

Abstract

A series of 4-bicyclic heteroaryl 1,2,3,4-tetrahydroisoquinoline inhibitors of the serotonin transporter (SERT), norepinephrine transporter (NET), and dopamine transporter (DAT) was discovered. The synthesis and structure–activity relationship (SAR) of these triple reuptake inhibitors (TRIs) will be discussed. Compound 10i (AMR-2), a very potent inhibitor of SERT, NET, and DAT, showed efficacy in the rat forced-swim and mouse tail suspension models with minimum effective doses of 0.3 and 1 mg/kg (po), respectively. At efficacious doses in these assays, 10i exhibited substantial occupancy levels at the three transporters in both rat and mouse brain. The study of the metabolism of 10i revealed the formation of a significant active metabolite, compound 13.
Keywords: Triple reuptake inhibitor, TRI, serotonin reuptake inhibitor, dopamine reuptake inhibitor, norepinephrine reuptake inhibitor, depression, antidepressant
Major depressive disorder (MDD) is the leading cause of disability in the U.S. among those aged 15–44.1 It affects approximately 14.8 million American adults, or about 6.7% of the U.S. population age 18 and older in a given year.2 Aside from the mental and physical suffering of patients, the economic burden of depression was 83.1 billion dollars in the US alone in 2000.3
Currently, nearly all antidepressants prescribed are serotonin or dual serotonin and norepinephrine reuptake inhibitors (SSRIs and SNRIs respectively), which inhibit central serotonin reuptake transporters (SERT) or both SERT and norepinephrine transporters (NET). Despite the therapeutic benefits of SSRIs and SNRIs, a significant proportion of patients show a suboptimal response.4,5
Dopamine is involved in centrally mediated reward responses.6 The inhibition of dopamine reuptake transporter (DAT) elevates synaptic dopamine levels.7 It has been suggested that the addition of an appropriate degree of DAT inhibition to SSRI or SNRI therapy might help depressed patients overcome anhedonia, a symptom of depressive disorders inadequately addressed by SSRI and SNRIs alone.6 Bupropion and methylphenidate are inhibitors of dopamine and norepinephrine transporters (DAT/NET), and they are used in the treatment of various psychiatric disorders.8,9 Several small clinical studies in MDD using combinations of bupropion or methylphenidate with an SNRI or SSRI have shown improved efficacy in patients refractory to SSRI, SNRI, or bupropion therapy alone.10−13 These clinical results suggest that a triple reuptake inhibitor (TRI), which blocks SERT, NET, and DAT simultaneously, might benefit patients experiencing an inadequate response to current antidepressant therapies.
A number of TRIs have been reported in the past decade.14−19 Structurally, the design of novel TRIs originated from our series of dual NET/DAT inhibitors,20 represented by compound 1. Following the pharmacological strategy outlined above, we sought structural modifications that would build in SERT activity and retain the NET/DAT reuptake profile of 1. We now report that the placement of bicyclic heterocycles on the 4-position of the tetrahydroisoquinoline (THIQ) core (Figure 1) accomplished this objective and provided compounds with potent TRI profiles. The structure–activity relationship (SAR) of the 4-phenyl THIQ series suggested that substitution on the 7-position of the THIQ often provided potency enhancements. Therefore, we also explored 7-substitution in the current series. In this letter, we report the synthesis and biological evaluation of this novel series of 4-bicyclic heteroaryl 1,2,3,4-THIQ derivatives.
Figure 1.

Design of THIQ TRIs.
Chemistry
A convenient synthetic route to 4-bicyclic heteroaryl THIQ derivatives without substitution on the 7-position is illustrated in Scheme 1.21 Commercially available isoquinolin-4-ylboronic acid or 4-bromoisoquinoline underwent Suzuki coupling with ArBr or ArB(OH)3 (or the corresponding boronate ester), respectively, to give the 4-aryl isoquinoline 2. A one-pot methylation followed by reduction yielded the desired 4-bicyclic heteroaryl 1,2,3,4-THIQs 3 (Table 1).
Scheme 1.

Table 1. Potency at SERT, DAT, and NET Expressed as IC50s or % Inhibition at 100 nM.
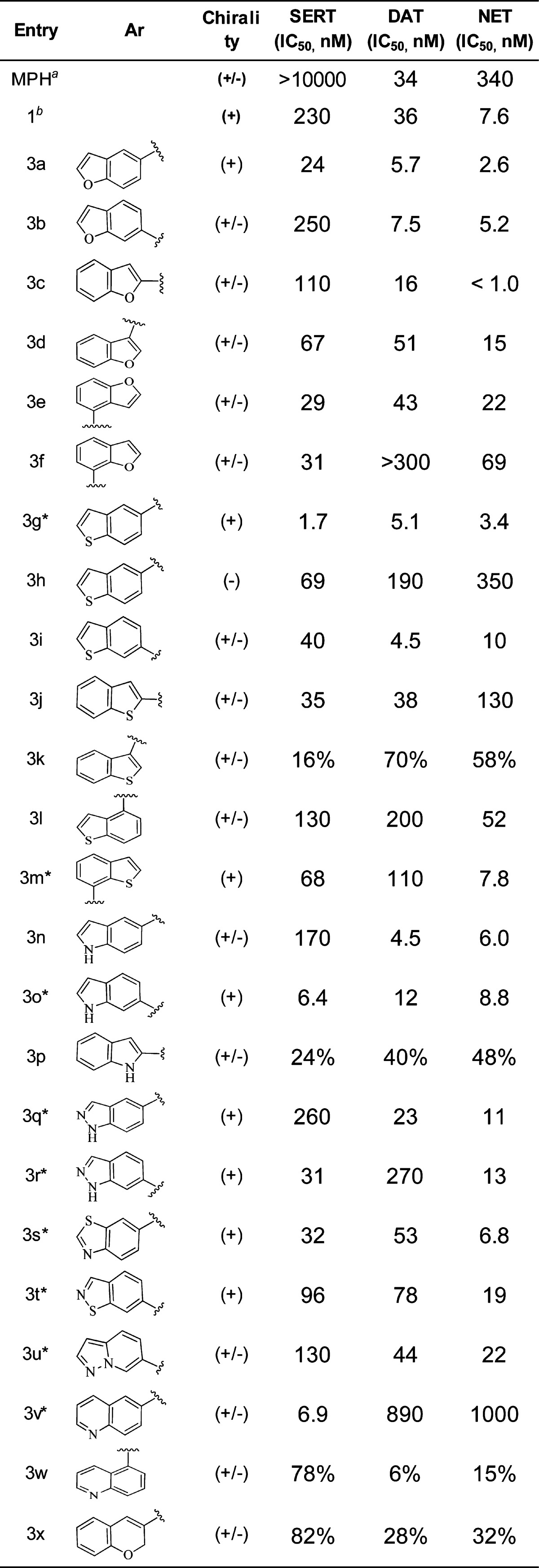
Inhibition values are Kis.23,20 Inhibition data (3a–3x) were obtained using assay protocol 2 except for data for compounds with “*”, which were measured using assay protocol 1 (see Supporting Information). % Inhibition at 100 nM measured using assay protocol 2. The two protocols gave similar results based on the examination of a small set of data generated using both protocols. All data were measured with at least n = 2.
Alternatively, the 4-quinolinyl analogues 3v and 3w (Table 1) were prepared by α-arylation of dihydroisoquinolinone (4a) with aryl bromide followed by reduction of the lactam 4 (Scheme 2).22
Scheme 2.
The general synthetic route for the majority of the 4-bicyclic heteroaryl 1,2,3,4-THIQs with substitution on the 7-position is illustrated in Scheme 3.20 Alkylation of benzylamines 5 with bromoacetophenones 4 yielded the desired ketones 6. Reduction of 6 with NaBH4 followed by a Friedel–Crafts cyclization using methanesulfonic acid gave 4-aryl THIQs 7 as the major regioisomer. For the Friedel–Crafts cyclization step, we found that both the desired regioselectivity and the yield were greater for the methoxy compounds (6b) versus the bromides (6a). The cyclization typically gave a 2:1 ratio of the desired 7-methoxy isomer 7 over the undesired 5-methoxy isomer in ∼60% isolated yield of 7b. The O-demethylation of 7b followed by the treatment with triflic anhydride provided the triflates 9. Further functionalization of 9 or bromides 7a, usually via palladium mediated coupling reactions, provided the final products 10. For compound 10a, 10b, or 10d, benzylamine 5 with the appropriate R1 as Y (i.e., −CH3, −OCH3, and −F, respectively) was used for the alkylation step (Scheme 3).21 Compounds 10f–10h were derived from 10e using straightforward transformations.21 The reduction of 10e with lithium aluminum hydride gave 10f. A reductive amination reaction of 10f with formalin and sodium cyanoborohydride afforded 10g. Compound 10h was obtained by the bis-alkylation of 10f with oxybis(ethane-2,1-diyl) bis(4-methylbenzenesulfonate) in the presence of sodium carbonate in refluxing acetonitrile. Compounds 3d, 3i, and 3l were also prepared using the route outlined in Scheme 3 since the corresponding aryl bromides or aryl boronic acids were not commercially available.21 Single enantiomers of compounds 3 and 10 typically were obtained by chiral high-performance liquid chromatography (HPLC) using ChiralpakAD or ChiralcelOD columns.
Scheme 3.

Results and Discussion
We first examined a variety of bicyclic heterocycles on the 4-position of the THIQ core. The inhibition activities for SERT, DAT, and NET are listed in Table 1. The inhibition data for known dual DAT/NET inhibitors methylphenidate (MPH)23 and our reported compound 1(20) are also included in Table 1. In general, a racemic compound was separated into single enantiomers if it showed potent triple inhibition or if a new structural moiety was introduced. To our surprise, while the 4-phenyl THIQs such as 1 possessed a dual NET/DAT inhibition profile, many of the 4-bicyclic heterocycle THIQs inhibited all three transporters with similar potency. In general, benzofuran, benzothiophene, and indole substituted THIQs (3a–3p) possessed the greatest activity across the three transporters. The point of connection on the pendant heteroaryls had a significant impact on the TRI profile: attachment to the 5- or 6-position on these three heterocycles was optimal for transporter potency. Benzothiophene 3g was particularly potent and demonstrated low nanomolar inhibition of all three transporters. Compound 3g was found to be a modestly potent CYP2D6 inhibitor (IC50 = 0.8 μM), which represents a potential drug–drug interaction liability. Consistent with our previous observation,20 there is a stereogenic preference in transporter binding activity. The (+)-enantiomers were found to be more potent than the (−)-enantiomers (exemplified by 3g and 3h) in all enantiomeric pairs examined. It is also interesting to note that both the indole and indazole compounds (3n and 3q) with the connecting point at the 5-position had dual DAT/NET activity and that compounds 3f and 3r had SERT/NET profiles. The indole and indazole compounds in general had weaker transporter and CYP2D6 inhibition activities compared to the benzofuran and benzothiophene analogues (data not shown). The [6.6]-fused heterocycles (3v–3x), however, demonstrated a more SERT-selective profile. From the SAR in Table 1, it was clear to us that both the choice of bicyclic heterocycle and the point of connection can have profound impact on the transporter inhibition profile.
Our SAR studies of the 4-phenyl series20 revealed that substitution on the 7-position of the THIQ scaffold often provided significant activity at all three transporters. Encouraged by the high potency of the benzothiophene compound 3g, we decided to explore the substitution of the 7-position with the objective of identifying compounds with TRI profiles and reduced CYP2D6 activity.
We prepared a number of analogues of 3g with various substitutions at the 7-position of the THIQ core (Table 2). As expected, these compounds frequently demonstrated good inhibition at the three transporters. A simple methyl substitution (10a) at the THIQ 7-position did not change the TRI profile compared to 3g. Methoxy or hydroxy substitution also had little impact on the potency at the three transporters (10b and 10c). However, 10a–c were still CYP2D6 inhibitors (IC50 <1 μM). Fluoro and cyano (10d and 10e) weakened the inhibition significantly, suggesting that electron withdrawing groups were not favored. Aminomethyl analogues 10f–10h showed very potent triple profiles. The morpholino analogue 10i had very potent inhibition at all three transporters with a much reduced CYP2D6 inhibition (IC50 = 11 μM). As observed in the 7-unsubstituted series (3), the (+)-enantiomers of 10 that were examined were more potent TRIs, but also more potent as CYP2D6 inhibitors when compared to the (−)-enantiomers. It is worth noting that the (−)-enantiomer of 10i, i.e., 10j, showed weaker, but significant activity at the three transporters. The absolute stereochemistry of 10i was determined to be (S) by X-ray crystallography as shown in Figure 2.
Table 2. Potency at SERT, DAT, and NET for 10a.
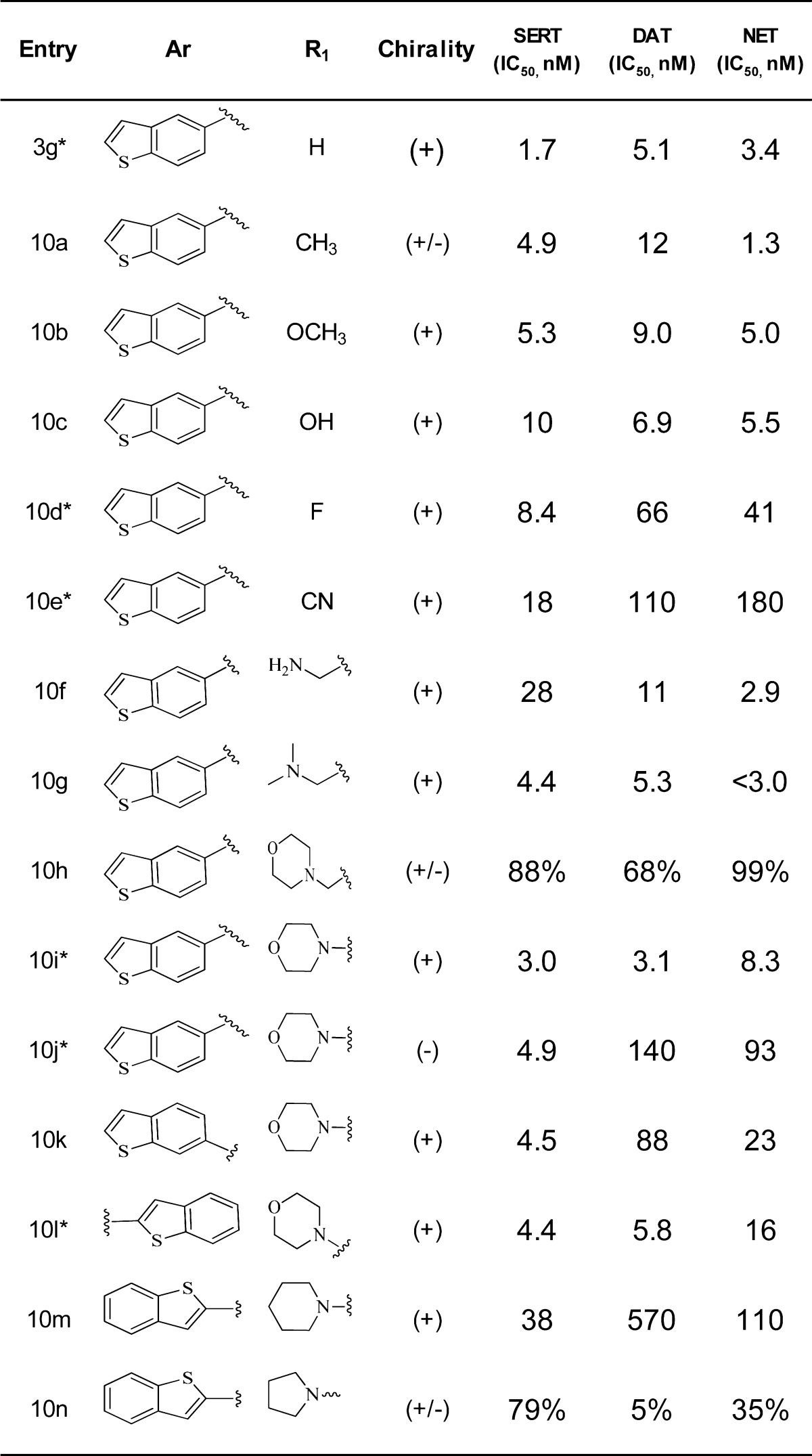
Inhibition data were obtained using assay protocol 2 except for compounds with “*”, which were measured using assay protocol 1 (see Supporting Information). Data for compounds 10h and 10n are inhibition at 100 nM. The two protocols gave similar results based on the examination of a small set of data generated using both protocols. All data were measured with at least n = 2.
Figure 2.
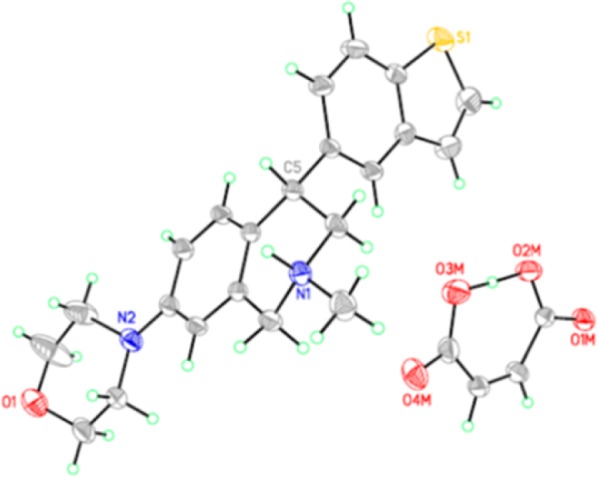
Single crystal structure of 10i (maleate salt).
The benzothiophen-6-yl analogue 10k showed weaker activity at DAT compared to the benzothiophen-5-yl 10i. The benzothiophen-2-yl analogue 10l, however, had a transporter inhibition profile very similar to that of 10i. Replacing the morpholine with piperidine (10m) and pyrrolidine (10n) resulted in an overall reduction of transporter inhibition potency.
On the basis of its most potent and balanced triple inhibition profile and weak CYP2D6 inhibition profile, compound 10i(24) was advanced for profiling in a number of in vitro and in vivo assays. An ex vivo binding assay was used to measure the transporter occupancy levels in rodent brain.25 Clinically effective doses of SSRIs and SNRIs are associated with significant SERT occupancy, as measured by positron emission tomography (PET).26,27 Following oral administration of 10i in the rat at 0.3, 1, and 3 mg/kg, ex vivo binding was significant and dose-dependent at SERT, NET, and DAT at 1 h postdosing (Table 3),28 with SERT and NET occupancies approaching full occupancy of the transporters at the highest dose.
Table 3. SERT, DAT, and NET Occupancies for 10i at Different Doses.
| compd 10i PO dose (mpk) | SERT occupancy | DAT occupancy | NET occupancy |
|---|---|---|---|
| 0.3 | 84 ± 6% | 23 ± 7% | 88 ± 3% |
| 1 | 90 ± 4% | 52 ± 5% | 94 ± 2% |
| 3 | 95 ± 8% | 77 ± 5% | 98 ± 1% |
Compound 10i was also tested in two rodent models used to predict clinical antidepressant activities. In the rat forced swim model, 10i significantly reduced immobility (a measure of despair) with a lowest effective dose of 1 mg/kg (Figure 3).
Figure 3.

Rat forced-swim test results. Asterisk-marked bars denote significance at p < 0.05 versus vehicle for duration of immobilities (n = 12–13). Immobility was measured at 1 h postdosing.
When compound 10i was tested in the mouse tail suspension model,28 it was found to significantly reduce the duration of immobility in a dose-dependent manner with a minimum effective dose of 1 mg/kg (Figure 4). In a mouse occupancy study, 10i showed 84% SERT, 82% NET, and 60% DAT occupancy 1 h after a dose of 1 mg/kg p.o., consistent with the rat transporter occupancy results.
Figure 4.
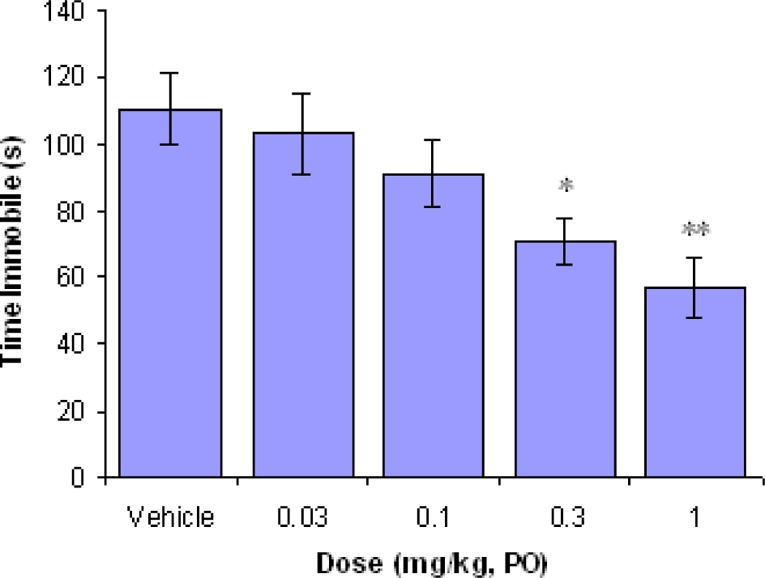
Mouse tail suspension test results for 10i. *p < 0.05 and **p < 0.01 versus vehicle for duration of immobilities (n = 10–12). Immobility was measured at 1 h postdosing.
Compound 10i was screened against a broad panel of receptors and enzymes and showed no significant activity at 1 μM. Compound 10i was also negative in the Ames test, which evaluates mutagenic potential. In an in vitro assay for evaluating the formation of reactive metabolites, no dansyl–glutathione adducts were detected. In the hERG patch-clamp assay, compound 10i exhibited an IC50 of approximately 1 μM.
The key pharmacokinetic parameters in rats and monkeys for compound 10i are summarized in Table 4. Compound 10i exhibited moderate clearance, extensive distribution (high Vd), and was readily absorbed with good oral bioavailability. A brain/plasma ratio of 3 was determined in rat 1 h after oral dosing indicating good CNS penetrance.
Table 4. Pharmacokinetic Parameters for 10i in Rat and Monkey.
Average of 3 male Sprague–Dawley rats; p.o. 10 mg/kg; i.v. 1 mg/kg.
Average of 3 male cynomolgus monkeys; p.o. 2 mg/kg; i.v. 0.5 mg/kg.
Metabolite profiling studies of compound 10i in human and rat liver microsomes and subsequent structural analysis of metabolites revealed a significant morpholine N,O-dealkylated metabolite 13 (prepared in Scheme 4). To confirm the proposed structure, compound 13 was synthesized starting with triflate 11(21) via a Buchwald coupling with oxazolidin-2-one to give 12, followed by hydrolysis (Scheme 4). A pharmacokinetic study of compound 10i in monkey revealed that 13 was formed in significant amounts (>50% based on AUCs) relative to the parent 10i. Compound 13 was found to be almost equally potent to parent compound 10i with IC50s for SERT, DAT, and NET of 2.5, 1.4, and 9.1 nM, respectively. In contrast to the observation of 13 in monkey, the formation of metabolite 13 in rat was not significant. To quickly assess the brain penetrance of this metabolite, 13 was administered intravenously to rat at 2 mg/kg. Significant concentrations (∼3–5 μM) of 13 were detected in rat brain tissue 15 min and 1 h after dosing, with brain to plasma ratios of 2.2 (15 min) and 3.6 (1 h). These findings suggested that 13 might contribute significantly to the in vivo pharmacology of 10i. Compound 13 was also a CYP2D6 inhibitor (IC50 = 1 μM) and therefore carries a potential drug–drug interactions liability. The formation of a significant amount of an active metabolite could potentially complicate the development of compound 10i.29 We have continued to explore compounds with more desirable metabolism profiles and will report our findings in due course.
Scheme 4.

In conclusion, we have discovered a novel series of potent 4-bicyclic heteroaryl THIQ triple monoamine reuptake inhibitors (TRIs). Varying the pendant 4-aryl group and the substituent at the 7-position on the THIQ scaffold resulted in potent TRIs with a range of triple inhibition profiles. A representative compound 10i demonstrated significant occupancy at SERT, NET, and DAT after oral dosing in an ex vivo binding assay and showed excellent efficacy in two animal models used to predict clinical antidepressant efficacy.
Supporting Information Available
Binding, ex vivo occupancy, and rat forced-swim and tail suspension study protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- The World Health Organization. The World Health Report 2004: Changing History, Annex Table 3: Burden of Disease in DALYs by Cause, Sex, and Mortality Stratum in WHO Regions, Estimates for 2002. WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Kessler R. C.; Chiu W. T.; Demler O.; Walters E. E. Prevalence, severity, and comorbidity of twelve-month DSM-IV disorders in the National Comorbidity Survey Replication (NCS-R). Arch. Gen. Psychiatry 2005, 626617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg P. E.; Kessler R. C.; Birnbaum H. G.; Leong S. A.; Lowe S. W.; Berglund P. A.; Corey-Lisle P. K. The economic burden of depression in the United States: how did it change between 1990 and 2000?. J. Clin. Psychiatry 2003, 64121465–1475. [DOI] [PubMed] [Google Scholar]
- Walter M. W. Monoamine reuptake inhibitors: highlights of recent research developments. Drug Dev. Res. 2005, 65, 97–118. [Google Scholar]
- Liu S.; Molino B. F. Recent developments in monoamine reuptake inhibitors. Annu. Rep. Med. Chem. 2007, 42, 13–26. [Google Scholar]
- D’Aquila P. S.; Collu M.; Gessa G. L.; Serra G. The role of dopamine in the mechanism of action of antidepressant drugs. Eur. J. Pharmacol. 2000, 405, 365–373. [DOI] [PubMed] [Google Scholar]
- Gerasimov M. R.; Franceschi M.; Volkow N. D.; Gifford A.; Gatley S. J.; Marsteller D.; Molina P. E.; Dewey S. L. Comparison between intraperitoneal and oral methylphenidate administration: A microdialysis and locomotor activity study. J. Pharmacol. Exp. Ther. 2000, 295, 51–57. [PubMed] [Google Scholar]
- Capp P. K.; Pearl P. L.; Conlon C. Methylphenidate HCl: therapy for attention deficit hyperactivity disorder. Expert Rev. Neurother. 2005, 53325–31. [DOI] [PubMed] [Google Scholar]
- Fava M.; Rush A. J.; Thase M. E.; Clayton A.; Stahl S. M.; Pradko J. F.; Johnston J. A. 15 years of clinical experience with bupropion HCl: from bupropion to bupropion SR to bupropion XL. Prim. Care Companion J. Clin. Psychiatry 2005, 73106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakostas G. I.; Worthington J. J. III; Iosifescu D. V.; Kinrys G.; Burns A. M.; Fisher L. B.; Homberger C. H.; Mischoulon D.; Fava M. The combination of duloxetine and bupropion for treatment-resistant major depression disorder. Depression Anxiety 2006, 23, 178–180. [DOI] [PubMed] [Google Scholar]
- Trivedi M. H.; Fava M.; Wisniewski S. R.; Thase M. E.; Quitkin F.; Warden D.; Ritz L.; Nierenberg A. A.; Lebowitz B. D.; Biggs M. M.; Luther J. F.; Shores-Wilson K.; Rush A. J. Medication augmentation after the failure of SSRIs for depression. New Engl. J. Med. 2006, 354, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Lavretsky H.; Park S.; Siddarth P.; Kumar A.; Reynolds C. F. III. Methylphenidate-enhanced antidepressant response to citalopram in the elderly: a double-blind, placebo-controlled pilot trial. Am. J. Geriatr. Psychiatry 2006, 142181–185. [DOI] [PubMed] [Google Scholar]
- Masand P. S.; Anand V. S.; Tanquary J. F. Psychostimulant augmentation of second generation antidepressants: A case series. Depression Anxiety 1998, 7289–91. [DOI] [PubMed] [Google Scholar]
- Skolnick P.; Popik P.; Janowsky A.; Beer B.; Lippa A. S. Antidepressant-like actions of DOV21,947: a “triple” reuptake inhibitor. Eur. J. Pharmacol. 2003, 461, 99–104. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Shaw A. M.; Boules M.; Briody S.; Robinson J.; Oliveros A.; Blazar E.; Williams K.; Zhang Y.; Carlier P. R.; Richelson E. Antidepressant-like pharmacological profile of a novel triple reuptake inhibitor, (1S,2S)-3-(methylamino)-2-(naphthalen-2-yl)-1-phenylpropan-1-ol (PRC200-SS). J. Pharmacol. Exp. Ther. 2008, 3272573–583. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Cavanni P.; Arban R.; Benedetti R.; Bertani B.; Bettati M.; Bettelini L.; Bonanomi G.; Braggio S.; Checchia A.; Davalli S.; Di Fabio R.; Fazzolari E.; Fontana S.; Marchioro C.; Minick D.; Negri M.; Oliosi B.; Read K. D.; Sartori I.; Tedesco G.; Tarsi L.; Terreni S.; Visentini F.; Zocchi A.; Zonzini L. 1-(Aryl)-6-[alkoxyalkyl]-3-azabicyclo[3.1.0]hexanes and 6-(aryl)-6-[alkoxyalkyl]-3-azabicyclo[3.1.0]hexanes: A new series of potent and selective triple reuptake inhibitors. J. Med. Chem. 2010, 53, 2534–2551. [DOI] [PubMed] [Google Scholar]
- Lucas M. C.; Weikert R. J.; Carter D. S.; Cai H. Y.; Greenhouse R.; Iyer P. S.; Lin C. J.; Lee E. K.; Madera A. M.; Moore A.; Ozboya K.; Schoenfeld R. C.; Steiner S.; Zhai Y.; Lynch S. M. Design, synthesis, and biological evaluation of new monoamine reuptake inhibitors with potential therapeutic utility in depression and pain. Bioorg. Med. Chem. Lett. 2010, 20, 5559–5566. [DOI] [PubMed] [Google Scholar]
- Shao L.; Hewitt M. C.; Malcolm S. C.; Wang F.; Ma J.; Campbell U. C.; Spicer N. A.; Engel S. R.; Hardy L. W.; Jiang Z. D.; Schreiber R.; Spear K. L.; Varney M. A. Synthesis and pharmacological characterization of bicyclic triple reuptake inhibitor 3-aryl octahydrocyclopenta[c]pyrrole analogues. J. Med. Chem. 2011, 54, 5283–5295. [DOI] [PubMed] [Google Scholar]
- Gopishetty B.; Hazeldine S.; Santra S.; Johnson M.; Modi G.; Ali S.; Zhen J.; Reith M.; Dutta A. Further structure–activity relationship studies on 4-((((3S,6S)-6-benzhydryltetrahydro-2H-pyran-3-yl)amino)methyl)phenol: Identification of compounds with triple uptake inhibitory activity as potential antidepressant agents. J. Med. Chem. 2011, 54, 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechulis A. D.; Beck J. P.; Curry M. A.; Wolf M. A.; Harms A. E.; Xi N.; Opalka C.; Sweet M. P.; Yang Z.; Vellekoop A. S.; Klos A. M.; Crocker P. J.; Hassler C.; Laws M.; Kitchen D. B.; Smith M. A.; Olson R. E.; Liu S.; Molino B. F. 4-Phenyl tetrahydroisoquinolines as dual norepinephrine and dopamine reuptake inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7219–7222. [DOI] [PubMed] [Google Scholar]
- US patent US 7,541,357 B2. Synthetic procedures and characterizations for all compounds in Tables 1 and 2 except for 3p, 3u, 3v, and 3w are described in this patent. Compound 3p was prepared using a Boc-protected 2-bromoindole via Scheme 1. Compound 3u was prepared using 6-bromopyrazolo[1,5-a]pyridine via Scheme 1.
- Hu M.; Guzzo P. R.; Zha C.; Nacro K.; Yang Y.-L.; Hassler C.; Liu S. A facile synthesis of 4-aryl-1,2,3,4-tetrahydroisoquinolines. Tetrahedron Lett. 2011, 53, 846–848. [Google Scholar]
- Bymaster F. P.; Katner J. S.; Nelson D. L.; Hemrick-Luecke S. K.; Threlkeld P. G.; Heiligenstein J. H.; Morin S. M.; Gehlert D. R.; Perry K. W. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 2002, 27, 699–711. [DOI] [PubMed] [Google Scholar]
- For the process development of 10i, see:Yang Q.; Ulysse L. G.; McLaws M. D.; Keefe D. K.; Haney B. P.; Zha C.; Guzzo P. R.; Liu S. Initial process development and scale-up of the synthesis of a triple Reuptake Inhibitor ALB 109780. Org. Process Res. Dev. 2012, 16, 499–506. [Google Scholar]
- Lengyel K.; Pieschl R.; Strong T.; Molski T.; Mattson G.; Lodge N. J.; Li Y.-W. Ex vivo assessment of binding site occupancy of monoamine reuptake inhibitors: Methodology and biological significance. Neuropharmacology 2008, 55, 63–70. [DOI] [PubMed] [Google Scholar]
- Myer J. H.; Wilson A. A.; Sagrati S.; Hussey D.; Carella A.; Potter W. Z.; Ginovart N.; Spencer E. P.; Cheok A.; Houle S. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: An [11C]DASB positron emission tomography study. Am. J. Psychiatry 2004, 161, 826–835. [DOI] [PubMed] [Google Scholar]
- Lundberg J.; Tiger M.; Landén M.; Halldin C.; Farde L. Serotonin transporter occupancy with TCAs and SSRIs: a PET study in patients with major depressive disorder. Int. J. Neuropsychopharmacol. 2012, 15, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow L. J.; Easton A.; Seager M.; Olson R.; Liu S.; Jones K.; La-Paglia M.; Heman K.; Li Y.-W.; Macor J.; Sargent B.; Houston J.; Molino B. F.; Zaczek R.. The Triple Monoamine Reuptake Inhibitor, AMR-2, Improves Attentional Set Shifting in a Rat Neurodevelopmental Model of Schizophrenia. American College of Neuropharmacology (ACNP), Dec. 4–8th, 2011, Hawaii, USA. [Google Scholar]
- Smith D. A.; Obach R. S. Metabolites in safety testing (MIST): Considerations of mechanisms of toxicity with dose, abundance, and duration of treatment. Chem. Res. Toxicol. 2009, 22, 267–279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.