

Abstract
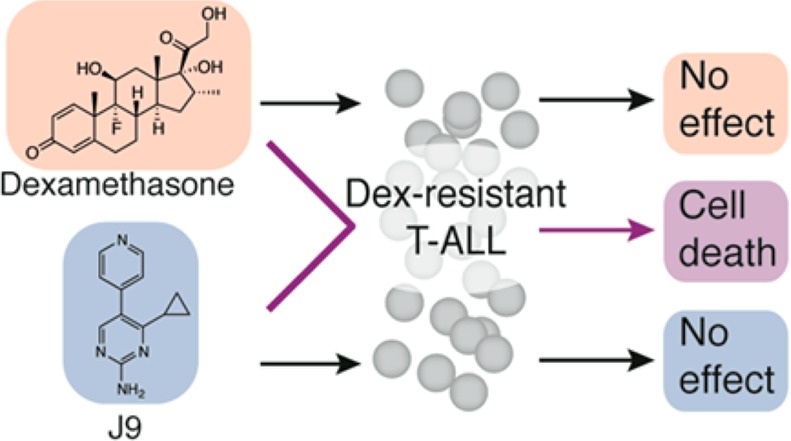
Glucocorticoids are one of the most utilized and effective therapies in treating T-cell acute lymphoblastic leukemia. However, patients often develop resistance to glucocorticoids, rendering these therapies ineffective. We screened 9517 compounds, selected for their lead-like properties, chosen from among 3 372 615 compounds, against a dexamethasone-resistant T-ALL cell line to identify small molecules that reverse glucocorticoid resistance. We synthesized analogues of the most effective compound, termed J9, from the screen in order to define the scaffold’s structure–activity relationship. Active compounds restored sensitivity to glucocorticoids through upregulation of the glucocorticoid receptor. This compound and mechanism may provide a strategy for overcoming glucocorticoid resistance in patients with T-ALL.
Keywords: T-cell acute lymphoblastic leukemia, dexamethasone, glucocorticoid resistance, NOTCH1
Acute lymphoblastic leukemia (ALL) is a hematological tumor that is characterized by aggressive invasion of the bone marrow by immature lymphoid progenitors. This disease is generally managed by aggressive chemotherapy with glucocorticoids (GCs).1 GCs such as prednisolone or dexamethasone are especially cytotoxic to ALL tumors, and the initial response to such treatments is associated with favorable disease prognosis.2,3 Conversely, primary glucocorticoid resistance confers a much higher risk of relapse and is particularly prevalent in T-ALL compared to B-precursor leukemias. Moreover, resistance to GC therapy is greatly enhanced upon relapse.4,5 In this context, reversing glucocorticoid resistance could potentially render these glucocorticoids effective in GC-resistant patients. As a biological tool, a small molecule that reverses such resistance could provide insight into the mechanism of glucocorticoid resistance and thus further address the problem of drug resistance in ALL.
The goal of our study was to find a small molecule with suitable pharmaceutical properties that could reverse glucocorticoid resistance in T-ALL. We utilized a human T-ALL cell line with a t(7;9)(q34;q34) translocation expressing a truncated and constitutively active NOTCH1 (CUTLL1).6 In the past decade, the prevalence of such activating NOTCH1 mutations in T-ALL cases has become apparent,7 and interestingly, the constitutive activity of NOTCH has been shown to cause GC resistance in normal developing thymocytes.8 Moreover, inhibition of NOTCH signaling can antagonize glucocorticoid resistance in T-ALL.9 CUTLL1 cells are highly NOTCH dependent and have previously been shown to be dexamethasone resistant and thus represent a good model in which to screen compounds.
We screened 9517 compounds that had been selected for lead-like properties in CUTLL1 cells in 384-well format.10,11 Starting from 3 372 615 compounds, we used computational filtering to choose compound molecular properties desired for lead compounds (Figure S1, Supporting Information).12 Cells treated with each compound were screened in parallel with cells treated with both 1 μM dexamethasone and 5.33 μg/mL of each test compound. Using a cutoff of 30% difference in viability between the two screening conditions, we identified J9, a compound that, in combination with dexamethasone, inhibited cell growth with an EC50 of 28 μM. J9 treatment alone was significantly less toxic to these cells, with an EC50 of 294 μM, suggesting it either restores sensitivity to dexamethasone or is synergistically lethal with dexamethasone. (Figure 1).
Figure 1.

Percent growth inhibition of CUTLL1 cells 48 h after treatment with J9 or J9 + dexamethasone (1 μM). Structure of compound J9 is shown on the right.
To confirm that J9 and dexamethasone cotreatment kills cells rather than merely decreases alamar blue activity, we examined levels of cleaved PARP1, a marker for apoptosis. We found that, as expected, treatment with either J9 or dexamethasone alone induced negligible levels of cleaved PARP1, as compared to cotreatment of J9 and Dex (Figure 2). This corresponds with our observations that J9 + Dex treatment can cause cell death in as little as 24 h, whereas an equivalent concentration of J9 alone causes minimal lethality to CUTLL1 cells. We were also interested in seeing whether J9 was lethal only in the presence of dexamethasone or acting through a more general cytotoxic mechanism. We treated CUTLL1 cells with J9 in combination with a variety of cytotoxic agents acting through diverse mechanisms and found that J9 did not increase the potency of any of these other compounds (Figure S2, Supporting Information). These data further show that J9 activity is specific to dexamethasone-treated cells, and it does not merely sensitize cells to any lethal agent.
Figure 2.

Levels of cleaved PARP after 24 h of treatment with J9 (25 μM), J9 + Dex, or J9 + Dex + RU486. Cleaved-PARP protein quantification (shown as fold change) shown on bottom. Protein levels of intracellular NOTCH (ICN) after 24 h of treatment with J9, J9 + Dex, or Dex + the gamma-secretase inhibitor Compound E (CE). Both Dex and RU486 were tested at 1 μM. CE was tested at 100 nM.
Since previously it was reported that gamma-secretase inhibitors act synergistically with dexamethasone to kill CUTLL1 cells, we tested whether J9 was acting as a gamma-secretase inhibitor.9 To address this question, we measured levels of intracellular NOTCH after treatment of J9 or J9 + Dex. We found that J9 or J9 + Dex treatment did not affect levels of activated NOTCH, as compared to treatment with a commercially available gamma-secretase inhibitor (Compound E), which completely abrogated NOTCH cleavage from the membrane (Figure 2). This suggests that J9 is not a gamma-secretase inhibitor, but rather has a distinct mechanism of action.
We were interested in determining whether treatment with J9 resensitized cells to dexamethasone or instead acted through a glucocorticoid-receptor-independent pathway. To investigate these hypotheses, we cotreated J9 + Dex with RU486 (Mifepristone), an estrogen and glucocorticoid receptor antagonist. The addition of RU486 significantly rescued cells from J9 + Dex-induced death, revealing the dependence of cytotoxicity of J9 + Dex treatment on the activity of the glucocorticoid receptor (Figure 3). We further investigated the importance of this receptor using CUTLL1 cells expressing high and low levels of exogenous glucocorticoid receptor (Figures S3 and S4, Supporting Information). Cells with higher levels of GR were significantly more susceptible to dexamethasone treatment, and we found that we could greatly enhance dexamethasone cytotoxicity by the addition of J9 to these cells. Conversely, cells with low GR levels were insensitive to dexamethasone treatment alone, and while they were sensitive to high concentrations of the J9 + Dex treatment, they were significantly less sensitive than either the overexpressed GR cells or CUTLL1 cells. The basal level of sensitivity that is exhibited by the low GR cells is most likely due to the incomplete knockdown of the GR, as measured by Western blot (Figures S3 and S4, Supporting Information). Neither cell line was sensitive to equal concentrations of J9 alone (Figure 4). To further understand these results, we performed Western blots comparing GR expression levels in each of these cell lines, which confirmed that GR protein level (either expressed exogenously or through treatment with high concentrations of J9) correlates with sensitivity to dexamethasone (Figure S4, Supporting Information). These data reinforce the notion that glucocorticoid receptor expression is required for the lethality of J9 + Dex cotreatment.
Figure 3.

Growth inhibition of J9 + Dex treatment compared to J9 + Dex + RU486 in CUTLL1 cells. Growth inhibition was measured after 48 h. Dex and RU486 were tested at 1 μM.
Figure 4.

Growth inhibition by J9 or J9 + Dex in cells expressing high and low levels of glucocorticoid receptor (GR). Cells treated with 1 μM dexamethasone.
It has been suggested that decreased expression of the glucocorticoid receptor is an important mechanism of resistance to dexamethasone, so we investigated whether J9 might increase glucocorticoid receptor (GR) levels and thus increase the effectiveness of dexamethasone treatment.13−15 We treated cells for 24 h with J9 alone or J9 + Dex and found that GR protein levels in J9 + Dex-treated cells were higher than treatment with dexamethasone alone. At concentrations less than the EC50, J9 alone did not significantly increase GR protein levels, a change that was also reflected in transcript levels after treatment (Figure 5).
Figure 5.

Glucocorticoid receptor (GR) protein levels normalized to α-tubulin (left) and relative mRNA levels after 24 h (right). Cells in the top panel were treated with 25 μM J9, while those in the bottom panel were treated with 50 μM J9.
To confirm these results, we examined GR protein levels after cotreatment of Dex + J9 + RU486, the estrogen receptor antagonist that rescues from Dex + J9-induced death. We found that the addition of RU486 to the J9 + Dex treatment prevented the increase in GR protein levels. We also conducted identical experiments to determine whether these changes could be confirmed at the transcript level. We performed RT-qPCR and found that mRNA levels after 24 h of J9 + Dex treatment were also increased, a phenomenon that could be abrogated by the addition of RU486 (Figure 5). We then treated cells with high concentrations of J9 alone, which we knew to be lethal, to see if this would induce a change in the glucocorticoid receptor. We found that the addition of 50 μM J9 did indeed increase expression of the GR as early as 6 h after treatment (Figures 5 and S5, Supporting Information). RT-qPCR analysis of transcript levels did not reflect this same change, suggesting a post-translational regulation of GR by J9.
As glucocorticoids have been shown to control the expression of a number of different genes, we were interested in exploring whether J9 + Dex treatment enriched for the same genes as GC treatment alone. Using microarray gene expression analysis, we compared CUTLL1 cells treated with dexamethasone alone, to cotreatment with Dex and J9. These data showed a marked increase in the upregulation and downregulation of Dex-regulated genes, suggesting an increase in dexamethasone activity upon cotreatment with J9. J9 alone had no effect on these genes (Figure 6B). We used gene set enrichment analysis (GSEA)16 to compare our results to a previously generated data set depicting changes in gene expression upon treatment of GC-sensitive ALL patients with GCs.17 This analysis revealed that genes upregulated and downregulated by treatment with J9 + Dex were highly enriched for genes upregulated and downregulated after GC treatment in the clinic, further confirming the relevance of J9 + Dex treatment in the context of ALL (Figure 6A).
Figure 6.

(a) GSEA enrichment plots of genes regulated by glucocorticoids in ALL patients undergoing glucocorticoid therapy, in CUTLL1 cells treated with Dex + J9 compared to CUTLL1 cells treated with dexamethasone alone. (b) Heat map representation of top enriched genes (leading edge) in CUTLL1 cells treated with dexamethasone + J9 compared with CUTLL1 cells treated with dexamethasone alone. Each column represents the expression data in CUTLL1 cells treated with vehicle only (DMSO), dexamethasone alone (1 μM), J9 alone (30 μM), and dexamethasone (1 μM) + J9 (30 μM), for 24 h (in triplicate). The scale bar shows a color-coded differential expression with red indicating higher levels and blue lower levels of expression.
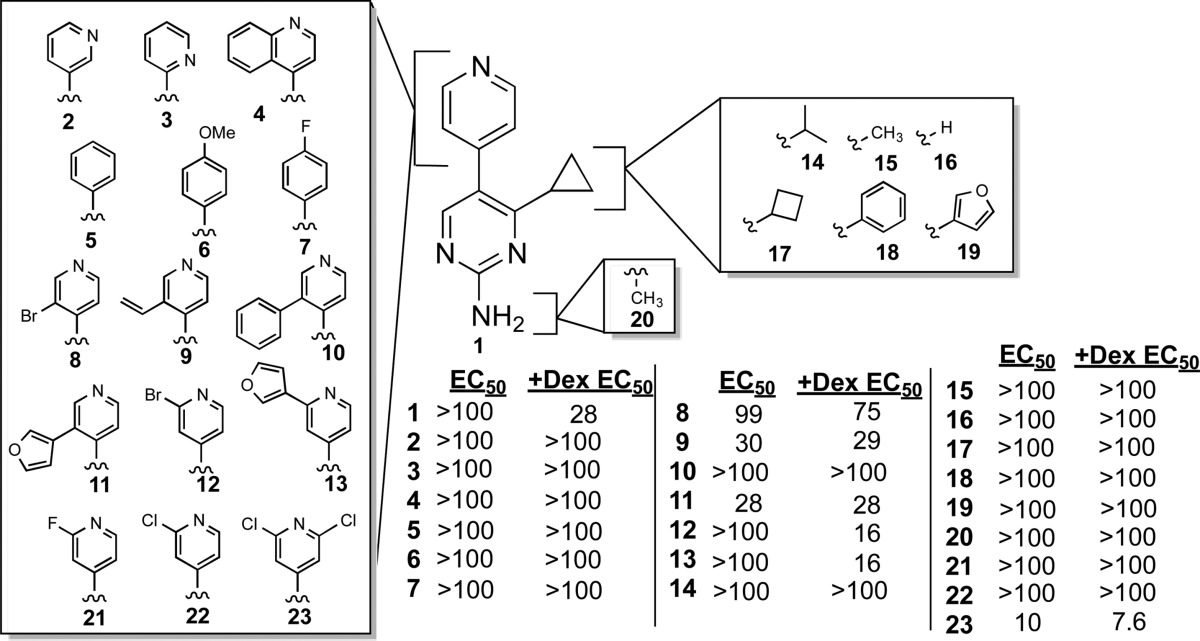
In order to understand further the structure–activity relationship (SAR) of J9, as well as to develop compounds with increased potency, we synthesized and tested a number of structural analogues with single-site modifications compared to J9 (Table 1). J9 itself was synthesized through the LDA-facilitated addition of 4-methylpicoline to ethyl cyclopropyl carboxylate (Scheme 1a). The construction of the pyrimidine core was then accomplished via aminoformylation of the resulting ketone with N,N-dimethylformamide dimethyl acetal (Scheme 1b) and subsequent cyclization with guanidine hydrochloride (Scheme 1c). These conditions proved to be robust and allowed for the formation of compounds 2–4, 8–13, and 14–23 in Table 1, using the corresponding ester, picoline, or amidine derivative. Compounds 9, 10, 11, and 13 were synthesized from compounds 8 or 12 though either a Suzuki or Stille coupling (Scheme 1d). Compounds 5 and 6 required an alternative method for making the cyclopropyl ketone. This was accomplished through converting cyclopropylmagnesium bromide to the corresponding cuprate (via CuBr) followed by the nucleophilic addition to a phenacetyl chloride derivative (see Supporting Information). Interestingly even changes as subtle as substituting the cylcopropyl ring for an isopropyl group (compound 14, Table 1) or movement of the nitrogen in the pyridine ring from the 4- to 3- position (compound 3, Table 1) resulted in complete loss of potency, suggesting a highly specific interaction. While any alterations made to the J9 core eliminated activity, we found that by substituting the 3 position of the pyridine ring with either bromine (J9A) or a furan ring (J9B), (compounds 12 and 13, Table 1, respectively) gave a 2-fold increase in potency relative to the parent compound (Figure 7), whereas the other substitutions we made to this position showed decreased potency and/or selectivity. The same alterations made at the 2 position (compounds 8 and 11, Table 1) resulted in a loss of selectivity for dexamethasone treatment. Further exploration of different moieties added to the 3-position will likely allow for further increased cytotoxicity in combination with dexamethasone without losing specificity.
Table 1. Structure–Activity Relationship Analysis of Compound J9.
Scheme 1.
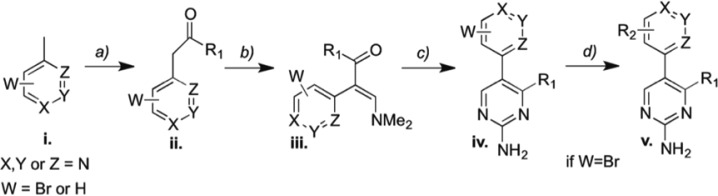
Figure 7.

Growth inhibition of analogues J9A and J9B treated for 48 h with and without dexamethasone (1 μm).
To verify that the two most potent analogues (J9A and J9B) were acting through the same mechanism as J9, we treated both J9A and J9B with Dex and RU486 to see if RU486 could reverse the lethality of J9A/B + Dex, as it did for J9 + Dex. Indeed we found that at lethal concentrations of J9A + Dex or J9B + Dex, treatment with RU486 decreased growth inhibition by up to 60% (J9A) or 50% (J9B) (Figure 8). These results suggest that these derivatives indeed act through the same mechanism as the parent compound and that further chemical modification could both enhance potency and maximize specificity.
Figure 8.

Growth inhibition after 24 h treatment of analogues J9A and J9B with Dex/J9/RU486. Both Dex and RU486 were tested at 1 μM.
In conclusion, we screened in a dexamethasone-resistant human T-ALL cell line a library of small molecules optimized for lead-like properties to identify those that reverse glucocorticoid resistance. From this screen, we found a compound, J9, which is lethal to CUTLL1 cells only in the presence of dexamethasone. Preliminary mechanistic studies revealed that the lethality of J9 + Dex treatment is dependent on expression of the glucocorticoid receptor and also results in increased abundance of the receptor. GSEA analysis further confirmed that cotreatment with dexamethasone and J9 enhances dexamethasone activity and affects gene expression in a manner consistent with GC-treatment in the clinic. These data also reveal genes that are regulated by J9 treatment alone, which may provide insight into the mechanism of action of this compound. Finally, we performed SAR studies in an effort to increase the potency of J9 and understand the components of the molecule that contribute to its specificity. These studies revealed that minor changes to this structure result in a lack of activity, suggesting a possible specific molecular recognition event. Derivatives of this scaffold could help further our understanding of the mechanisms behind glucocorticoid resistance and could potentially lead to drugs that resensitize GC-resistant T-ALL cells to glucocorticoid therapy.
Acknowledgments
B.R.S. is an Early Career Scientist of the Howard Hughes Medical Institute. M-Y. K. is a fellow of the Leukemia and Lymphoma Society. We acknowledge the assistance of Dr. John Decatur, and the use of Columbia Chemistry NMR core facility instruments provided by NSF grant CHE 0840451 and NIH grant 1S10RR025431-01A1. We thank Dr. Y. Itagaki for assistance with MS analyses.
Glossary
ABBREVIATIONS
- ALL
acute lymphoblastic leukemia
- T-ALL
T-cell acute lymphoblastic leukemia
- GC
glucocorticoid
- GR
glucocorticoid receptor
- Dex
dexamethasone
- CE
compound E
- LDA
lithium di-isopropyl amide
- PARP
poly ADP ribose polymerase
Supporting Information Available
Experimental procedures for biological assays and syntheses of compounds. Details of LOC library construction and additional figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∇ (A.M.C.) Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, United States.
This work was funded by grants from the US National Institutes of Health (NIH R01CA097061, R01GM085081, R01 CA161061, and RC2 CA148308), the Swim Across America Foundation, the Leukemia and Lymphoma Society, and the Chemotherapy Foundation.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Goldberg J. M.; Silverman L. B.; Levy D. E.; Dalton V. K.; Gelber R. D.; Lehmann L.; Cohen H. J.; Sallan S. E.; Asselin B. L. Childhood T-cell acute lymphoblastic leukemia: the Dana–Farber Cancer Institute acute lymphoblastic leukemia consortium experience. Am. J. Clin. Oncol. 2003, 21193616–3622. [DOI] [PubMed] [Google Scholar]
- Beesley A. H.; Firth M. J.; Ford J.; Weller R. E.; Freitas J. R.; Perera K. U.; Kees U. R. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. Br. J. Cancer 2009, 100121926–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dördelmann M.; Reiter A.; Borkhardt A.; Ludwig W. D.; Götz N.; Viehmann S.; Gadner H.; Riehm H.; Schrappe M. Prednisone response is the strongest predictor of treatment outcome in infant acute lymphoblastic leukemia. Blood 1999, 9441209–1217. [PubMed] [Google Scholar]
- Kaspers G. J.; Pieters R.; Klumper E.; De Waal F. C.; Veerman A. J. Glucocorticoid resistance in childhood leukemia. Leuk. Lymphoma 1994, 133–4187–201. [DOI] [PubMed] [Google Scholar]
- Kaspers G. J. L.; Wijnands J. J. M.; Hartmann R.; Huismans L.; Loonen A. H.; Stackelberg A.; Henze G.; Pieters R.; Hählen K.; Van Wering E. R.; Veerman A. J. P. Immunophenotypic cell lineage and in vitro cellular drug resistance in childhood relapsed acute lymphoblastic leukaemia. Eur. J. Cancer 2005, 4191300–1303. [DOI] [PubMed] [Google Scholar]
- Palomero T.; Barnes K. C.; Real P. J.; Glade Bender J. L.; Sulis M. L.; Murty V. V.; Colovai A. I.; Balbin M.; Ferrando A. A. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to γ-secretase inhibitors. Leukemia 2006, 2071279–1287. [DOI] [PubMed] [Google Scholar]
- Weng A. P.; Ferrando A. A.; Lee W.; Morris J. P. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 3065694269–271. [DOI] [PubMed] [Google Scholar]
- Deftos M. L.; He Y. W.; Ojala E. W.; Bevan M. J. Correlating notch signaling with thymocyte maturation. Immunity 1998, 96777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real P. J.; Tosello V.; Palomero T.; Castillo M.; Hernando E.; de Stanchina E.; Sulis M. L.; Barnes K.; Sawai C.; Homminga I.; Meijerink J.; Aifantis I.; Basso G.; Cordon-Cardo C.; Ai W.; Ferrando A. γ-Secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 2008, 15150–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A.; Merz K. M. Prediction of aqueous solubility of a diverse set of compounds using quantitative structure-property relationships. J. Med. Chem. 2003, 46173572–3580. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 461–33–26. [DOI] [PubMed] [Google Scholar]
- Dixon S. J.; Lemberg K. M.; Lamprecht M. R.; Skouta R.; Zaitsev E. M.; Gleason C. E.; Patel D. N.; Bauer A. J.; Cantley A. M.; Yang W. S.; Morrison B.; Stockwell B. R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 14951060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploner C.; Schmidt S.; Presul E.; Renner K.; Schröcksnadel K.; Rainer J.; Riml S.; Kofler R. Glucocorticoid-induced apoptosis and glucocorticoid resistance in acute lymphoblastic leukemia. J. Steroid Biochem. Mol. Biol. 2005, 932–5153–160. [DOI] [PubMed] [Google Scholar]
- Schmidt S.; Irving J. A. E.; Minto L.; Matheson E.; Nicholson L.; Ploner A.; Parson W.; Kofler A.; Amort M.; Erdel M.; Hall A.; Kofler R. Glucocorticoid resistance in two key models of acute lymphoblastic leukemia occurs at the level of the glucocorticoid receptor. FASEB J. 2006, 20142600–2602. [DOI] [PubMed] [Google Scholar]
- Geley S.; Hartmann B. L.; Hala M.; Strasser-Wozak E. M.; Kapelari K.; Kofler R. Resistance to glucocorticoid-induced apoptosis in human T-cell acute lymphoblastic leukemia CEM-C1 cells is due to insufficient glucocorticoid receptor expression. Cancer Res. 1996, 56215033–5038. [PubMed] [Google Scholar]
- Subramanian A.; Tamayo P.; Mootha V. K.; Mukherjee S.; Ebert B. L.; Gillette M. A.; Paulovich A.; Pomeroy S. L.; Golub T. R.; Lander E. S.; Mesirov J. P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005, 1024315545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S.; Rainer J.; Riml S.; Ploner C.; Jesacher S.; Achmüller C.; Presul E.; Skvortsov S.; Crazzolara R.; Fiegl M.; Raivio T.; Jänne O. A.; Geley S.; Meister B.; Kofler R. Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood 2006, 10752061–2069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.