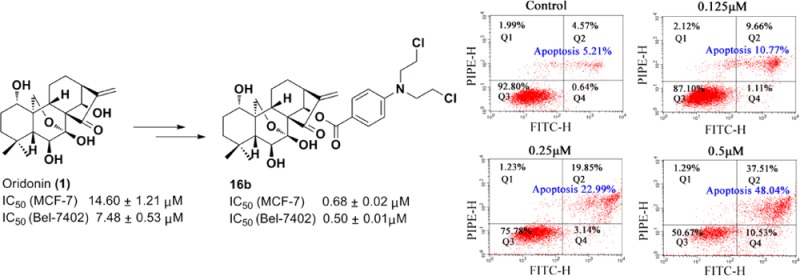
A series of novel hybrids from natural product oridonin and nitrogen mustards were designed and synthesized to obtain more efficacious and less toxic antitumor agents. The antiproliferative evaluation showed that most conjugates were more potent than their parent compounds oridonin and clinically used nitrogen mustards against four human cancer cell lines (K562, MCF-7, Bel-7402, and MGC-803). Furthermore, the representative compounds 16a–c exhibited antiproliferative activities against the multidrug resistant cell lines (SW620/AD300 and NCI-H460/MX20). It was shown that the most effective compound 16b possesses a strong inhibitory activity with an IC50 value 21-fold lower than that of oridonin in MCF-7 cells and also exhibits selective cytotoxicity toward the cancer cells. Intriguingly, compound 16b has been demonstrated to significantly induce apoptosis and affect cell cycle progression in human hepatoma Bel-7402 cells.
Keywords: Oridonin, antiproliferative activities, nitrogen mustards, combination principle, drug-resistant, apoptosis
Cancer is a major global health problem, representing the second-leading cause of death worldwide.1 Effective anticancer agents with novel scaffolds or new mechanisms of action are urgently needed for the highly aggressive and drug-resistant cancer.
Nitrogen mustards are among the DNA alkylating agents that are most widely used in cancer chemotherapy.2 However, the lack of drug-specific affinity toward cancer cells necessitates the use of a large dose of a drug to achieve high local concentration, systemic toxicity leading to many side effects, and the failure of chemotherapy due to acquired drug resistance limited the use of nitrogen mustards in clinics.3 In this regard, it is necessary to carry out chemical modifications on the nitrogen mustards to acquire new agents with enhanced anticancer activity, reduced systemic toxicity, and high selectivity for chemotherapy of cancer.4
Combination principle is used widely in drug design.5 There have been a great deal of reports describing the hybrids between natural products and anticancer drugs,6 in which the individual molecules are commonly connected via a chemically stable linker.7 For example, prednimustine (Figure 1), the prednisolone ester of chlorambucil, not only exhibits a similar profile to that of chlorambucil in murine tumors8 but also demonstrates activity against chlorambucil-resistant tumors. Also included within this group are estramustine phosphate sodium and uramustine (Figure 1).9
Figure 1.

Structures of chlorambucil and available anticancer drugs designed by using combination principle.
Natural diterpenoids with unique chemical skeletons and interesting activities have attracted our attention and aroused our curiosity for intensive study. Oridonin (1) is a widely distributed ent-kaurene in the Rabdosia plants and has recently attracted much attention due to its extensive biological activities.10 Although oridonin exhibited unique, safe, broad antitumor activity, the development of oridonin for cancer therapy was hampered by its relatively moderate potency. Therefore, developing novel oridonin derivatives by rational modification to improve its anticancer activity profile is in urgent need.11,12
In our previous studies, we disclosed that some novel 1-O- and 14-O-derivatives of oridonin showed excellent cytotoxicities against several human cancer cell lines in vitro and in vivo. It was found that the modification on the 14-hydroxy of oridonin did not diminish the cytotoxicities against a panel of human cancer cell lines.13 These findings prompted us to introduce different kinds of nitrogen mustards to the 14-hydroxy of oridonin, including chlorambucil, melphalan, and formylmerphalan. In addition, the benzoic acid mustard was also chosen due to its low toxicity.14,15
Herein, in this presentation, we describe the design and synthesis of a series of novel oridonin-coupled nitrogen mustard conjugates. It is hoped that the oridonin/nitrogen mustard hybrids could exhibit potent synergistic anticancer effect, leading to a higher antiproliferative efficacy, a broader therapeutic scope, and a lower systemic toxicity.
First, the oridonin analogues (2 and 5) were synthesized as shown in Scheme 1 according to our previous protocols.16 The oxidation of 1 with Jones reagent afforded ketone 2. The treatment of 1 with 2,2-dimethoxypropane in the presence of TsOH provided ketal 3 in 85% yield. Compound 3 upon reaction with Ac2O/DMAP/TEA led to acetylated compound 4 in the yield of 95%. Deprotection of 4 gave the corresponding alcohol 5 in almost quantitative yield. Then the nitrogen mustard intermediates were prepared as depicted in Scheme 2. The selective protection of the primary amine of melphalan (6) afforded Boc-protected melphalan (7) or formylmerphalan (8). Esterification of 6 carried out in dry methanol in the presence of SOCl2 led to ester 9 in quantitative yield, and subsequent reaction of 9 with succinic anhydride or glutaric anhydride provided melphalan derivatives 10 and 11, respectively. The benzoic acid mustard (15) was prepared according to the literature procedure.14 At last, the target oridonin-coupled nitrogen mustard conjugates (16a–f, 17a–c, and 18a–c) were obtained through the synthetic route outlined in Scheme 3.
Scheme 1. Synthesis of Oridonin Analogues 2 and 5.

Reagents and conditions: (a) Jones reagent, acetone, 0 °C, 10 min, 88%; (b) 2,2-dimethoxypropane, acetone, TsOH, 56 °C, 20 min, 85%; (c) Ac2O, TEA, DMAP, rt, 2 h, 95%; (d) 10% HCl, THF (1:1), rt, 30 min, 98%.
Scheme 2. Synthesis of Melphalan Derivatives 7, 8, 10, and 11 and Benzoic Acid Mustard 15.

Reagents and conditions: (a) Boc2O, TEA, dioxane, rt, 3 h, 95% for compound 7; Ac2O, HCOOH, 50 °C, 5 h, 67% for compound 8; (b) SOCl2, MeOH, reflux, 12 h, 93%; (c) corresponding anhydrides, DMAP, DCM, rt, 18 h, 93% for compound 10; 24 h, 91% for compound 11; (d) ethanol, H2SO4, reflux, 5 h, 94%; (e) ethylene oxide, H2O, HCOOH, rt, 24 h, 81%; (f) (i) POCl3, 50 °C, 0.5 h; (ii) 10% HCl, 12 h, 81% over two steps.
Scheme 3. Synthesis of Oridonin Nitrogen Mustard Derivatives 16a–f, 17a–c, and 18a–c.

Reagents and conditions: (a) 7, 8, 10, 11, 15, chlorambucil, EDCI, DMAP, DCM, rt, 12–24 h, 60%–82%; (b) 10, 15, chlorambucil, EDCI, DMAP, DCM, rt, 9–16 h, 71%–83%; (c) 10, 15, chlorambucil, EDCI, DMAP, DCM, rt, 12–18 h, 72–78%.
The oridonin-coupled nitrogen mustard derivatives were screened for their in vitro antiproliferative activities against four human cancer cell lines, human leukemia K562 cells, breast adenocarcinoma MCF-7 cells, human hepatocellular carcinoma Bel-7402 cells, and MCG-803 human gastric cancer cells, by the standard MTT methods, and the results are summarized in Table 1. The IC50 values revealed that all the target compounds were more potent than positive control drug chlorambucil and melphalan whatever the cell line considered, and most of the conjugates exhibited more potent inhibitory activities against four cancer cell lines than their parent oridonin. As shown in Table 1, although melphalan showed more potent cytotoxicity than chlorambucil and benzoic acid mustard, compounds 16a and 16d–f incorporated oridonin with melphalan showed less activity than compounds 16b–c that are incorporated with chlorambucil or benzoic acid mustard. Considering compounds 16a, 17a, and 18a, varying only the substitution on C-1 position of oridonin, the IC50 values of 16a were higher than those of 17a and 18a in all tested cell lines. However, when conjugated with chlorambucil or benzoic acid mustard, the modification on the C-1 position of oridonin had no impact on the cytotoxicities of these conjugates; 16b–c, 17b–c, and 18b–c all had relatively high antiproliferative activities against four cancer cell lines. It is interesting that the benzoic acid mustard has less cytotoxicity, while when incorporated with oridonin, conjugate 16b with OH at C-1, is the most potent hybrid among all the target compounds, with the IC50 values at 1.12 ± 0.07, 0.68 ± 0.02, 0.50 ± 0.01, and 1.09 ± 0.14 μM for K562, MCF-7, Bel-7402, and MGC-803, respectively.
Table 1. IC50a Values (μM) of Synthetic Oridonin Nitrogen Mustard Derivatives (16a–f, 17a–c, and 18a–c) against Human Cancer Cell Linesb.
| compd | K562 | MCF-7 | Bel-7402 | MGC-803 |
|---|---|---|---|---|
| oridonin | 4.76 ± 0.32 | 14.60 ± 1.21 | 7.48 ± 0.53 | 5.69 ± 0.37 |
| chlorambucil | 60.12 ± 5.48 | 80.54 ± 4.73 | 49.31 ± 2.87 | 133.64 ± 11.25 |
| melphalan | 33.37 ± 1.16 | 19.58 ± 1.02 | 24.76 ± 1.35 | 36.88 ± 1.21 |
| benzoic acid mustard (15) | 142.67 ± 10.23 | 152.15 ± 8.66 | >200 | 146.20 ± 9.73 |
| 16a | 9.01 ± 0.78 | 14.37 ± 1.24 | 8.35 ± 0.74 | 9.30 ± 0.18 |
| 16b | 1.12 ± 0.07c | 0.68 ± 0.02c | 0.50 ± 0.01c | 1.09 ± 0.14c |
| 16c | 2.28 ± 0.69c | 1.32 ± 0.05c | 1.43 ± 0.09c | 2.02 ± 0.27c |
| 16d | 5.74 ± 0.45 | 12.90 ± 0.96 | 7.91 ± 0.85 | 6.92 ± 0.45 |
| 16e | 1.41 ± 0.10c | 3.22 ± 0.45c | 2.36 ± 0.67c | 6.72 ± 0.58 |
| 16f | 7.03 ± 0.34 | 14.58 ± 0.28 | 3.84 ± 0.32c | 7.10 ± 0.33 |
| 17a | 1.73 ± 0.09c | 3.63 ± 0.15c | 2.56 ± 0.51c | 4.71 ± 0.16 |
| 17b | 1.35 ± 0.12c | 0.91 ± 0.03c | 2.41 ± 0.23c | 3.76 ± 0.53c |
| 17c | 1.10 ± 0.15c | 0.79 ± 0.01c | 1.37 ± 0.06c | 2.25 ± 0.19c |
| 18a | 1.66 ± 0.22c | 2.88 ± 0.19c | 1.41 ± 0.11c | 3.39 ± 0.27c |
| 18b | 1.17 ± 0.14c | 0.67 ± 0.01c | 0.90 ± 0.03c | 1.60 ± 0.08c |
| 18c | 2.37 ± 0.19c | 2.25 ± 0.08c | 2.53 ± 0.47c | 3.95 ± 0.52c |
IC50: concentration that inhibits 50% of cell growth.
MTT method: drug exposure was for 72 h (means ± SD, n = 3).
*p < 0.001 vs oridonin group.
Multidrug resistance (MDR) is one of the major reasons for the failure of cancer chemotherapy.17 One of the major mechanisms in cancer cells that give rise to MDR is the overexpression of ATP-binding cassette (ABC) transporters.18 To investigate whether these nitrogen mustard-fused oridonin derivatives are effective on drug-resistant ABCB1-overexpressing SW620/AD300 and ABCG2-overexpressing NCI-H460/MX20 cells, representative compounds 16a–c, which are coupled with melphalan, benzoic acid mustard, and chlorambucil, respectively, were selected to test their antiproliferative activities against the drug-resistant and parental sensitive cells by using MTT methods. As shown in Table 2, oridonin had moderate anticancer activities against both sensitive and drug-resistant cells. Unfortunately, when conjugated with melphalan, the antiproliferative activity of compound 16a decreased, especially on the NCI-H460/MX20 cells, which did not exhibit any cytotoxicity even at a concentration of 100 μM. This result indicated that 16a may be a substrate of multidrug transporter BCRP (breast cancer resistance protein, also called ABCG2). To our delight, compounds 16b and 16c, the 14-OH modified oridonin ester of benzoic acid mustard or chlorambucil, not only displayed potent antiproliferative activities against cancer cells but also demonstrated activities against drug-resistant cancer cells. Particularly, hybrid 16b exhibited the most potent antiproliferative activities against SW620, SW620/AD300, NCI-H460, and NCI-H460/MX20 cells with IC50 values at 1.96 ± 0.11, 1.86 ± 0.06, 2.35 ± 0.14, and 2.91 ± 0.12 μM, respectively. Therefore, it clearly illustrated that multidrug transport P-glycoprotein (P-gp/ABCB1) did not affect antiproliferative activities of these compounds. As a result, the design of hybrids from natural product and nitrogen mustards appears to be a viable strategy to address the problem of MDR in cancer therapy.
Table 2. IC50a Values (μM) of Representative Compounds 16a–c in the Drug-Resistant and Parental Sensitive Cellsb.
| compd | SW620 | SW620/AD300 | NCI-H460 | NCI-H460/MX20 |
|---|---|---|---|---|
| oridonin | 6.26 ± 0.33 | 4.67 ± 0.17 | 17.14 ± 1.06 | 21.71 ± 0.98 |
| 16a | 13.95 ± 0.16 | 14.92 ± 1.28 | 28.41 ± 0.76 | >100 |
| 16b | 1.96 ± 0.11 | 1.86 ± 0.06 | 2.35 ± 0.14 | 2.91 ± 0.12 |
| 16c | 2.70 ± 0.07 | 2.28 ± 0.09 | 4.81 ± 0.11 | 7.26 ± 0.28 |
IC50: concentration that inhibits 50% of cell growth.
MTT cytotoxicity assay was assessed in pairs of parental and transporter-overexpressing cell lines: SW620 and ABCB1-overexpressing SW620/AD300 cells; NCI-H460 and ABCG2-overexpressing NCI-H460/MX20 cells.
Nonselective cytotoxicity is also the main effect that limits the use of optimal doses in most conventional chemotherapeutic regiments.19 In an attempt to estimate the toxicity of conjugates 16b and 16c compared with chlorambucil or benzoic acid mustard, we tested the toxicities of the compounds 1, 15, 16b, 16c, and chlorambucil on the human normal liver cells L-02 and human liver cancer cells Bel-7402. Cell apoptosis induced by these compounds is both concentration and time dependent; the survival curves of 16b and 16c in Bel-7402 and L-02 cells after a 72 h treatment showed that 90.10% kill was achieved for 16b in Bel-7402 at the concentration of 2.5 μM, but only 25.1% human normal liver L-02 cells were killed at this concentration. For compound 16c, 90.4% Bel-7402 cells were killed at 5 μM, while only 17.36% normal liver cells were killed. As seen in Table 3, we found that conjugate 16b has approximately 8-fold higher selectivity for Bel-7402 cells compared with normal L-02 cells, which was even much higher than that of oridonin. The data suggests that the hybrids from natural product and nitrogen mustards would be safer for the cancer patients receiving chemotherapy.
Table 3. IC50a Values (μM) of Compounds 1, 15, 16b, 16c, and Chlorambucil against Human Liver Cancer Cells Bel-7402 and Human Normal Liver Cells L-02.
| compd | Bel-7402 | L-02 | SIb |
|---|---|---|---|
| oridonin | 7.48 ± 0.53 | 17.78 ± 0.64 | 2.38 |
| chlorambucil | 49.31 ± 1.87 | 105.78 ± 8.22 | 2.14 |
| 15 | >200 | >100 | |
| 16b | 0.50 ± 0.01 | 4.03 ± 0.36 | 8.06 |
| 16c | 1.43 ± 0.09 | 9.62 ± 0.23 | 6.73 |
IC50: concentration that inhibits 50% of cell growth.
SI: selective index (IC50 on normal cells/IC50 on tumor cells).
Drug combinations are widely used for the treatment of cancer to increase the efficiency and reduce side effects.20 Oridonin has been reported to be combined with anticancer drugs to treat the cancer patients in clinical practice.21 In order to investigate whether conjugates 16b and 16c exhibited their cytotoxicities alone or through the synergistic effect, we tested the potency of equimolar concentration of oridonin, benzoic acid mustard, or 16b alone, and the combination of equimolar oridonin and benzoic acid mustard on MCF-7 cells. The results showed that the combination of the oridonin and benzoic acid mustard was not superior to conjugate 16b alone at any concentration. The same phenomenon could also be observed on the compound 16c (see Supporting Information Figure S1).
As shown in Figure 2, the IC50 values of 1:1 combination of oridonin and benzoic acid mustard or chlorambucil was 11-fold or 6-fold higher than that of 16b or 16c on MCF-7 cells, respectively. These results indicated that hybrids 16b and 16c were more potent than oridonin or an equimolar mixture of its components nitrogen mustard and oridonin. Therefore, the efficacy of 16b and 16c in breast cancer could not be mimicked by the simple direct combination.
Figure 2.

(a) IC50 values (μM) of the hybrid (16b), oridonin, benzoic acid mustard, and oridonin + benzoic acid mustard against the MCF-7 cells; (b) IC50 values (μM) of the hybrid (16c), oridonin, chlorambucil, and oridonin + chlorambucil against the MCF-7 cells.
To determine whether the suppression of cell growth by compound 16b is caused by a cell-cycle effect, the DNA content of cell nuclei was detected by flow cytometry. As shown in Figure 3, compound 16b could influence cell cycle progression at low micromolar concentrations. When Bel-7402 cells were treated with different concentrations of compound 16b (0.125, 0.25, or 0.5 μM), the percentage of cells in G2 fraction decreased from 19.52% to 4.46% associated with a percentage increase of G1-phase cells (39.66% to 51.98%, see Supporting Information Table S1).
Figure 3.
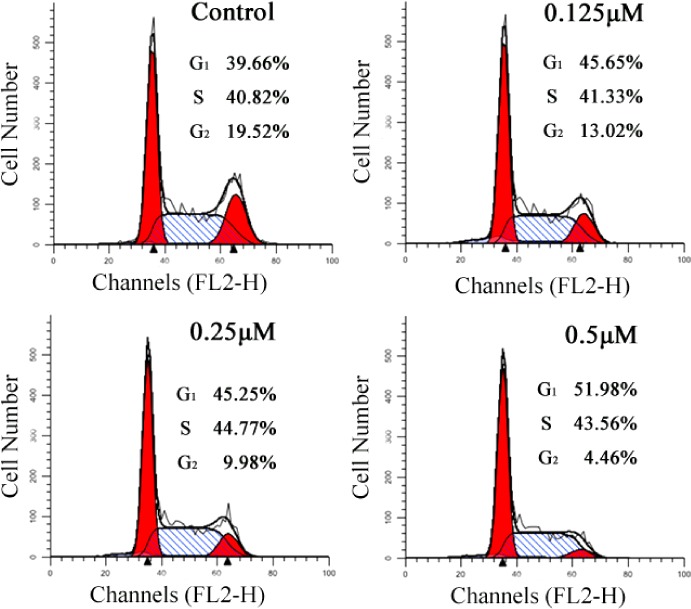
Effects of hybrid 16b on cell cycle of Bel-7402 cells.
To clarify whether the loss of cancer cell viability promoted by conjugate 16b is associated with apoptosis, an annexin V-FITC/propidium iodide (PI) binding assay was performed. Bel-7402 cells were treated with vehicle alone or with various concentrations (0.125, 0.25, or 0.5 μM) of compound 16b for 48 h and then stained with FITC-annexin V and propidium iodide (PI). The percentages of apoptotic Bel-7402 cells were determined by flow cytometry. As shown in Figure 4, conjugate 16b caused significant induction of apoptosis in a concentration-dependent manner. When treated with 0.125, 0.25, and 0.5 μM compound 16b for 48 h; the percentages of apoptotic cells were 10.77%, 22.99%, and 48.08% (Q2 + Q4), respectively, compared with 5.21% of vehicle control. This result demonstrated that the antiproliferative activity of hybrid 16b could be attributed to the induction of apoptosis in Bel-7402 cells.
Figure 4.
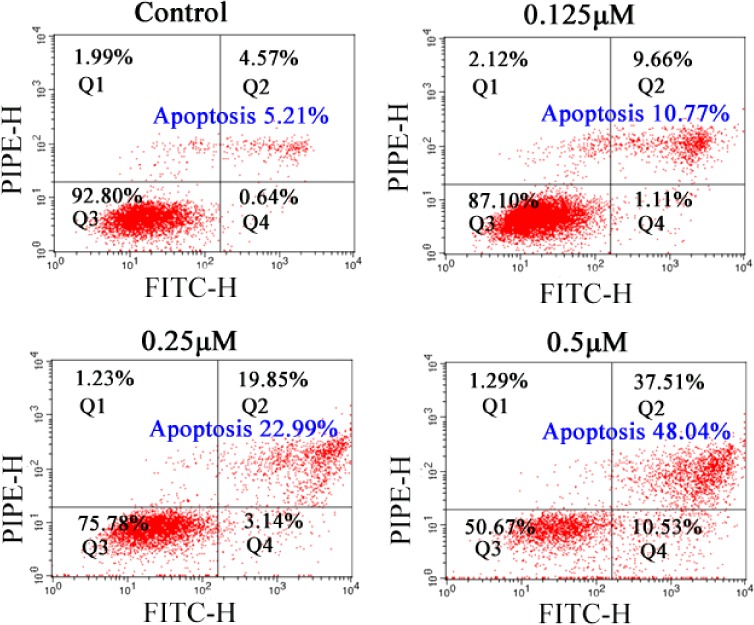
Effects of hybrid 16b on cell apoptosis.
In conclusion, the findings arising from the studies described above open a possible approach to the development of hybrids as potential anticancer agents. In this effort, a series of novel oridonin-coupled nitrogen mustard conjugates were designed and synthesized by following the “combination principle” and their anticancer activities were evaluated against four human cancer cell lines. All the target compounds showed more effective antiproliferative activity than positive control melphalan and chlorambucil. Among them, compound 16b was the most potent hybrid with IC50 values 0.68 ± 0.02 and 0.50 ± 0.01 μM against MCF-7 and Bel-7402 cells, respectively. The promising compounds 16b and 16c also exhibited potent antiproliferative activity against drug-resistant cells. Furthermore, it was found that conjugate 16b has an approximately 8-fold higher selectivity for cancer cells than normal cells, which was higher than those of parent oridonin and clinically used nitrogen mustard drugs. Importantly, it was also found that the antiproliferative activity of the most promising compound 16b could be attributed to the induction of cell cycle arrest and apoptosis in cancer cells. Collectively, 16b could be considered as the promising lead compound for the design of more efficacious and less toxic chemotherapeutic agents to enhance the efficacy of chemotherapy in cancer patients.
Acknowledgments
The authors thank Drs. S. E. Bates and R. W. Robey (NIH, Maryland, USA) for the cell lines H460, H460/MX20, SW620, and SW620/AD300. The authors thank COUP-US.com (New Jersey, USA) for free editing of the article.
Supporting Information Available
Synthetic methods and characterization of target compounds; procedures for pharmacological activities. This material is available free of charge via the Internet at http://pubs.acs.org.
This work is supported by the National Natural Science Foundation (No. 81373280) and Huahai Pharmaceutical Graduate Innovation Fund (CX13B-002HH).
The authors declare no competing financial interest.
Supplementary Material
References
- Bray F.; Ren J. S.; Masuyer E.; Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int. J. Cancer 2008, 132, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Povirk L. F.; Shuker D. E. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. 1994, 318, 205–226. [DOI] [PubMed] [Google Scholar]
- Sanderson B. J.; Shield A. J. Mutagenic damage to mammalian cells by therapeutic alkylating agents. Mutat. Res. 1996, 355, 41–57. [DOI] [PubMed] [Google Scholar]
- Zarytova V. F.; Ivanova E. M.; Chasovskikh M. N. Synthesis of steroid-containing oligonuleotides and their alkylating derivatives. Bioorg. Khim. 1990, 16, 610–616. [PubMed] [Google Scholar]
- Yasobu N.; Kitajima M.; Kogure N.; Shishido Y.; Matsuzaki T.; Nagaoka M.; Takayama H. Design, synthesis, and anti-tumor activity of 4-halocolchicines and their pro-drugs activated by cathepsin B. ACS Med. Chem. Lett. 2011, 2, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker M. Hybrid molecules incorporating natural products: applications in cancer therapy, neurodegenerative disorders and beyond. Curr. Med. Chem. 2011, 18, 1464–1475. [DOI] [PubMed] [Google Scholar]
- Saha P.; Debnath C.; Bérubé G. Steroid-linked nitrogen mustards as potential anticancer therapeutics: a review. J. Steroid Biochem. Mol. Biol. 2013, 137, 271–300. [DOI] [PubMed] [Google Scholar]
- Hartley-Asp B.; Gunnarsson P. O.; Liljekvist J. Cytotoxicity and metabolism of prednimustine, chlorambucil and prednisolone in a Chinese hamster cell line. Cancer Chemother. Pharmacol. 1986, 16, 85–90. [DOI] [PubMed] [Google Scholar]
- Perry C. M.; McTavish D. Estramustine phosphate sodium. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in prostate cancer. Drugs Aging 1995, 7, 49–74. [DOI] [PubMed] [Google Scholar]
- Sun H. D.; Huang S. X.; Han Q. B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006, 23, 673–698. [DOI] [PubMed] [Google Scholar]
- Ding C. Y.; Zhang Y. S.; Chen H. J.; Yang Z. D.; Wild C.; Ye N.; Ester C. D.; Xiong A. L.; White M. A.; Shen Q.; Zhou J. Oridonin ring A-based diverse donstructions of enone functionality: identification of novel dienone analogues effective for highly aggressive breast cancer by inducing apoptosis. J. Med. Chem. 2013, 56, 8814–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C. Y.; Zhang Y. S.; Chen H. J.; Yang Z. D.; Wild C.; Chu L. L.; Liu H. L.; Shen Q.; Zhou J. Novel nitrogen-enriched oridonin analogues with thiazole-fused A-ring: protecting group-free synthesis, enhanced anticancer profile, and improved aqueous solubility. J. Med. Chem. 2013, 56, 5048–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Y.; Yang J. Y.; Ran Q.; Wang L.; Liu J.; Wang Z. X.; Wu X. M.; Hua W. Y.; Yuan S. T.; Zhang L. Y.; Shen M. Q.; Ding Y. F. Synthesis and biological evaluation of novel 1-O-and 14-O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 4741–4744. [DOI] [PubMed] [Google Scholar]
- Luo W.; Zhao Y. M.; Wang Y. X.; Xie S. Q.; Zhao J.; Wang C. J. Synthesis and antitumor activity of benzoic nitrogen mustard derivatives. Acta Pharm. Sin. 2007, 42, 1327–1329. [PubMed] [Google Scholar]
- Zheng Q. Z.; Zhang F.; Cheng K.; Yang Y.; Chen Y.; Qian Y.; Zhang H. J.; Li H. Q.; Zhou C. F.; An S. Q.; Jiao Q. C.; Zhu H. L. Synthesis, biological evaluation and molecular docking studies of amide-coupled benzoic nitrogen mustard derivatives as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 880–886. [DOI] [PubMed] [Google Scholar]
- Wang L.; Li D. H.; Xu S. T.; Cai H.; Yao H. Q.; Zhang Y. H.; Jiang J. Y.; Xu J. Y. The conversion of oridonin to spirolactone-type or enmein-type diterpenoid: synthesis and biological evaluation of ent-6,7-seco-oridonin derivatives as novel potential anticancer agents. Eur. J. Med. Chem. 2012, 52, 242–250. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Sun Y. L.; Patel A.; Kumar P.; Chen Z. S. Role of ABC transporters in cancer chemotherapy. Chin. J. Cancer 2012, 31, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hileman E. O.; Liu J.; Albitar M.; Keating M. J.; Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [DOI] [PubMed] [Google Scholar]
- Li J.; Wang Y.; Zhu Y.; Oupický D. Recent advances in delivery of drug-nucleic acid combinations for cancer treatment. J. Controlled Release 2013, 172, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu H. Q.; Luo J.; Chen H.; Zhang J. H.; Li H. H.; Guo H. C.; Wang Z. H.; Lin S. Z. Oridonin enhances antitumor activity of gemcitabine in pancreatic cancer through MAPK-p38 signaling pathway. Int. J. Oncol. 2012, 41, 949–598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.