

Abstract

Novel substituted 2,3-dihydrobenzofuran-7-carboxamide (DHBF-7-carboxamide) and 2,3-dihydrobenzofuran-3(2H)-one-7-carboxamide (DHBF-3-one-7-carboxamide) derivatives were synthesized and evaluated as inhibitors of poly(ADP-ribose)polymerase-1 (PARP-1). A structure-based design strategy resulted in lead compound 3 (DHBF-7-carboxamide; IC50 = 9.45 μM). To facilitate synthetically feasible derivatives, an alternative core was designed, DHBF-3-one-7-carboxamide (36, IC50 = 16.2 μM). The electrophilic 2-position of this scaffold was accessible for extended modifications. Substituted benzylidene derivatives at the 2-position were found to be the most potent, with 3′,4′-dihydroxybenzylidene 58 (IC50 = 0.531 μM) showing a 30-fold improvement in potency. Various heterocycles attached at the 4′-hydroxyl/4′-amino of the benzylidene moiety resulted in significant improvement in inhibition of PARP-1 activity (e.g., compounds 66–68, 70, 72, and 73; IC50 values from 0.718 to 0.079 μM). Compound 66 showed selective cytotoxicity in BRCA2-deficient DT40 cells. Crystal structures of three inhibitors (compounds (−)-13c, 59, and 65) bound to a multidomain PARP-1 structure were obtained, providing insights into further development of these inhibitors.
Introduction
Poly(ADP-ribose)polymerase-1 (PARP-1) is an abundant, ubiquitously expressed nuclear protein that is responsible for the maintenance and repair of DNA damage.1 PARP-1 is the founding member of an expanded family of related proteins with similar enzymatic activity. PARP-1 utilizes NAD+ as a substrate to synthesize linear and branched chains of poly(ADP-ribose) (PAR). These PAR chains covalently bind to the acceptor residues. Poly(ADP)ribosylation (PARylation) occurs mainly on PARP-1 itself (automodification) but also on number of nuclear target proteins (heteromodification) such as histones, p53, topoisomerase I/II, DNA polymerases, and DNA ligases.1,2 In response to DNA-strand breaks, PARylation by PARP-1 is stimulated nearly 500-fold.3 PARylation is involved in a number of important cellular processes including DNA damage repair, genomic stability, and regulation of transcription and cell death.4,5 The synthesis of highly negatively charged PAR chains leads to chromatin decondensation and dynamic nucleosome remodeling6 that facilitates accessibility of base excision repair (BER) proteins such as XRCC1, DNA ligase III, and DNA polymerase β (pol β) to sites of damaged DNA.7−9 The presence of PAR is both transient and dynamic, as hydrolase enzymes such as poly(ADP-ribose)glycohydrolase (PARG) quickly degrade PAR into ADP-ribose units.10
With such prominent and pivotal roles in the maintenance of genomic stability, PARP-1 inhibitors have been developed to target certain diseases such as cancer. PARP-1 inhibitors sensitize tumor cells to certain DNA alkylating agents (e.g., temozolomide, cisplatin), topoisomerase I poisons (e.g., irinotecan), and other chemotherapeutic agents and are being used in clinical trials as potentiators of chemo- and radiotherapy.5,11,12 The finding that double-strand DNA break repair-deficient cancer cells carrying defective or mutated BRCA1 and BRCA2 respond to PARP inhibitors (synthetic lethality) has paved the way for PARP inhibitors as single agents therapy in oncology.13,14 Some of the clinically proven PARP-1 inhibitors such as olaparib have been shown to exhibit single agent activity for tumors with mutations in BRCA1- and BRCA2-dependent DNA double-strand break repair mechanisms (homologous recombination, HR).15−18 In addition, PARP-1 inhibitors may also be effective at targeting other DNA repair defects in tumors with “BRCAness” phenotypes.19 For instance, lack of tumor suppressor gene PTEN (phosphatase and tensin homologue) demonstrate HR defects in human tumor cell lines, making them responsive to PARP-1 inhibitors.20 Taken together, PARP-1 inhibitors offer exciting therapeutic potential as molecular targeted agents in the field of oncology.
Previously, we reported N-1 substituted indazole-3-carboxamide21 and 2,8-disubstituted quinazolin-4(3H)-one22 derivatives as PARP-1 inhibitors. With the aims of structural novelty and improved potency of these scaffolds, we designed the previously unexplored pharmacophores 2,3-dihydrobenzofuran-7-carboxamide (DHBF-7-carboxamide) and 2,3-dihydrobenzofuran-3(2H)-one-7-carboxamide (DHBF-3-one-7-carboxamide) that are targeted to the NAD+ binding site of PARP-1. We obtained crystal structure data of these scaffolds to help further drug development. Inhibitor bound crystal structures were of the PARP-1 multidomain complex with DNA,23 demonstrating the first report of a PARP inhibitor bound to DNA-damage activated PARP-1.
Results and Discussion
Structure-Based Drug Design
A considerable number of PARP inhibitors have been described in the literature over the past several years.24,25 The structures of the selected PARP inhibitors (ABT-888,26 MK-4827,27 AG014699,28 and AZD-228129) in clinical trials are shown in Chart 1. It is well established that these agents target the NAD+ binding site by occupying the nicotinamide pocket. Inhibitors that target this site contain a cyclic or acyclic carboxamide pharmacophore, which makes π–π stacking interactions with adjacent tyrosine residues, Tyr896 and Tyr907, and form three key hydrogen bonds with Gly863 and Ser904 of the PARP-1 catalytic site. Optimization efforts that gave rise to these clinical candidates improved potency by restricting the amide conformation of the carboxamide through intramolecular hydrogen bonding27,30,31 or by locking the amide into a ring.22,28,29 With this guiding concept, we hypothesized that the DHBF-7-carboxamide (3) would establish an intramolecular hydrogen bond leading to the formation of a 6:5:6 pseudotricyclic ring system with improved potency.
Chart 1. PARP Inhibitors Currently Being Evaluated in the Clinic.

Similar to other PARP-1 inhibitors, docking of DHBF-7-carboxamide (compound 3) within the catalytic site of human PARP-1 (PDB code 4L6S)32 (Figure 1) revealed that the carbonyl oxygen atom formed a hydrogen bond with the side chain hydroxyl group of Ser904 (C=O···HO-Ser904, 1.91 Å) and backbone −NH of Gly863 (C=O···HN-Gly863, 2.09 Å) while the carboxamide hydrogen atom entered into hydrogen bonding interaction with the backbone carbonyl oxygen atom of Gly863 (NH···O=C-Gly863, 1.92 Å). We observed formation of an intramolecular hydrogen bond between the carboxamide hydrogen atoms and the ether oxygen atom of the DHBF-7-carboxamide that results in the formation of pseudotricyclic ring and restriction of the carboxamide moiety. The aromatic ring of DHBF was stabilized through a π–π stacking interaction with the nearly coplanar electron rich phenyl ring of Tyr907, as seen with other PARP-1 inhibitors. To further develop this lead series, we carried out SAR analysis, stereochemical characterization, and small molecule and macromolecular X-ray crystallographic analysis of the DHBF-7-carboxamide and DHBF-3-one-7-carboxamide derivatives. Further biochemical evaluation of PARP-1 activity and cytotoxicity analysis of BRCA2-deficient DT40 cells demonstrated the therapeutic potential of these series as PARP-1 inhibitors.
Figure 1.

XP-Glide predicted binding mode of compound 3 within the active site of PARP-1, represented in ribbon form, with the interacting amino acids represented as sticks and atoms colored according to the following: carbon, beige; hydrogen, white; nitrogen, blue; oxygen, red. The inhibitor is shown as ball and stick with the same color scheme depicted above except that carbons are represented as orange. Dotted red lines indicate intra- and intermolecular hydrogen bonding interaction, whereas the dotted yellow lines indicate the distance between the two atoms/groups/centroids in angstroms. The centroids are marked as green stars. The image was generated using PyMOL, version 1.6.0.
Synthesis
Dihydrobenzofuran derivatives were synthesized in good yields with diminutive variations in reported procedures.33−36 The carboxylation at the 7-position of the 2,3-dihydrobenzofuran (DHBF, compound 1) was achieved by lithiation with n-butyllithium (n-BuLi) in TEMED/hexane at room temperature under nitrogen atmosphere, followed by addition of dry ice and acidification with concentrated HCl33 (Scheme 1). The resultant carboxylic acid derivative 2 was converted to the carboxamide via a mixed-anhydride method to get the lead compound 3. Bromination of 3 using a solution of bromine in acetic acid at elevated temperature of 80 °C provided compound 4.34 Nitration33 of 3 with nitric acid and trifluoroacetic acid at low temperature yielded 5 in good yield. The nitro group of 5 was further reduced to amine using tin chloride dihydrate in ethyl acetate under refluxing conditions to yield compound 6.
Scheme 1. Synthesis of 5-Substituted 2,3-Dihydrobenzofuran-7-carboxamide Derivatives.

Reagents and conditions: (a) (i) n-BuLi, TEMED, hexane, rt, 4 h; (ii) dry ice, overnight; conc HCl; (b) (i) i-BuOCOCl, NMM, THF, −20 °C, 4 h; (ii) 30% NH4OH, rt, 1 h; (c) Br2, CH3COONa, CH3COOH, 80 °C, 3 h; (d) TFA, HNO3, 0 °C to rt, 3 h; (e) SnCl2·2H2O, EtOAc, reflux, 4 h; (f) H2SO4, MeOH, reflux, 3 h.
In order to modify the 2-position of DHBF, we used an alternative synthetic route to synthesize the 2-substituted and the 2,5-disubstituted DHBF-7-carboxamide derivatives from the corresponding salicylic acid derivatives to avoid the low yielding carboxylation step in the later stages of the synthetic protocol. First, the salicylic acid derivatives were converted to corresponding methyl esters using thionyl chloride in the presence of methanol (Scheme 2). This was followed by O-allylation, carried out using allyl bromide and potassium carbonate, to yield compounds 9a–c. The 3,3-sigmatropic Claisen rearrangement was carried out by heating compounds 9a–c neatly at elevated temperatures to yield ortho-rearranged products 10a–c.35 The furan ring was constructed by reacting the rearranged product with zirconium chloride in dichloromethane to yield the corresponding methyl 2-methyl-DHBF-7-carboxylate derivatives 11a–c.36 Hydrolysis of the methyl ester using sodium hydroxide provided corresponding acids 12a–c that were converted to carboxamides 13a–c under mixed anhydride conditions and aqueous ammonia (as discussed above). Nitration of compound 13a (where R = H) followed by reduction was carried out essentially as discussed above to obtain 14 and 15, respectively.
Scheme 2. Synthesis of 2,5-Disubstituted 2,3-Dihydrobenzofuran-7-carboxamides.

Reagents and conditions: (a) SOCl2, MeOH, reflux, 12 h; (b) allyl bromide, K2CO3, NaI, DMF, rt, 12 h; (c) neat, 160–190 °C, 2 h; (d) ZrCl4, DCM, rt, 10 h; (e) NaOH, MeOH, reflux, 2 h; (f) (i) i-BuOCOCl, NMM, THF, −20 °C, 4 h; (ii) 30% NH4OH, rt, 1 h; (g) when R = H, TFA, HNO3, 0 °C to rt, 3 h; (h) SnCl2·2H2O, EtOAc, reflux, 4 h.
Since the enantiomers bind differently within the active site, one of the enantiomers is presumed to be more potent when compared to the other. Therefore, we decided to resolve the 2-position enantiomers resulted during the construction of DHBF ring and evaluate the activity of the pure enantiomers. The 2-position enantiomers (compounds (+)-13a, (−)-13a, (+)-13c, and (−)-13c) were resolved by preparative chiral HPLC, using an amylose based chiral stationary phase Chiralpak 1A (4.6 mm × 250 mm) at GVK Biosciences Pvt. Ltd., Hyderabad, India. To determine the absolute configuration at the 2-position of the representative eutomer, the X-ray crystal structure of 12a was elucidated using the diastereomeric salt formation approach.37−40 The absolute configuration from direct determination based on anomalous scattering from the light atoms was in agreement with the starting materials. On the basis of the analysis of the crystals of (−)-enantiomer of 12a with (S)-(−)-α-methylbenzylamine, it was revealed that the (−)-enantiomer bears the R-configuration (Figure 2). On the basis of this finding, the (+)-enantiomer was assigned as the S-enantiomer.
Figure 2.
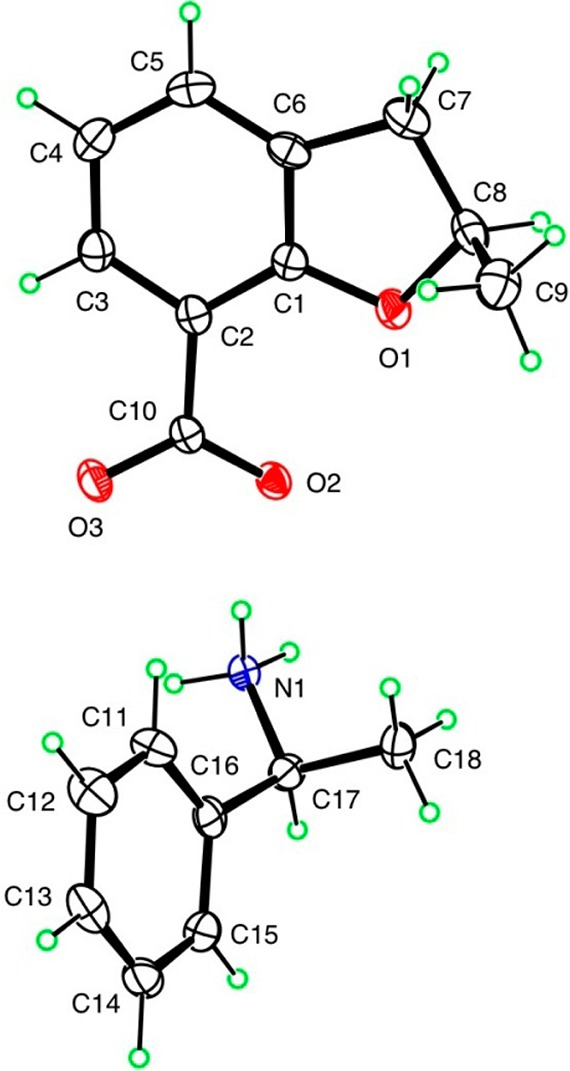
ORTEP diagram of salt of (R)-(−)-12a with (S)-(−)-α-methylbenzylamine.
To synthesize the 5-fluoro derivative of the DHBF-7-carboxamide, we first attempted to launch the 5-fluoro substituent by a reported method41 in which nitration at the 5-position of the DHBF was carried out (as mentioned previously), followed by its reduction using a Pd/C catalyst, to yield 5-amino-DHBF compound 18a (Scheme 3). The diazonium tetrafluoroborate salt of 18a was prepared by adding aqueous sodium nitrite solution into a mixture of 5-amino-DHBF, hydrochloric acid, and tetrafluoroboric acid in THF at −15 °C. The resulting precipitates were refluxed in xylene to obtain 5-fluoro-DHBF compound 19a. Unfortunately, the lithiation-mediated 7-carboxylation of compound 19a did not yield the desired compound (5-fluoro-DHBF-7-carboxylic acid, 19b). An alternative strategy was applied in which we used DHBF-7-carboxylic acid 2 as a starting material instead of DHBF to elude the low yielding carboxylation step. The 5-position nitration followed by reduction was performed by the same procedure described for compound 18a to yield 18b. Attempts to replace the amino group of compound 18b with a fluoro group (by the aforementioned method) yielded 5-fluoro-DHBF-7-carboxylic acid (compound 19b) in a very low yield. To overcome this, compound 2 was first converted to the corresponding methyl ester 16 by refluxing it in methanol and a catalytic amount of sulfuric acid as shown in Scheme 1. Subsequent nitration at the 5-position of 16 followed by hydrogenation (as mentioned above) yielded methyl 5-amino-DHBF-7-carboxylate 18c in good yield which was fluorinated using the aforementioned procedure to get the desired compound 19c in good yield. Compound 19c was hydrolyzed to yield 19b, which was then converted to carboxamide using the mixed-anhydride condition to obtain the desired product 20 (Scheme 3).
Scheme 3. Synthesis of 5-Fluoro-2,3-dihydrobenzofuran-7-carboxamide.

Reagents and conditions: (a) TFA, HNO3, 0 °C to rt, 3 h; (b) H2/Pd/C, EtOH, 60 psi, rt, 2–18 h; (c) (i) THF, HCl, HBF4, rt to −15 °C, NaNO2, 30 min; (ii) xylene, reflux, 2 h; (d) R=H, (i) n-BuLi, TEMED, hexane, rt, 4 h; (ii) dry ice, overnight; (e) when R = -COOCH3; NaOH, MeOH, reflux, 3 h; (f) (i) i-BuOCOCl, NMM, THF, −20 °C, 4 h; (ii) 30% NH4OH, rt, 1 h.
The 4-amino-2-methyl-DHBF-7-carboxamide 28 was synthesized using the 4-nitrosalicylic acid 21 as the precursor, since the direct electrophilic substitution onto the DHBF-7-carboxamide ring favored the 5-position and attempts to use 4-aminosalicylic acid methyl ester as the starting material yielded mono-N- and di-N-allylated methyl ester derivatives in the allylation step (data not shown). The nitro group in compound 21 acted as a surrogate, which was reduced to an amino group in the latter steps of the synthetic scheme. The ring construction was accomplished as outlined in Scheme 4. The esterification of the acid 21 was accomplished by the aforementioned method wherein the acid was refluxed in the presence of thionyl chloride and methanol. The resulting ester 22 was allylated onto the phenolic hydroxyl with allyl bromide utilizing the conditions mentioned previously. The 3,3-sigmatropic Claisen rearrangement of the allyl derivative 23 was performed using carbitol as a solvent, since the aforementioned procedure of neat heating of the compound above 160 °C yielded the rearranged product 24 in a very low yield. Cyclization of the rearranged nitro analogue followed by hydrolysis did not yield the desired nitro acid (NiAc) derivative (Scheme 4). We therefore decided to change the synthetic plan by reducing the nitro group of 25 to an amine (compound 26) followed by hydrolysis using sodium hydroxide, yielding acid intermediate 27. The resulting acid 27 was converted to carboxamide 28 by modified mixed-anhydride conditions for carboxamide synthesis, wherein N-methylmorpholine was added first followed by isobutyl chloroformate to prevent the N-acylation. The anhydride, once formed, was quickly treated with dry ammonia gas, resulting in the formation of the target carboxamide rac-28 (Scheme 4). The rac-28 was resolved by means of preparative chiral chromatography similar to the method developed for racemates 13a and 13c.
Scheme 4. Synthesis of 4-Amino-2-methyl-2,3-dihydrobenzofuran-7-carboxamide.

Reagents and conditions: (a) SOCl2, MeOH, reflux, 12 h; (b) allyl bromide, K2CO3, NaI, DMF, rt, 12 h; (c) carbitol, 170–180 °C, 2 h; (d) ZrCl4, DCM, rt, 10 h; (e) NaOH, MeOH, reflux; (f) H2/Pd/C, EtOH, 60 psi, rt, 4 h; (g) NaOH, MeOH, reflux, 18 h; (h) (i) NMM, i-BuOCOCl, THF, −20 °C, 20 min; (ii) dry NH3, rt, 45 min.
A successful synthesis of DHBF-3-one-7-carboxamide (compound 36) (Scheme 5) was carried out after trying two unsuccessful strategies, which involved (i) carboxylation of DHBF-3-one and (ii) oxidation of 7-methyl group of 7-methyl-DHBF-3-one, none of which yielded the desired penultimate intermediate DHBF-3-one-7-carboxylic acid 35. Finally, in order to resolve the issue of ring oxidation, which occurred in the latter case, we decided to oxidize the aromatic methyl group prior to the ring construction. This was met by first esterifying 3-methylsalicylic acid to obtain 30 followed by O-alkylation with ethyl bromoacetate to yield diester intermediate 31, which was then hydrolyzed to yield the diacid derivative 32 and was further oxidized to 2-(carboxymethoxy)isophthalic acid (compound 33). Cyclization of 33 to 3-acetoxybenzofuran-7-carboxylic acid (34) was accomplished by treating 33 with the 1:3:542 proportions of sodium acetate, acetic acid, and acetic anhydride, respectively, under refluxing conditions. The resulting compound was hydrolyzed using a mixture of 1:10:40 42 proportions of hydrochloric acid (11 N)/water/methanol, generating a 3-oxo derivative 35. The resulting acid was finally converted to an amide 36 by the aforementioned mixed-anhydride technique.
Scheme 5. Synthesis of 3-Oxo-2,3-dihydrobenzofuran-7-carboxamide.

Reagents and conditions: (a) SOCl2, MeOH, reflux, 12 h; (b) BrCH2COOEt, K2CO3, NaI, DMF, rt, 12 h; (c) KOH, MeOH, reflux, 4 h; (d) KMnO4, water, reflux, 2 h; (e) NaOAc, (CH3CO)2O, CH3COOH, reflux, 5 h; (f) HCl (11 N)/H2O/MeOH, reflux, 1 h; (g) (i) i-BuOCOCl, NMM, THF, −20 °C, 4 h; (ii) 30% NH4OH, rt, 1 h.
The benzaldehydes required for the synthesis of target compounds 50–71 were either commercially available or prepared (37–49) as shown in Schemes 6 and 7. The 3- or 4-hydroxybenzaldehydes were alkylated with commercially available 2-chloroethylmorpholine/piperidine in the presence of potassium carbonate under refluxing conditions to obtain 37–40 (Scheme 6). The 4-(2-chloroethoxy)benzaldehyde 41 was synthesized by reacting 4-hydroxybenzaldehyde with bromochloroethane in the presence of potassium carbonate and refluxing acetonitrile. Compound 41 was then reacted with N-methylpiperazine to yield the desired benzaldehyde 42 (Scheme 6). Benzaldehyde derivative 43 was prepared similar to that of 41 using bromochloropropane instead of bromochloroethane (Scheme 7). Compound 43 was then treated with morpholine to obtain the desired aldehyde intermediate 44. The acyl derivative 46 was synthesized by performing diminutive variations in the reported conditions (Scheme 7).43,44 First, the morpholine was reacted with chloroacetyl chloride in the presence of triethylamine in dichoromethane at 0 °C, which generated N-2-chloroacetylmorpholine (45). Compound 45 was then treated with 4-hydroxybenzaldehyde by following the aforementioned alkylation technique (using potassium carbonate as a base) to yield 46. The aldehyde 47 was synthesized by alkylating 4-hydroxybenzaldehyde with epibromhydrin under alkylation conditions (Scheme 7). Compound 48 was synthesized by epoxide ring-opening, which was mediated by nucleophilic attack of morpholine onto the electropositive methylene group of the epoxide ring of 47 (Scheme 7). Compound 49 was synthesized by alkylating commercially available 1-(4-fluorophenyl)piperazine with compound 41 using potassium carbonate as a base (Scheme 7).
Scheme 6. Synthesis of Substituted Benzaldehydes 37–40 and 42.

Reagents and conditions: (a) K2CO3, 4-(2-chloroethyl)morpholine (for compounds 37 and 39) and 4-(2-chloroethyl)piperidine (for compounds 38 and 40), CH3CN, reflux, 6 h; (b) K2CO3, KI, bromochloroethane, CH3CN, reflux, 12 h; (c) K2CO3, KI, N-methylpiperazine, CH3CN, reflux, 2–6 h.
Scheme 7. Synthesis of Substituted Benzaldehydes 44 and 46–49.

Reagents and conditions: (a) K2CO3, bromochloropropane, CH3CN, reflux, 3 h; (b) K2CO3, morpholine, CH3CN, reflux, 6 h; (c) chloroacetyl chloride, DCM, triethylamine, 0 °C to rt, 3 h; (d) K2CO3, 4-hydroxybenzaldehyde, CH3CN, reflux, 6 h; (e) epibromhydrin, K2CO3, CH3CN, reflux, 6 h; (f) morpholine, K2CO3, CH3CN, reflux, 6 h; (g) 4-fluorophenylpiperazine, K2CO3, CH3CN, reflux, 48 h.
Knoevenagel condensation45 of compound 36 with various commercially available and in house synthesized benzaldehyde derivatives under refluxing toluene in the presence of ammonium acetate yielded target compounds 50–73, which were finally washed with water (to remove excess of ammonium acetate catalyst), dried, and obtained in moderate yields as shown in Scheme 8.
Scheme 8. Synthesis of Target Compounds 50–73.

Reagents and conditions: (a) NH4OAc, commercially available/synthesized R-CHO, toluene, reflux, 1–24 h.
The benzaldehydes required for making the target sulfonamide analogues 72 and 73 were synthesized by reacting 72a (obtained by reducing 4-nitrobenzaldehyde using tin chloride dihydrate in ethyl acetate) with chloroethanesulfonyl chloride (to obtain compound 72b) and chloropropanesulfonyl chloride (to obtain compound 72c) at 0 °C in the presence of triethylamine in dichloromethane. The resulting benzaldehydes 72b and 72c were then reacted with N-methylpiperazine to yield intermediates 72d and 72e, respectively (Scheme 9).
Scheme 9. Synthesis of Substituted Benzaldehyde Intermediates 72d and 72e Required for Respective Synthesis of Target Compounds 72 and 73.

Reagents and conditions: (a) SnCl2·2H2O, ethyl acetate, reflux, 4 h; (b) chloroethanesulfonyl chloride (for compound 72b) or chloropropanesulfonyl chloride (for compound 72c), DCM, triethylamine, 0 °C to rt, 6 h; (c) N-methylpiperazine, K2CO3, CH3CN, reflux, 6 h.
Structure–Activity Relationship
All synthesized analogues were evaluated for PARP-1 inhibitory activity. The clinical candidates, veliparib (ABT-888) and olaparib (AZD-2281), were used as reference standards in the PARP-1 enzyme inhibitory assay. Our initial lead compound 3 demonstrated moderate activity with an IC50 value of 9.45 μM (Table 1). From the docking model of 3 into the PARP-1 active site (Figure 1), it was observed that the carboxamide group undergoes the aforementioned interactions with Gly863 (NH···O=C-Gly863, 1.92 Å; C=O···HN-Gly863, 2.09 Å) and Ser904 (C=O···HO-Ser904, 1.91 Å), and the aromatic ring was stabilized through π–π stacking interaction with the nearly coplanar electron rich phenyl ring of Tyr907 (centroidligand–centroidTyr907 = 4.3 Å, Figure 1). Initially, we synthesized derivatives of 3 modified at the synthetically feasible 5-position. Substitution with bromo, nitro, and amino at the 5-position of 3 yielded compounds 4, 5, and 6, which were concluded to be inactive with IC50 values above 25 μM. Because of the unfavorable contribution of substituents at the 5-position of 3, we next focused onto the 2-position of the 2,3-DHBF scaffold. To probe the 2-position of 3, we followed a new synthetic route as depicted in Scheme 2. The 2-methyl analogue rac-13a was obtained in racemic form and showed an IC50 value of 10.44 μM. Compound rac-13a offered an opportunity to investigate PARP-1 inhibitory activity of each enantiomer. The rac-13a was resolved into pure enantiomers by preparative chiral chromatography (GVK Biosciences Pvt. Ltd., India). Both (R)-(−) and (S)-(+) isomers of 13a exhibited comparable IC50 values (6.34 and 8.44 μM, respectively). The 5-methyl, 5-nitro, and 5-amino derivatives (compounds 13b, 14, and 15, respectively) proved to be inactive, similar to the earlier series of compounds (4, 5, and 6). Collectively, this indicates that the active site region around the 5-position of the DHBF scaffold bears steric restrictions (e.g., compounds 13a vs 13b, Table 1).
Table 1. Structures and PARP-1 Inhibition Data for the Synthesized Compounds 3–28.

| compd | R1 | R2 | R3 | IC50 (μM)b |
|---|---|---|---|---|
| 3 | -H | -H | -H | 9.45 ± 0.25 |
| 4 | -Br | -H | -H | >25 |
| 5 | -NO2 | -H | -H | >25 |
| 6 | -NH2 | -H | -H | >25 |
| rac-13a | -H | -CH3 | -H | 10.44 ± 0.96 |
| (−)-13a | -H | -CH3 | -H | 6.34 ± 1.08 |
| (+)-13a | -H | -CH3 | -H | 8.44 ± 1.05 |
| 13b | -CH3 | -CH3 | -H | >25 |
| rac-13c | -F | -CH3 | -H | 2.45 ± 0.65 |
| (−)-13c | -F | -CH3 | -H | 1.53 ± 0.12 |
| (+)-13c | -F | -CH3 | -H | 3.62 ± 0.76 |
| 14 | -NO2 | -CH3 | -H | >25 |
| 15 | -NH2 | -CH3 | -H | >25 |
| 20 | -F | -H | -H | 2.12 ± 0.37 |
| rac-28 | -H | -CH3 | -NH2 | 4.65 ± 0.25 |
| 281 a | -H | -CH3 | -NH2 | 2.43 ± 0.42 |
| 282 a | -H | -CH3 | -NH2 | 5.33 ± 0.16 |
| ILCc | 6.80 ± 0.57 | |||
| ABT-888c | 0.006 ± 0.001 | |||
| AZD-2281c | 0.007 ± 0.001 |
281 and 282 represent the resolved enantiomers of the racemate 28 by chiral HPLC technique.
IC50 values were determined by at least two independent experiments done in triplicate.
Indazole lead compound (ILC) reported in ref (21) and clinical candidates ABT-888 and AZD-2281 were included as reference standards for comparative purposes.
Examination of previous X-ray crystallography data revealed that small substituents, e.g., fluoro, are well tolerated at the 6-position (5-position equivalent in the DHBF scaffold) of the benzimidazole-4-carboxamide scaffold.12 Similarly, addition of a fluoro group at the 5-position of 2H-indazole-7-carboxamide46 and at the 8-position of 1,3,4,5-tetrahydrobenzonaphthyridin-6-one47 (analogous to the 5-position in the DHBF scaffold) showed 2- to 3-fold improvement in activity. To obtain the 5-fluoro substituted derivative of compound 3 (5-fluoro-DHBF-7-carboxamide, compound 20), we developed a new synthetic route (Scheme 3). As expected from the previous finding, the 5-fluoro derivative 20 (IC50 = 2.12 μM) was found to be ∼5-fold more potent than our initial lead 3 (Table 1). To examine the effect of the 5-fluoro substitution on 2-methyl-DHBF-7-carboxamide (13a), we synthesized rac-13c (IC50 = 2.45 μM) and found it to be 4-fold more potent than 13a. The compound rac-13c was resolved and enantiomers were evaluated in vitro for PARP-1 inhibition. The (+)-13c enantiomer had an IC50 value of 3.62 μM, whereas (−)-13c exhibited an IC50 value of 1.53 μM. The electron deficit phenyl ring of compound (−)-13c (contributed to by the electronegative nature of the fluorine group) was predicted to generate a stronger π–π stacking interaction with the phenyl ring of Tyr907 compared to the unsubstituted analogue 13a (Table 1).
To further develop this scaffold, we obtained cocrystal structure data of (−)-13c bound to the catalytic domain of PARP (Figure 3E). From this structure it appeared the 4-position of (−)-13c was in proximity to the γ-carboxylate group of Glu988 (an essential residue involved in PAR synthesis). Therefore, we chose to synthesize the 4-amino derivative rac-28 to capture electrostatic interactions with the carboxylate group of Glu988. The 4-amino-2-methyl-DHBF-7-carboxamide rac-28 (IC50 = 4.65 μM) had a 2-fold increase in inhibition over rac-13a, thus underscoring the positive role of the 4-amino group. Chiral resolution of this racemate yielded individual enantiomers (compounds 281 and 282) that were discriminated by enzymatic activity as evident from their respective IC50 values of 2.43 and 5.33 μM (Table 1).
Figure 3.

Crystal structure of inhibitors in the active site of PARP-1. (A) Structural representation of the PARP-1 activated complex, which highlights the NAD+ binding site (active site) where most PARP inhibitors bind. (B–D) Fo – Fc electron density difference map (blue mesh) of PARP-1 crystals soaked with 1–5 mM (−)-13c, 59, or 65 (PBD code 4OPX, 4OQA, or 4OQB, respectively), contoured to 2.5σ in which density is calculated in the absence of ligand. (E–G) Compounds bound to the NAD+ site of PARP-1 make π–π stacking interactions between Tyr907 and Tyr896 and H-bond interactions with the backbone of Gly863 and Ser904 consistent with the traditional benzamide pharmacophore of most PARP inhibitors. Compounds 59 and 65 extend out of the nicotinamide pocket and make further interactions in the ADP-ribose pocket of the NAD+ binding site.
Extensive analysis of the substituent effect onto the 2-, 4- and 5-positions of the DHBF-7-carboxamide core revealed that the 2-position was the most promising site for various substitutions owing to the steric restriction at the 5-position of DHBF-7-carboxamide. Cocrystal structure data (Figure 3E) revealed an empty pocket adjacent to the 2-position of the DHBF scaffold, which could likely tolerate large substitutions. Since substitution at the 2-position of the DHBF scaffold presented significant synthetic challenges, we proposed an alternative core, DHBF-3-one-7-carboxamide (compound 36, IC50 = 16.2 μM, Table 2), which is analogous to the DHBF-7-carboxamide with a robust synthetic advantage of presenting an electrophilic 2-position (being connected to the carbonyl group on one side and ethereal oxygen atom on the other side) amenable to Knoevenagel condensation with benzaldehydes. Knoevenagel condensation of compound 36 with substituted benzaldehydes resulted in the desired 2-position substituted compounds (Table 2). The exocyclic double bond of the benzylidene group in these target compounds (Table 2) is stereogenic; however, it has previously been reported that condensation at the 2-position of DHBF-3-one with different aldehydes yields the Z-isomer.42,48 In agreement with literature NMR reports, the olefinic β-proton in these compounds was found to be around 7.03 ppm, which indicates the formation of the Z-isomer (calculated chemical shift for the same proton of the E-isomer was found to be ∼6.19 ppm).
Table 2. Structures and PARP-1 Inhibition Data for Target Compounds 36 and 50–73.


IC50 values were determined by at least two independent experiments done in triplicate.
Indazole lead compound (ILC) reported in ref (21) and clinical candidates ABT-888 and AZD-2281 were included as reference standards for comparative purposes.
In order to study the steric compliance at the 2-position of the DHBF-3-one-7-carboxamide scaffold, initial compounds probed the active site pocket by substituting hydrophobic groups such as phenyl (compound 50), 3-phenoxyphenyl (compound 51), and p-chlorophenyl (compound 52). PARP-1 inhibitory data of this series revealed that extensive hydrophobic substitution at the 2-position marginally improved potency (IC50 values of 12.02, 4.65, and 6.09 μM, respectively; Table 2). However, the added hydrophobicity contributed by these substitutions inevitably increased the magnitude of log P, diminishing the druglike properties of the molecules. Structural data revealed that the benzylidene ring of the DHBF-3-one-7-carboxamide scaffold was located in proximity to polar side groups, namely, Glu763, Asp766, and Tyr889 (Figure 3F). This indicated that the polar substituents on the benzylidene ring could in turn favor PARP-1 inhibition. With this hypothesis, we aimed at synthesizing analogues bearing polar groups on the benzylidene ring. Compound 53 with a vanillyl (4′-hydroxy-3′-methoxyphenyl) group was found to exhibit an IC50 value of 3.62 μM (Table 2). The moderate inhibition by compound 54 (IC50 = 3.30 μM) also suggested that substitutions made onto the 4′-position of the benzylidene ring were well tolerated. Structural data suggested that a nearby vacant pocket was likely accessible from the 4′-position of the benzylidene ring, without any potential for a steric clash (Figure 3F). In order to further delineate the contribution of the hydroxyl group at each position of the benzylidene moiety, we synthesized analogues bearing hydroxyl groups at 2′-position (55), 3′-position (56), and 4′-position (57). Among them, 56 and 57 were found to have improved potency with IC50 values of 2.14 and 0.813 μM, respectively (Table 2). Poor inhibition of PARP-1 by compound 55 also corroborated the importance of 4′-position substitution onto the benzylidene ring. This further suggested that the hydroxyl group at the 4′-position of the benzylidene ring is important to increase potency. We also synthesized dihydroxy substituted analogues, bearing hydroxyl groups at the 3′-position and either the 4′- (compound 58) or 5′-position (compound 60). Among the dihydroxy analogues (compounds 58, 59, 60, and 61), compound 58 bearing 3′,4′-dihydroxy substitution and compound 59 with 2′,4′-dihydroxy groups demonstrated IC50 values of 0.531 and 0.753 μM, respectively. Structural data of 59-PARP-1 revealed that the 4′-hydroxyl group substituted onto the benzylidene ring was directed toward the side chain of Asp766 (Figure 3F). In addition, methylation of the 4′-hydroxyl group of compound 57 led to compound 54 which showed 4-fold reduced inhibitory activity. Collectively, these data clearly underline that polar hydroxyl groups at the 4′-position are beneficial for PARP-1 inhibition. Interestingly, poor inhibition by compounds 60 and 61 indicated that neither the 3′-position nor 5′-position was a significant substituent position for PARP-1 inhibition.
According to the structural data, the 4′-hydroxyl group of compound 59 was found to point toward the vacant hydrophilic pocket formed by the side chains of Asp766, Asn767, Asp770, Ser864, Asn868, and Arg878 (Figure 3F). With an intention to extend substitutions into this pocket, we decided to attach a basic nitrogen heterocycle at this position, wherein the hydroxyl group would act as a good handle for synthetic feasibility. Toward this end, we synthesized an analogue bearing piperidine (compound 64) linked to the 4′-position of 57 by an ethyl linker. Our next strategy was aimed at incorporation of an electronegative oxygen atom at the C4-position of the piperidine ring, which involved replacing the piperidine ring (64) with a morpholine (65). However, this approach did not lead to the desired result, since compound 65 (IC50 = 2.07 μM) was found to be 4-fold less potent compared to 64 (IC50 = 0.544 μM). However, a potential interaction between the morpholine ring oxygen atom and the side chain of Arg878 was suggested by studying the X-ray cocrystal structure of compound 65 bound to the catalytic domain in the PARP-1 (Figure 3G). In the case of 66 (IC50 = 0.114 μM), the electron rich N-methylpiperazine ring was observed to be in proximity to the side chain of Arg878 based on molecular docking (Figure 4A). Further, the terminal nitrogen atom of the piperazine moiety was predicted to undergo an electrostatic interaction with the side chain carboxyl group of Asp770 (NH+···–OOC-Asp770, 1.89 Å). These data hinted toward a favorable role of basic groups at the 4′-substitued saturated heterocycles (Figure 4A). Additionally, we wanted to test if the above substituents at the 3′-position of the benzylidene ring could also bind favorably to this hydrophilic pocket. This was experimentally verified by synthesizing analogues bearing a morpholine ring (compound 62) or piperidine ring (compound 63) linked by an ethyl linker to the 3′-position of compound 56. However, substitution at the 3′-position was found to be detrimental for PARP-1 inhibition (compounds 62 and 63 vs 64 and 65, respectively).
Figure 4.

Predicted binding mode of compounds 66 (A) and 72 (B) within the active site of PARP-1. The color scheme for the active site amino acid residues and the inhibitor is the same as in Figure 1. The centroids are generated as green stars. The yellow dotted lines indicate the distance between the two atoms/groups/centroids (in angstroms). The image was generated in PyMOL, version 1.6.0.
Up to this point, we tested the efficacy of inhibitors bearing ethyl linkers at different positions of benzylidene (compounds 62–66). In an attempt to evaluate the effect of different linkers, we synthesized analogues 67, 68, and 70. With an intention to extend the substitution deeper into the hydrophilic pocket, we extended the linker by one carbon atom (compound 67) to bring the oxygen of the morpholine ring in compound 65 closer to the side chain of Arg878. Compound 67 was found to be nearly 3 times more potent (IC50 = 0.718 μM) compared to the corresponding ethyl analogue 65 (IC50 = 2.07 μM), likely owing to the stronger interaction with the guanidine group of Arg878 (Table 2). In order to increase the ligand–PARP-1 interactions, we aimed to introduce polar groups onto the linker positions, with the potential for interaction with the polar amino acid residues present in the hydrophilic pocket. Introduction of a hydroxyl group onto the second carbon atom of the propyl linker (compound 70) led to a significant improvement in the potency with an IC50 of 0.176 μM. Another advantage of polar substitutions is that they also improved the solubility properties of this compound. The replacement of the ethyl group of compound 65 with the acetyl group (compound 68) led to a 9-fold improvement in potency (IC50 = 0.223 μM). This observation revealed that polar groups incorporated onto the linker significantly improved potency. In order to substantiate our hypothesis that substituted nitrogen containing heterocycles play a significant role in PARP-1 inhibition, we synthesized and tested a truncated analogue with an oxiranylmethoxy moiety (compound 69, IC50 = 1.21 μM). It did not completely lose potency owing to the contribution by polar epoxide ring (Table 2).
Several fragments have been linked to nicotinamide mimicking pharmacophores and are widely reported in the literature as significantly improving PARP-1 inhibition.49 In an attempt to check the usefulness of one such fragment known to give significant inhibition, we tested the impact of linking 1-(4-fluorophenyl)piperazine via ethyl bridge at the 4′-position of the benzylidene moiety. This analogue (compound 71) gave a significant inhibition with an IC50 value of 0.445 μM, demonstrating that our studies correlate with literature reports.
In an attempt to improve the enzyme inhibitory activity of 66, we have successfully replaced the ethoxy linker in 66 with ethanesulfonamide (compound 72, IC50 = 0.079 μM) and propanesulfonamide (compound 73, IC50 = 0.113 μM) linkers. It was anticipated that the sulfonamide groups will provide a sharp angle, mimic the phosphate group of NAD+ (substrate of PARP), and place the piperazine moiety deeper into the adenine-ribose binding pocket of PARP-1 active site. Two of the most potent inhibitors 66 and 72 not only are water-soluble but also present a group amenable for the preparation of pharmaceutical salts. Predicted binding model of 72 within the active site of PARP-1 is shown in Figure 4B. In contrast to compound 66, compound 72 showed three additional hydrogen bonds between the sulfonamide group and the side chains of Asp766, Ser864, and Asn868. Similar to compound 66, sulfonamide analogue 72 was able to reach the adenine-ribose binding pocket, as evident from the docking studies (Figure 4B), wherein the N-methylpiperazine moiety of 72 is in the vicinity of adenine-ribose binding residues Asp770 and Arg878. Taken together, successful replacement of ethoxy linker with ethanesulfonamide further showcases the potential of developing highly active DHBF-3-one scaffold inhibitors.
Selective Killing of BRCA2-Deficient Cells
Since catalytic PARP inhibition is known to induce synthetic lethality in BRCA1- or BRCA2-deficient cells, we tested one of the potent inhibitors (compound 66) for sensitivity against wild-type, PARP-1-deficient, and BRCA2-deficient DT40 cells. This compound was compared to veliparib, a leading clinical PARP inhibitor with high potency. Consistent with previous reports,50 veliparib was not cytotoxic in wild-type and PARP-1-deficient cells up to 10 μM, while it was selectively cytotoxic in BRCA2-deficient cells with an IC90 of 5.0 μM (Figure 5A). Compound 66 also selectively killed BRCA2-deficient cells with an IC90 of 5.2 μM while being minimally cytotoxic in wild-type and PARP-1-deficient cells (Figure 5B). The weak cytotoxicity of compound 66 in wild-type and PARP-1-deficient cells compared to veliparib suggests off-target effects beside PARP-1. PARP inhibitors can have promiscuous inhibitory activity extending to PARP-1–4 and tankyrases25 and can have PARP-1-independent cytotoxicity.51,52 Hence, we conclude that compound 66 has similar cellular potency as veliparib in BRCA2-deficient cells.
Figure 5.

Cell viability of DT40 cells after continuous exposure to (A) veliparib or (B) compound 66 for 72 h. Cellular ATP concentration was used to measure cellular viability. The viability of untreated cells was set as 100%. Error bars represent standard deviation (n = 3). Circle: wild-type cell. Triangle: PARP1-deficient cells. Square: BRCA2-deficient cells. Invisible error bars are encompassed within the symbol sizes.
Structural Studies
To further validate our SAR approach and docking studies, we obtained crystal structure data of both scaffolds bound to the recent structure of PARP-1 in complex with DNA (Figure 3A).23 The diffraction limit of these crystals restricts the level of detail obtained from the PARP-1/compound complexes as a result of large multidomain protein; however, the data allowed us to confidently model the major features of their binding poses within the catalytic domain. Furthermore, we obtained data with three compounds of various sizes but based on the same scaffold, which helped confirm the placement of the inhibitors (Figure 3A). Consistent with the docking studies, the benzamide portion of the DHBF scaffold stacks between two tyrosine residues and makes hydrogen bonding interactions with Gly863 and Ser904 (Figure 3E). Compound 59 appears to reach outside of the traditional nicotinamide pocket with its benzylidene modification to further interact with Tyr889 (Figure 3F). It is interesting to note that modification of the 5′-position of the benzylidene ring would cause a steric clash with Tyr889, which is consistent with the complete loss of potency observed with compounds containing a 5′-position modification (60 and 61). Scaffolds with larger modifications reach deeper into the adenine-ribose binding region of the active site, as seen with compound 65 (Figure 3G). The observed interaction of 65 with Arg878 is only speculative because of the poor density in this region. However, the structures clearly explain why the 4′-position modifications are superior in potency compared to the 3′-position modifications, since the 3′-position would lead to significant steric clash in the NAD+ binding site.
Conclusions
A novel series of DHBF-7-carboxamide and DHBF-3-one-7-carboxamide derivatives were designed, synthesized, and evaluated for PARP-1 inhibition. Substituents larger than fluorine at the 5-position of the DHBF scaffold were found to be detrimental for PARP-1 inhibition. The 2-position methyl substitution is well tolerated in the DHBF-7-carboxamide scaffold, yielding enantiomers that bind differently in the active site. The molecules were resolved and tested for PARP-1 inhibitory activity concluding levorotatory analogues to be the eutomers ((−)-13a and (−)-13c). Synthesizing the DHBF-3-one-7-carboxamide derivatives demonstrated an added advantage of an ease of substitution at the electrophilic 2-position. An initial set of lead compounds 57, 58, and 59 revealed that substituting the hydrophilic groups onto the 4′-position of the benzylidene ring was important for potency. Alkylating the 4′-hydroxyl group of compound 57 with the basic heterocycles linked by a two-carbon spacer generated compounds 64 and 66 with significantly improved PARP-1 inhibitory activity. Crystal structure determination confirmed that these compounds target the nicotinamide binding pocket of the active site and reach out into the adenine-ribose binding region, resulting in increased potency. Extending the side chain on the 4′-position of the benzylidene ring as well as modification of the linker proved to have a significant effect on PARP-1 inhibition, as evident from the inhibition by compounds 67–71. Also, significant inhibition by 71 highlighted that our studies corroborated with literature reports.49 The replacement of ethoxy linker in 66 with aminosulfonylethyl and aminosulfonylpropyl linkers, respectively, resulted in improved inhibitors 72 and 73. Compound 66 was selectively active in BRCA2-deficient cells and comparable to veliparib. Overall, compound 66 was identified as one of the potent compounds in the series with an IC50 of 0.114 μM in an enzyme assay and an IC90 of 5.2 μM against BRCA2-deficient DT40 cells. Compounds 66 and 72 will serve as promising leads for future SAR studies.
Experimental Section
Chemistry. Synthesis: General
All chemicals and solvents were purchased from Sigma–Aldrich (St. Louis, MO), AK Scientific (Union City, CA), Oakwood Laboratories (West Columbia, SC), and Alfa Aesar (Ward Mill, MA) and were used as received. The clinical candidates ABT-888 and AZD-2281 were purchased from the Selleckchem library (Houston, TX). Melting points were determined in open capillary tube on a Thomas-Hoover capillary melting point apparatus and reported as uncorrected values. 1H NMR spectra were recorded on a Bruker AM-400 spectrometer. Chemical shifts are reported as δ (ppm) relative to the tetramethylsilane as an internal standard. Coupling constants (J) are expressed in hertz (Hz). Proton peak multiplicity is reported as s (singlet), d (doublet), t (triplet), q (quintet), sext (sextet), dd (doublet of doublets), ddt (doublet of doublet of triplet), td (triplet of doublets), m (multiplet), and bs (broad singlet). All compounds were routinely checked by thin layer chromatography (TLC) and 1H NMR. TLC was performed on silica gel fluorescent coated plates obtained from Analtech, Inc., Newark, DE. Flash chromatography purification was performed using silica gel (0.060–0.200 mm) obtained from Dynamic Adsorbents, Norcross, GA. Optical rotation of the chiral compounds dissolved in methanol was measured using PerkinElmer 241 polarimeter. Elemental analysis (C, H, N) was performed by Atlantic Microlab, Inc., (Norcross, GA) for all compounds screened in the biological assays, and the results are within ±0.40% of theoretical values. The purity of the synthesized target compounds was determined to be ≥95% by elemental analysis.
Chiral HPLC Analysis
The chiral column used to resolve the enantiomers (CHIRALPACK 1A) had amylose tris(3,5-dimethylphenylcarbamate) as the chiral auxiliary. Optimum separation was achieved for both compounds when the flow rate of the mobile phase was set to 1.0 mL/min. The samples (compounds 13a and 13c) were dissolved in ethanol and eluted using an isocratic mobile phase (n-hexane/ethanol 85:15) at an ambient temperature of 25 °C. Compound 28 was resolved by using an isocratic mobile phase (n-hexane/isopropyl alcohol 70:30). Retention times for the individual enantiomers were reported as tR (in minutes), whereas the ee (enantiomeric excess) values were calculated based on the UV absorption (254 nm) areas for the two enantiomers, respectively. The purity of the resolved enantiomers was determined by measuring the area under the curve (AUC) of the LC–MS spectra of the individual enantiomers and represented as percent AUC of the molecular ion peak relative to the other peaks in the spectra.
Synthesis of 2,3-Dihydrobenzofuran-7-carboxylic Acid (2)
To a solution of n-butyllithium (2.5 M in hexane, 2 mL, 5.0 mmol) in 5 mL of hexane at room temperature was added (0.58 g, 5.0 mmol) N,N,N′,N′-tetramethylethylenediamine (TEMED), followed by a solution of 2,3-dihydrobenzofuran (1, 0.3 g, 2.5 mmol) in hexane (5 mL). The mixture was stirred under nitrogen at room temperature for 4 h and then poured into dry ice. After being stirred at room temperature overnight, the mixture was diluted with 15 mL of water and the layers were separated. The aqueous layer was acidified with hydrochloric acid to pH 1, cooled, and the precipitates were collected on a filter. The crude product was reprecipitated using ethyl acetate/hexane to give compound 2 (0.21 g, yield 52%) as white solid: mp 163–165 °C (lit. 167–169 °C);331H NMR (400 MHz, DMSO-d6, TMS) δ 3.18 (t, J = 8.8 Hz, 2H), 4.59 (t, J = 8.8 Hz, 2H), 6.86 (t, J = 8.8 Hz, 1H), 7.41 (d, J = 7.4 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 12.62 (s, 1H).
Synthesis of 2,3-Dihydrobenzofuran-7-carboxamide (3)
To a solution of compound 2 (0.3 g, 1.83 mmol) in 7 mL of anhydrous tetrahydrofuran (THF) were added isobutyl chloroformate (0.3 g, 2.2 mmol) and N-methylmorpholine (NMM) (0.22 g, 2.2 mmol) under nitrogen atmosphere at −20 °C, and the mixture was stirred for 4 h. Then to this mixture was added 5 mL of aqueous ammonia (30%), and the mixture was stirred at room temperature for 1 h. The organic layer was separated, and the aqueous layer was extracted with (2 × 10 mL) of THF. The combined organic layers were dried over sodium sulfate and concentrated under vacuum. The resulting residue was purified by column chromatography using ethyl acetate/methanol (95:5) as an eluent, and the product was obtained as white crystals (0.23 g, yield 75%): mp 185–187 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.23 (t, J = 8.6 Hz, 2H), 4.68 (t, J = 8.8 Hz, 2H), 6.92 (t, J = 7.5 Hz, 1H), 7.29 (s, 1H), 7.39 (d, J = 7.2 Hz, 1H), 7.58 (s, 1H), 7.61 (d, J = 7.9 Hz, 1H). Anal. Calcd for C9H9NO2·1/6CH3OH: C, 65.34; H, 5.74; N, 8.31. Found: C, 65.52; H, 5.51; N, 8.41.
Synthesis of 5-Bromo-2,3-dihydrobenzofuran-7-carboxamide (4)
To a mixture of DHBF-7-carboxamide 3 (0.2 g, 1.23 mmol) and sodium acetate (0.18 g, 2.21 mmol) in 3 mL of acetic acid was added bromine solution (0.25 g, 1.60 mmol) in 2 mL of acetic acid. The reaction mixture was heated at 80 °C for 3 h and then poured onto ice and filtered. Reprecipitation from ethyl acetate afforded the desired compound 4 as a white solid (0.24 g, yield 80%): mp 195–197 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.25 (t, J = 8.8 Hz, 2H), 4.71 (t, J = 8.8 Hz, 2H), 7.26 (s, 1H), 7.57 (d, J = 1.9 Hz, 1H), 7.66 (d, J = 2.3 Hz, 1H), 7.73 (s, 1H). Anal. Calcd for C9H8BrNO2·1/3CH3OH: C, 44.32; H, 3.69; N, 5.54. Found: C, 44.52; H, 3.43; N, 5.24.
Synthesis of 5-Nitro-2,3-dihydrobenzofuran-7-carboxamide (5)
To an ice-cooled solution of DHBF-7-carboxamide 3 (0.2 g, 1.23 mmol) in 5 mL of trifluoroacetic acid was added 0.4 mL of nitric acid dropwise. After 30 min, the ice bath was removed and the mixture was stirred at room temperature for 3 h, and then the mixture was poured into ice–water. The resulting precipitates were collected on a filter to give a crude product which was reprecipitated from ethyl acetate to yield compound 5 (0.17 g, yield 65%) as a pale yellow solid: mp 239–241 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.35 (t, J = 8.4 Hz, 2H), 4.48 (t, J = 8.6 Hz, 2H), 7.42 (s, 1H), 7.92 (s, 1H), 8.26 (s, 1H), 8.47 (s, 1H). Anal. Calcd for C9H8N2O4·1/6H2O: C, 51.18; H, 3.79; N, 13.27. Found: C, 51.37; H, 3.76; N, 13.10.
Synthesis of 5-Amino-2,3-dihydrobenzofuran-7-carboxamide (6)
To a solution of compound 5 (0.2 g, 0.96 mmol) in 7 mL of ethyl acetate was added tin chloride dihydrate (1.3 g, 5.77 mmol), and the mixture was refluxed for 4 h. After completion of the reaction, mixture was diluted (add 15 mL of ethyl acetate), cooled, and poured into the saturated sodium bicarbonate solution (20 mL). The organic layer was separated and the aqueous layer was washed with ethyl acetate (2 × 20 mL). The combined organic layers were dried over sodium sulfate and evaporated to give crude product, which was purified by column chromatography (ethyl acetate/methanol 95:5) to yield compound 6 as a pale yellow solid (0.09 g, yield 55%): mp 141–143 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.11 (t, J = 8.2 Hz, 2H), 4.55 (t, J = 8.8 Hz, 2H), 4.76 (bs, 2H), 6.67 (d, J = 1.4 Hz, 1H), 6.87 (d, J = 2.2 Hz, 1H), 7.17 (s, 1H), 7.42 (s, 1H). Anal. Calcd for C9H10N2O2: C, 60.66; H, 5.66; N, 15.72. Found: C, 60.76; H, 5.76; N, 15.57.
Synthesis of Methyl 5-Methylsalicylate (8b)
To a solution of 5-methylsalicylic acid (3.0 g, 19.7 mmol) in methanol was added dropwise thionyl chloride (2.81 g, 23.70 mmol), and then the reaction mixture was refluxed for 12 h. The reaction mixture was then cooled to room temperature, and the solvent was evaporated under vacuum. The resulting oil was diluted with water (20 mL) followed by extracting it with ethyl acetate (3 × 20 mL). The organic layers were combined, dried over sodium sulfate, and evaporated under vacuum to yield a yellow liquid, which was further purified by column chromatography (n-hexane/ethyl acetate 95:5) to give the desired product 8b as a pale yellow oil (2.95 g, yield 90%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.26 (s, 3H), 3.92 (s, 3H), 6.87 (d, J = 8.5 Hz, 1H), 7.24 (d, J = 8.7 Hz, 1H), 7.61 (s, 1H), 10.57 (s, 1H).
Synthesis of Methyl 5-Fluorosalicylate (8c)
8c was obtained by the same procedure as mentioned for compound 8b using 5-fluorosalicylic acid (2.96 g, 19.0 mmol) as starting material, as pale yellow crystalline solid (2.74 g, yield 85%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.96 (s, 3H), 6.94 (dd, J = 9.0, 4.6 Hz, 1H), 7.18 (td, J = 8.5, 3.3 Hz, 1H), 7.50 (dd, J = 8.8, 3.3 Hz, 1H), 10.52 (s, 1H).
Synthesis of Methyl 2-(Allyloxy)benzoate (9a)
To a solution of methyl salicylate (3.0 g, 19.7 mmol) in 10 mL of DMF were added allyl bromide (2.63 g, 21.7 mmol), potassium carbonate (2.99 g, 21.7 mmol), and sodium iodide (3.25 g, 21.7 mmol). The reaction mixture was stirred for 12 h at room temperature and then poured into water (50 mL) and extracted with ethyl acetate (3 × 50 mL). The organic layers were combined, dried over sodium sulfate, and concentrated under vacuum to yield yellowish brown oil which was further purified by column chromatography (n-hexane/ethyl acetate 90:10) to get the desired product 9a as pale yellow oil (3.52 g, yield 92%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.79 (s, 3H), 4.63 (m, 2H), 5.25 (dd, J = 10.6, 1.7 Hz, 1H), 5.46 (dd, J = 17.2, 1.7 Hz, 1H), 6.02 (ddt, J = 17.4, 10.7, 4.7 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 7.51 (td, J = 7.8, 1.7 Hz, 1H), 7.63 (dd, J = 7.7, 1.6 Hz, 1H).
Synthesis of Methyl 2-(Allyloxy)-5-methylbenzoate (9b)
9b was synthesized according to compound 9a using compound 8b (3.25 g, 19.6 mmol) as the starting material, as pale yellow oil (3.51 g, yield 87%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.30 (s, 3H), 3.89 (s, 3H), 4.58 (m, 2H), 5.29 (dd, J = 10.6, 1.2 Hz, 1H), 5.50 (dd, J = 17.2, 1.4 Hz, 1H), 6.06 (ddt, J = 17.2, 10.6, 4.8 Hz, 1H), 6.85 (d, 1H), 7.23 (dd, J = 8.4, 2.2 Hz, 1H), 7.62 (s, 1H).
Synthesis of Methyl 2-(Allyloxy)-5-fluorobenzoate (9c)
9c was synthesized according to compound 9a using compound 8c (3.06 g, 18.0 mmol) as the starting material, as colorless-light yellow oil (3.21 g, yield 85%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.89 (s, 3H), 4.58 (m, 2H), 5.29 (dd, J = 11.0, 1.8 Hz, 1H), 5.50 (dd, J = 17.4, 1.8 Hz, 1H), 6.05 (ddt, J = 17.4, 10.6, 4.6 Hz, 1H), 6.93 (dd, J = 7.8, 1.8 Hz, 1H), 7.14 (dd, J = 7.8, 1.8 Hz, 1H), 7.51 (dd, J = 7.8, 1.8 Hz, 1H).
Synthesis of Methyl 3-Allyl-2-hydroxybenzoate (10a)
Compound 9a (3.00 g, 15.62 mmol) was neatly heated at 160 °C for 2 h, which resulted in the crude brownish oil that was purified by flash chromatography (n-hexane/ethyl acetate 95:5) to get compound 10a as pale yellow oil (2.61 g, yield 87%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.36 (d, J = 6.5 Hz, 2H), 3.90 (s, 3H), 5.04 (m, 2H), 6.02 (ddt, J = 16.8, 10.2, 6.5 Hz, 1H), 6.90 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 7.3 Hz, 1H), 7.67 (dd, J = 7.8, 1.7 Hz, 1H), 10.92 (s, 1H).
Synthesis of Methyl 3-Allyl-2-hydroxy-5-methylbenzoate (10b)
Compound 9b (2.09 g, 10.19 mmol) was neatly heated at 190 °C for 4 h to obtain crude brownish oil that was further purified by flash chromatography to get compound 10b as pale yellow oil (1.73 g, yield 83%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.25 (s, 3H), 3.39 (d, J = 6.8 Hz, 2H), 3.93 (s, 3H), 5.06 (m, 1H), 5.09 (m, 1H), 6.00 (ddt, J = 16.0, 10.0, 6.6 Hz, 1H), 7.14 (s, 1H), 7.51 (s, 1H), 10.85 (s, 1H).
Synthesis of Methyl 3-Allyl-5-fluoro-2-hydroxybenzoate (10c)
Compound 9c (2.6 g, 12.38 mmol) was neatly heated at 190 °C for 4 h to obtain crude brownish oil that was further purified by flash chromatography to get compound 10c as pale yellow oil (2.13 g, yield 82%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.41 (d, J = 6.6 Hz, 2H), 3.94 (s, 3H), 5.10 (m, 1H), 5.13 (m, 1H), 6.05 (ddt, J = 17.0, 10.4, 6.6 Hz, 1H), 7.09 (dd, J = 8.8, 3.3 Hz, 1H), 7.38 (dd, J = 8.6, 3.3 Hz, 1H), 10.83 (s, 1H).
Synthesis of Methyl 2-Methyl-2,3-dihydrobenzofuran-7-carboxylate (11a)
To a solution of compound 10a (2.0 g, 10.41 mmol) in dichloromethane (20 mL), cooled to 0 °C, zirconium(IV) chloride (2.91 g, 12.48 mmol) was added in portions, and the reaction mixture was stirred at room temperature for 10 h. After completion, the reaction was quenched by addition of cold water (10 mL) and the organic layer was separated. The aqueous layer was extracted with (3 × 10 mL) dichloromethane. The combined organic phase was washed with water, dried over sodium sulfate, and concentrated to give the crude product, which was purified by flash chromatography (n-hexane/ethyl acetate 80:20) to get the desired product 11a as pale yellow oil (1.70 g, yield 85%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.40 (d, J = 6.3 Hz, 3H), 2.78 (dd, J = 15.7, 7.6 Hz, 1H), 3.32 (dd, J = 15.7, 9.0 Hz, 1H), 3.78 (s, 3H), 5.0 (sext, J = 7.0 Hz, 1H), 6.87 (t, J = 7.3 Hz, 1H), 7.40 (dd, J = 7.3, 1.3 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H).
Synthesis of Methyl 2,5-Dimethyl-2,3-dihydrobenzofuran-7-carboxylate (11b)
11b was synthesized according to the above procedure for 11a using compound 10b (1.5 g, 7.28 mmol) as starting material, as colorless oil (1.24 g, yield 83%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.50 (d, J = 6.2 Hz, 3H), 2.27 (s, 3H), 2.77 (dd, J = 15.6, 7.4 Hz, 1H), 3.28 (dd, J = 15.6, 8.8 Hz, 1H), 3.88 (s, 3H), 5.04 (sext, J = 7.0 Hz, 1H), 7.12 (s, 1H), 7.50 (s, 1H).
Synthesis of Methyl 5-Fluoro-2-methyl-2,3-dihydrobenzofuran-7-carboxylate (11c)
11c was synthesized according to 11a using compound 10c (1.2 g, 5.71 mmol) as starting material and obtained as colorless oil (1.0 g, yield 83%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.52 (d, J = 6.2 Hz, 3H), 2.82 (dd, J = 16.0, 7.4 Hz, 1H), 3.32 (dd, J = 16.0, 9.0 Hz, 1H), 3.89 (s, 3H), 5.09 (sext, J = 7.0 Hz, 1H), 7.04 (d, J = 7.4 Hz, 1H), 7.39 (dd, J = 9.6, 2.8 Hz, 1H).
Synthesis of 2-Methyl-2,3-dihydrobenzofuran-7-carboxylic Acid (12a)
To a solution of compound 11a (1.50 g, 7.81 mmol) in methanol was added sodium hydroxide (0.94 g, 23.43 mmol), and the mixture was refluxed for 2 h. Upon completion, the mixture was cooled and the solvent was evaporated under reduced pressure. The resulting white solid was dissolved in a small amount of water and cooled to 0 °C. To this, concentrated hydrochloric acid was added dropwise until pH 1 to get white precipitates, which were filtered and dried under vacuum to get the desired compound rac-12a as white solid (1.17 g, yield 85%): mp 121–123 °C (lit. 125–127 °C);531H NMR (400 MHz, DMSO-d6, TMS) δ 1.40 (d, J = 6.2 Hz, 3H), 2.78 (dd, J = 16.0, 7.8 Hz, 1H), 3.31 (dd, J = 15.9, 8.8 Hz, 1H), 4.97 (sext, J = 7.0 Hz, 1H), 6.85 (t, J = 7.6 Hz, 1H), 7.37 (dd, J = 7.1, 1.1 Hz, 1H), 7.55 (d, J = 7.7 Hz, 1H), 12.56 (bs, 1H).
The 2-methyl enantiomers obtained were resolved to elucidate the absolute stereochemistry at the 2-position of 12a, wherein the acid derivative was heated with equimolar proportion of either of the (−)-brucine dihydrate in acetone or (S)-(−)-α-methylbenzylamine or (R)-(+)-α-methylbenzylamine in methanol for crystallization. The superior quality crystals obtained were used to elucidate the absolute stereochemistry at Department of Chemistry, Louisiana State University, Baton Rouge, LA, by X-ray crystallographic analysis. Furthermore, the absolute stereochemistry of the compound was correlated with the optical rotation performed by quenching the crystals (having stereochemistry determined) with diluted hydrochloric acid. The quenched fractions, upon extraction in ethyl acetate, were subjected to optical rotation using a polarimeter at a concentration of 0.1 g/mL in methanol. The specific rotation for each enantiomer was calculated from the observed rotation, which was found to be [α]25D +14.3° and −13.8°, respectively. The X-ray crystallographic analysis of the diastereomeric salt of the (−)-enantiomer of 12a with (S)-(−)-α-methylbenzylamine revealed that the (−)-enantiomer bears the R-configuration (Figure 2). On the basis of this finding, the (+)-enantiomer was assigned as the S-enantiomer.
Synthesis of 2,5-Dimethyl-2,3-dihydrobenzofuran-7-carboxylic Acid (12b)
12b was prepared according to the procedure used to synthesize compound 12a using compound 11b (1.50 g, 7.28 mmol) as starting material and obtained as a white solid (1.21 g, yield 87%): mp 146–148 °C (lit. 149–150 °C);541H NMR (400 MHz, DMSO-d6, TMS) δ 1.38 (d, J = 6.1 Hz, 3H), 2.22 (s, 3H), 2.73 (dd, J = 16.0, 7.6 Hz, 1H), 3.27 (dd, J = 15.6, 8.9 Hz, 1H), 4.93 (sext, J = 7.0 Hz, 1H), 7.19 (s, 1H), 7.35 (s, 1H), 12.48 (s, 1H).
Synthesis of 5-Fluoro-2-methyl-2,3-dihydrobenzofuran-7-carboxylic Acid (12c)
12c was prepared according to the procedure used to synthesize compound 12a using compound 11c (1.30 g, 6.19 mmol) as starting material and obtained as pale yellow solid (1.03 g, yield 85%): mp 128–130 °C (lit. 129–131 °C);531H NMR (400 MHz, DMSO-d6, TMS) δ 1.39 (d, J = 6.3 Hz, 3H), 2.95 (dd, J = 16.4, 7.9 Hz, 1H), 3.32 (dd, J = 15.9, 9.0 Hz, 1H), 5.00 (sext, J = 7.0 Hz, 1H), 7.24 (dd, J = 9.9, 2.9 Hz, 1H), 7.32 (dd, J = 8.0, 3.0 Hz, 1H), 12.89 (s, 1H).
Synthesis of 2-Methyl-2,3-dihydrobenzofuran-7-carboxamide (13a)
The acid rac-12a (1.10 g, 6.18 mmol) was converted to carboxamide using mixed-anhydride method as followed for compound 3 to yield the amide as white solid (0.87 g, yield 80%): mp 124–125 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.43 (d, J = 6.1 Hz, 3H), 2.83 (dd, J = 15.4, 7.1 Hz, 1H), 3.36 (dd, J = 15.4, 9.0 Hz, 1H), 5.08 (sext, J = 7.0 Hz, 1H), 7.25 (s,1H), 7.35 (d, J = 6.8 Hz, 1H), 7.57 (s, 1H), 7.62 (d, J = 7.8 Hz, 1H). Anal. Calcd for C10H11NO2: C, 67.78; H, 6.26; N, 7.90. Found: C, 67.56; H, 6.37; N, 7.80.
The racemate amide (rac-13a) was resolved by means of preparative chiral chromatography technique at GVK Biosciences Pvt. Ltd., Hyderabad, India. Optical rotations of the individual enantiomers measured were found to be [α]25D +8.3°, −8.1°. tR = 7.68 min (dextro), 7.04 min. (levo); LC–MS purity = 99.79% (dextro), 98.42% (levo); ee = 90.22% (dextro), 97.30% (levo).
Synthesis of 2,5-Dimethyl-2,3-dihydrobenzofuran-7-carboxamide (13b)
13b was synthesized by mixed-anhydride method according to compound 3 using 12b (1.00 g, 5.21 mmol) as starting material to yield a white solid (0.74 g, yield 75%): mp 152–153 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.42 (d, J = 6.3 Hz, 3H), 2.24 (s, 3H), 2.95 (dd, J = 15.9, 7.4 Hz, 1H), 3.32 (dd, J = 15.7, 8.8 Hz, 1H), 5.04 (sext, J = 7.0 Hz, 1H), 7.17 (s, 1H), 7.21 (s, 1H), 7.42 (s, 1H), 7.54 (s, 1H). Anal. Calcd for C11H13NO2: C, 69.09; H, 6.85; N, 7.32. Found: C, 68.81; H, 6.97; N, 7.20.
Synthesis of 5-Fluoro-2-methyl-2,3-dihydrobenzofuran-7-carboxamide (13c)
13c was synthesized by mixed-anhydride method according to compound 3 using rac-12c (1.30 g, 6.63 mmol) as starting material to yield a white solid (0.90 g, yield 70%): mp 137–139 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.44 (d, J = 6.1 Hz, 3H), 2.84 (dd, J = 16.2, 8.1 Hz, 1H), 3.37 (dd, J = 16.2, 8.8 Hz, 1H), 5.10 (sext, J = 7.0 Hz, 1H), 7.29 (m, 3H), 7.76 (s, 1H). Anal. Calcd for C10H10FNO2·1/6CH3OH: C, 60.89; H, 5.32; N, 6.98. Found: C, 60.62; H, 5.32; N, 6.98.
The amide enantiomers were resolved by means of chiral chromatography technique at GVK Biosciences Pvt. Ltd., Hyderabad, India. Optical rotations of the individual enantiomers measured were found to be [α]25D +14.2°, −12.3°. tR = 7.02 min (dextro) and 6.56 min (levo); LC–MS purity = 99.48% (dextro), 99.69% (levo); ee = 90.38% (dextro), 93.25% (levo).
Synthesis of 2-Methyl-5-nitro-2,3-dihydrobenzofuran-7-carboxamide (14)
14 was synthesized according to compound 5 using rac-13a (1.20 g, 6.78 mmol) as starting material to give a pale yellow solid (0.98 g, yield 65%): mp 204–205 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.50 (d, J = 6.5 Hz, 3H), 2.95 (dd, J = 16.4, 7.6 Hz, 1H), 3.49 (dd, J = 16.4, 8.8 Hz, 1H), 5.29 (sext, J = 7.0 Hz, 1H), 7.91 (s, 1H), 7.39 (s, 1H), 8.23 (s, 1H), 8.48 (d, J = 2.6 Hz, 1H). Anal. Calcd for C10H10N2O4: C, 54.05; H, 4.54; N, 12.61. Found: C, 53.89; H, 4.48; N, 12.46.
Synthesis of 5-Amino-2-methyl-2,3-dihydrobenzofuran-7-carboxamide (15)
15 was obtained according to the procedure for compound 6 using 14 (0.60 g, 2.70 mmol) as starting material, yielding a pale yellow solid (0.26 g, yield 50%): mp 152–154 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.39 (d, J = 6.2 Hz, 3H), 2.71 (dd, J = 15.7, 7.6 Hz, 1H), 3.22 (dd, J = 15.7, 8.6 Hz, 1H), 4.75 (s, 2H), 4.92 (sext, J = 7.0 Hz, 1H), 6.62 (s,1H), 6.86 (s, 1H), 7.18 (s, 1H), 7.42 (s, 1H). Anal. Calcd for C10H12N2O2: C, 62.49; H, 6.29; N, 14.57. Found: C, 62.27; H, 6.45; N, 13.56.
Synthesis of Methyl 2,3-Dihydrobenzofuran-7-carboxylate (16)
To a suspension of 2 (3.00 g, 18.29 mmol) in methanol was added a catalytic amount of sulfuric acid, and the mixture was refluxed for 3 h. After completion of the reaction, the reaction mixture was cooled and concentrated under vacuum. To the crude mass, water (20 mL) was added and compound was extracted in ethyl acetate (3 × 20 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to get crude solid, which was purified by flash chromatography to get desired product 16 as white crystalline solid (2.93 g, yield 90%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.21 (t, J = 8.8 Hz, 2H), 3.89 (s, 3H), 4.70 (t, J = 8.8 Hz, 2H), 6.86 (t, J = 7.6 Hz, 1H), 7.33 (d, J = 7.0 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H).
Synthesis of 5-Nitro-2,3-dihydrobenzofuran (17a)
17a was synthesized according to compound 5 using compound 1 (1.10 g, 9.16 mmol) as starting material, as pale yellow solid (1.18 g, yield 78%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.28 (t, J = 8.8 Hz, 2H), 4.72 (t, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 1H), 8.06 (dd, J = 8.8, 2.5 Hz, 1H), 8.13 (s, 1H).
Synthesis of 5-Nitro-2,3-dihydrobenzofuran-7-carboxylic Acid (17b)
17b was synthesized according to compound 5 using compound 2 (1.20 g, 7.32 mmol) as starting material, as pale yellow solid (1.07 g, yield 70%): mp 245–247 °C (lit. 249–251.5 °C);331H NMR (400 MHz, DMSO-d6, TMS) δ 3.30 (t, J = 8.6 Hz, 2H), 4.81 (t, J = 8.6 Hz, 2H), 8.29 (s, 1H), 8.44 (s, 1H), 13.37 (bs, 1H).
Synthesis of Methyl 5-Nitro-2,3-dihydrobenzofuran-7-carboxylate (17c)
17c was synthesized according to compound 5 using compound 16 (1.60 g, 8.99 mmol) as starting material, yielding yellow solid (1.52 g, yield 76%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.32 (t, J = 8.6 Hz, 2H), 3.84 (s, 1H), 4.84 (t, J = 8.8 Hz, 2H), 8.31 (s, 1H), 8.43 (s, 1H).
Synthesis of 5-Amino-2,3-dihydrobenzofuran (18a)
To a suspension of 17a (3.00 g, 18.18 mmol) in ethanol (50 mL) was added 10% palladium on carbon (0.20 g), and the mixture was stirred under hydrogen atmosphere at 60 psi for 18 h. The mixture was then filtered through Celite bed and concentrated to afford a gray solid, which was further purified by column chromatography to get the desired compound 18a as brownish-black solid (2.17 g, yield 90%): mp 77–79 °C (lit. 80–81 °C);541H NMR (400 MHz, CDCl3, TMS) δ 3.11 (t, J = 8.6 Hz, 2H), 3.30 (bs, 2H), 4.48 (t, J = 8.6 Hz, 2H), 6.45 (d, J = 8.2 Hz, 1H), 6.59 (m, 2H).
Synthesis of 5-Amino-2,3-dihydrobenzofuran-7-carboxylic Acid (18b)
18b was obtained by the aforementioned procedure for compound 18a using 17b (1.3 g, 6.22 mmol) as starting material (reaction completed in 2 h), as brownish black solid (1.24 g, yield 82%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.05 (t, J = 8.6 Hz, 2H), 4.44 (t, J = 8.6 Hz, 2H), 4.60 (bs, 2H), 6.72 (s, 1H), 6.80 (s, 1H), 12.39 (bs, 1H).
Synthesis of Methyl 5-Amino-2,3-dihydrobenzofuran-7-carboxylate (18c)
18c was obtained by the aforementioned procedure for compound 18a using 17c (1.2 g, 5.40 mmol) as starting material (reaction completed in 2 h), as crystalline pale yellow solid (0.87 g, yield 84%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.16 (t, J = 8.7 Hz, 2H), 3.49 (bs, 2H), 3.89 (s, 3H), 4.65 (t, J = 8.7 Hz, 2H), 6.79 (s, 1H), 7.06 (s, 1H).
Synthesis of 5-Fluoro-2,3-dihydrobenzofuran (19a)
To a solution compound 18a (2.00 g, 14.49 mmol) in THF (50 mL) was added 1.5 mL of concentrated aqueous hydrochloric acid in several portions. To the resulting white precipitates was added dropwise tetrafluoroboric acid (2.7 mL). The mixture was then chilled (−15 °C), and to it was added sodium nitrite (1.10 g, 15.94 mmol) in water (5 mL). Initially the suspension turned deep gray and was homogenized, followed by precipitation after some time. The mixture was stirred for 30 min at −15 °C, and then the solid was collected by filtration and washed with cold water, cold ethanol, and cold ether. The solid was dried by vacuum filtration to afford 5-diazonium-2,3-DHBF tetrafluoroborate salt, which was used without further purification.
A suspension of the above diazonium tetrafluoroborate salt in xylene (10 mL) was refluxed for 2 h. The mixture was then cooled and diluted with 20 mL of saturated aqueous sodium bicarbonate and then extracted with 3 × 20 mL of ethyl acetate. The combined organic layers were washed with 50 mL of aqueous sodium bicarbonate, 50 mL of brine, dried over sodium sulfate, filtered, and concentrated under vacuum to afford crude oil. The crude oil was purified by gradient flash chromatography (n-hexane/ethyl acetate 100:0 and than 99.5:0.5) to afford compound 19a as pale yellow oil (0.61 g, yield 30%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.16 (t, J = 8.6 Hz, 2H), 4.55 (t, J = 8.6 Hz, 2H), 6.66 (dd, J = 8.6, 4.2 Hz, 1H), 6.76 (t, J = 9.0 Hz, 1H), 6.87 (d, J = 7.8 Hz, 1H).
Synthesis of Methyl 5-Fluoro-2,3-dihydrobenzofuran-7-carboxylate (19c)
19c was obtained by the aforementioned procedure for compound 19a using 18c (2.10 g, 10.88 mmol) as starting material with a small modification (instead of filtering off the diazonium tetrafluoroborate salt). The mixture was concentrated under reduced pressure and the resulting mass was refluxed in xylene to yield compound 19c as colorless solid (0.30 g, yield 14%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.23 (t, J = 8.8 Hz, 2H), 3.91 (s, 3H), 4.73 (t, J = 8.8 Hz, 2H), 7.08 (d, J = 7.3 Hz, 1H), 7.38 (dd, J = 9.6, 2.7 Hz, 1H).
Synthesis of 5-Fluoro-2,3-dihydrobenzofuran-7-carboxylic Acid (19b)
19b was obtained from ester hydrolysis of compound 19c using sodium hydroxide (0.25 g, 1.27 mmol) as mentioned for synthesis of compounds 12a–c; however, the mixture was refluxed for 3 h (instead of 2 h), and the product was obtained as white solid (0.20 g, yield 87%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.20 (t, J = 8.8 Hz, 2H), 4.62 (t, J = 8.8 Hz, 2H), 7.25 (dd, J = 9.8 Hz, 3.0 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 12.91 (bs, 1H).
Synthesis of 5-Fluoro-2,3-dihydrobenzofuran-7-carboxamide (20)
20 was synthesized from compound 19b (0.15 g, 0.82 mmol) following the exact procedure used to synthesize compound 3 and obtained as pale yellow solid (0.12 g, yield 80%): mp 188–190 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.25 (t, J = 8.8 Hz, 2H), 4.71 (t, J = 8.8 Hz, 2H), 7.30 (m, 3H), 7.74 (s, 1H). Anal. Calcd for C9H8FNO2: C, 59.67; H, 4.45; N, 7.73. Found: C, 59.67; H, 4.51; N, 7.55.
Synthesis of Methyl 4-Nitrosalicylate (22)
22 was synthesized using 4-nitrosalicylic acid as the starting material as per the aforementioned method for compound 8b (3.00 g, 16.40 mmol) and obtained as bright yellow solid (2.90 g, yield 90%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.95 (s, 3H), 5,87 (s, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 11.31 (s, 1H).
Synthesis of Methyl 2-(Allyloxy)-4-nitrobenzoate (23)
23 was synthesized using compound 22 (2.50 g, 12.70 mmol) following the same protocol as that of compound 9a, as a bright yellow solid (2.62 g, yield 87%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.86 (s, 3H), 4.81 (d, J = 6.9 Hz, 2H), 5.30 (dd, J = 13.5, 4.2 Hz, 1H), 5.47 (dd, J = 11.3, 5.6 Hz, 1H), 6.04 (ddt, J = 17.0, 10.4, 6.8 Hz, 1H), 7.87 (s, 2H), 7.89 (s, 1H).
Synthesis of Methyl 3-Allyl-2-hydroxy-4-nitrobenzoate (24)
24 was synthesized using compound 23 (1.00 g, 4.22 mmol) with the diminutive variation in the procedure of compound 10a where the O-allylated product was heated in carbitol at an elevated temperature of 170–180 °C for 2 h and obtained as a dark brown oil (0.30 g, yield 30%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.49 (d, J = 6.1 Hz, 2H), 3.95 (s, 3H), 4.99 (dd, J = 11.30, 6.74 Hz, 2H), 5.88 (ddt, J = 17.0, 10.4, 6.8 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 11.27 (s, 1H).
Synthesis of Methyl 2-Methyl-4-nitro-2,3-dihydrobenzofuran-7-carboxylate (25)
25 was obtained from compound 24 (0.85 g, 3.58 mmol) as per the procedure mentioned for compound 11a in form of pale brown solid (0.55 g, yield 65%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.46 (d, J = 6.2 Hz, 3H), 3.26 (dd, J = 11.3, 5.1 Hz, 1H), 3.81 (dd, J = 10.1, 6.5 Hz, 1H), 3.84 (s, 3H), 5.18 (sext, J = 7.0 Hz, 1H), 7.67 (d, J = 8.8 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H).
Synthesis of Methyl 4-Amino-2-methyl-2,3-dihydrobenzofuran-7-carboxylate (26)
26 was obtained from compound 25 (1.10 g, 4.64 mmol) using the exact procedure as mentioned for compound 18a and obtained as bright yellow powder (0.82 g, yield 85%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.40 (d, J = 6.2 Hz, 3H), 2.48 (dd, J = 15.1, 7.8 Hz, 1H), 3.02 (dd, J = 15.3, 9.2 Hz, 1H), 3.71 (s, 3H), 4.95 (sext, J = 7.1 Hz, 1H), 5.62 (s, 2H), 6.2 (d, J = 8.5 Hz, 1H), 7.40 (d, J = 8.6 Hz. 1H).
Synthesis of 4-Amino-2-methyl-2,3-dihydrobenzofuran-7-carboxylic Acid (27)
27 was synthesized by hydrolyzing compound 26 (0.60 g, 2.89 mmol) in the same way as that the method followed for compound 12a and obtained in form of pale brown solid (0.41 g, yield 73%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.38 (d, J = 6.2 Hz, 3H), 2.54 (dd, J = 15.1, 7.8 Hz, 1H), 3.07 (dd, J = 15.3, 9.2 Hz, 1H), 4.94 (sext, J = 7.1 Hz, 1H), 5.66 (s, 2H), 6.16 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 13.1 (s, 1H).
Synthesis of 4-Amino-2-methyl-2,3-dihydrobenzofuran-7-carboxamide (28)
To a well stirred solution of compound 27 (0.30 g, 1.55 mmol) in anhydrous THF was added sequentially N-methylmorpholine (0.3 mL) followed by isobutyl chloroformate (0.3 mL) at −20 °C under nitrogen atmosphere. The resulting reaction mixture was then allowed to stir for 30 min to yield the anhydride through which was passed dry ammonia gas under anhydrous condition. The mixture was then brought to room temperature with intermittent monitoring by thin layer chromatography. Upon completion, the reaction mixture was purified by column chromatography (ethyl acetate/methanol 95:5), resulting in a pale yellow solid (0.12 g, yield 40%): mp 245–248 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.42 (d, J = 6.3 Hz, 3H), 2.52 (dd, J = 15.1, 9.2 Hz, 1H), 3.09 (dd, J = 15.6, 9.1 Hz, 1H), 5.03 (sext, J = 7.1 Hz, 1H), 5.63 (s, 2H), 6.15 (d, J = 8.6 Hz, 1H), 6.91 (s, 1H), 7.06 (s, 1H), 7.36 (d, J = 8.6 Hz, 1H).
The amide was resolved by means of preparative chiral chromatography technique at GVK Biosciences Pvt. Ltd., Hyderabad, India. Individual enantiomers were resolved and characterized for purity and retention times, with tR = 6.93 min (compound 281) and 8.12 min (compound 282); LC–MS purity = 99.07% (compound 281), 96.58% (compound 282); ee = 94.00% (compound 281), 94.46% (compound 282).
Synthesis of Methyl 2-Hydroxy-3-methylbenzoate (30)
30 was obtained by the same procedure mentioned for compound 8b using compound 29 (3.00 g, 19.73 mmol) as starting material and methanol as a solvent to yield compound 30 as pale yellow oil (2.00 g, yield 60%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.40 (t, J = 7.2 Hz, 3H), 2.26 (s, 3H), 4.39 (q, J = 7.1 Hz, 2H), 6.77 (t, J = 7.6 Hz, 1H), 7.30 (d, J = 7.2 Hz, 1H), 7.70 (d, J = 7.6 Hz, 1H), 11.10 (s, 1H).
Synthesis of Methyl 2-(2-Ethoxy-2-oxoethoxy)-3-methylbenzoate (31)
To a solution of 30 (3.0 g, 18.0 mmol) in 10 mL of DMF were added ethyl bromoacetate (4.51 g, 27.0 mmol), potassium carbonate (2.72 g, 19.8 mmol), and sodium iodide (2.96 g, 19.8 mmol). The reaction mixture was stirred for 12 h at room temperature and then poured into water (50 mL) and extracted with ethyl acetate (3 × 50 mL). The organic layers were combined, dried over sodium sulfate, and concentrated under vacuum to yield yellowish brown oil which was further purified by column chromatography (n-hexane/ethyl acetate 90:10) to get the desired product 31 as pale yellow oil (2.20 g, yield 92%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.32 (t, J = 7.2 Hz, 3H), 1.37 (t, J = 7.2 Hz, 3H), 2.35 (s, 2H), 4.29 (q, J = 7.5 Hz, 2H), 4.35 (q, J = 7.5 Hz, 2H), 4.58 (s, 2H), 7.08 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 7.4 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H).
Synthesis of 2-(Carboxymethoxy)-3-methylbenzoic Acid (32)
32 was obtained in the same way as the protocol followed for compound 12a using 3 equiv of potassium hydroxide when added to compound 31 under refluxing conditions, yielding 32 as pale yellow solid (1.34 g, yield 80%): mp 202–204 °C (lit. 207 °C);551H NMR (400 MHz, DMSO-d6, TMS) δ 2.28 (s, 3H), 4.49 (s, 2H), 7.10 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.54 (d, J = 7.6 Hz, 1H), 12.97 (s, 2H).
Synthesis of 2-(Carboxymethoxy)isophthalic Acid (33)
To a suspension of compound 32 (2.0 g, 9.5 mmol) in 20 mL of water was added KMnO4 (7.57 g, 47.6 mmol) gradually, and the resulting reaction mixture was refluxed for 2 h. Upon completion, the reaction mixture was filtered and filtrate was concentrated under vacuum. The resulting white solid was dissolved in minimum amount of water and cooled to 0 °C. To this solution, concentrated HCl (11 N) was added dropwise until pH 1 was obtained. The resulting precipitates were filtered and dried to get the desired compound 33 as white solid (0.68 g, yield 30%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 4.56 (s, 2H), 7.29 (t, J = 7.6 Hz, 1H), 7.84 (d, J = 7.6 Hz, 2H), 13.45 (bs, 3H).
Synthesis of 3-Acetoxybenzofuran-7-carboxylic Acid (34)
To a mixture of 33 (1.0 g, 4.17 mmol) and sodium acetate (0.34 g, 4.17 mmol) were added 3 mL of acetic acid and 5 mL of acetic anhydride, and the resulting mixture was refluxed for 5 h. Upon completion, the reaction mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over magnesium sulfate and concentrated under vacuum to obtain a yellow mass, which was purified by column chromatography (n-hexane/ethyl acetate 90:10) to get the desired compound 34 as pale yellow solid (0.55 g, yield 60%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.39 (s, 3H), 7.39 (t, J = 7.6 Hz, 1H), 7.80 (d, J = 7.6 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 8.29 (s, 1H), 13.38 (bs, 1H).
Synthesis of 3-Oxo-2,3-dihydrobenzofuran-7-carboxylic Acid (35)
Compound 34 (0.5 g, 2.27 mmol) was dissolved in a 10 mL mixture of HCl/H2O/MeOH (1:10:40) and was refluxed for 1 h. Upon completion, the reaction mixture was concentrated under vacuum. The resulting solid obtained was filtered off and washed with water to yield the desired compound as an orange solid (0.24 g, yield 60%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 4.89 (s, 2H), 7.23 (t, J = 7.6 Hz, 1H), 7.87 (d, J = 7.5 Hz, 1H), 8.18 (d, J = 7.7 Hz, 1H), 13.24 (bs, 1H).
Synthesis of 3-Oxo-2,3-dihydrobenzofuran-7-carboxamide (36)
36 was obtained by the same procedure mentioned for compound 3 using compound 35 (2.00 g, 11.23 mmol) as a starting material to yield compound 36 as yellow solid (0.70 g, yield 35%): mp 194–196 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 4.93 (s, 2H), 7.26 (t, J = 7.6 Hz, 1H), 7.44 (s, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.86 (s, 1H), 8.14 (d, J = 7.4 Hz, 1H). Anal. Calcd for C9H7NO3: C, 61.02; H, 3.98; N, 7.91. Found: C, 61.35; H, 3.62; N, 7.86.
Synthesis of 3-(2-Morpholin-4-ylethoxy)benzaldehyde (37)
To a well stirred solution of 3-hydroxybenzaldehyde (0.5 g, 4.09 mmol) in acetonitrile were added 4-(2-chloroethyl)morpholine (0.61 g, 4.09 mmol) and anhydrous potassium carbonate (0.56 g, 4.09 mmol), and the mixture was refluxed for 6 h. Upon completion, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was dried over anhydrous magnesium sulfate and concentrated under vacuum. The resulting crude product obtained upon workup was finally purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield the desired compound as dark brown oil (0.59 g, yield 62%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.17–2.32 (m, 4H), 2.47 (t, J = 5.4 Hz, 2H), 3.35–3.39 (m, 4H), 3.82 (t, J = 5.5 Hz, 2H), 6.87 (s, 1H), 7.02–7.09 (m, 1H), 7.09–7.25 (m, 2H), 9.62 (s, 1H).
Synthesis of 3-(2-Piperidin-1-ylethoxy)benzaldehyde (38)
38 was synthesized by following the same protocol as for compound 37 by reacting 3-hydroxybenzaldeyde (0.3 g, 2.45 mmol) and 1-(2-chloroethyl)piperidine (0.36 g, 2.45 mmol) using potassium carbonate (0.50 g, 3.70 mmol) as a base under refluxing conditions for 6 h. The resulting crude product was purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield a pale yellow oil (0.39 g, yield 72%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.24–1.43 (m, 5H), 2.01–2.34 (m, 5H), 2.47 (t, J = 6.0 Hz, 2H), 3.83 (t, J = 6.0 Hz, 2H), 6.89 (s, 1H), 7.05–7.29 (m, 3H), 9.65 (s, 1H).
Synthesis of 4-(2-Morpholin-4-ylethoxy)benzaldehyde (39)
39 was synthesized in the same way as mentioned for compound 37, by mixing equimolar proportions of 4-hydroxybenzaldehyde (0.5 g, 4.09 mmol) and 4-(2-chloroethyl)morpholine (0.61 g, 4.09 mmol) under refluxing conditions in the presence of potassium carbonate (0.56 g, 4.09 mmol). The mixture was refluxed for 6 h. The resulting crude product obtained upon workup was purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to provide pale brown oil (0.67 g, yield 70%): bp >300 °C (lit. 394 °C);561H NMR (400 MHz, CDCl3, TMS) δ 2.36–2.50 (m, 4H), 2.65 (t, J = 5.3 Hz, 2H), 3.55 (t, J = 3.9 Hz, 4H), 4.01 (t, J = 5.3 Hz, 2H), 6.84 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 7.8 Hz, 2H), 9.69 (s, 1H).
Synthesis of 4-(2-Piperidin-1-ylethoxy)benzaldehyde (40)
40 was synthesized by following the same method as mentioned for compound 37 by reacting 4-hydroxybenzaldehyde (0.3 g, 2.45 mmol) and 1-(2-chloroethyl)piperidine (0.36 g, 2.45 mmol) in refluxing conditions under the influence of potassium carbonate (0.50 g, 3.70 mmol) as a base. The mixture was refluxed for 6 h. The resulting crude product was purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield pale yellow oil (0.37 g, yield 68%): bp >300 °C (lit. 378 °C);561H NMR (400 MHz, CDCl3, TMS) δ 1.38 (q, J = 6.3 Hz, 2H), 1.46 (J = 5.7 Hz, 2H), 1.59 (q, J = 5.5 Hz. 4H), 1.66–1.80 (m, 2H), 2.71 (t, J = 6.0 Hz, 2H), 3.70 (t, J = 6.8 Hz, 2H), 7.70 (d, J = 8.1 Hz, 2H), 7.00 (d, J = 8.2 Hz, 2H), 9.80 (s, 1H).
Synthesis of 4-(2-Chloroethoxy)benzaldehyde (41)
To a well stirred solution of 4-hydroxybenzaldehyde (0.3 g, 2.45 mmol) in acetonitrile was added bromochloroethane (0.35 g, 2.45 mmol), followed by subsequent addition of potassium carbonate (0.41 g, 3.0 mmol) and potassium iodide (0.41 g, 2.45 mmol) under reflux. The reaction was allowed to run for 12 h. The reaction progress was monitored by thin layer chromatography. Upon completion, the reaction mass was diluted with water and extracted thrice with ethyl acetate (3 × 50 mL). The combined organic layers were dried over magnesium sulfate and concentrated under vacuum to afford a crude product, which was further purified by column chromatography (n-hexane/ethyl acetate 90:10) to afford compound 41 as a pale yellow oil which upon prolonged standing turned solid (0.32 g, yield 72%): mp 29–32 °C (lit. 31 °C);571H NMR (400 MHz, CDCl3, TMS) δ 3.75 (t, J = 5.6 Hz, 2H), 4.19 (t, J = 5.6 Hz, 2H), 6.90 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 9.76 (s, 1H).
Synthesis of 4-[2-(4-Methylpiperazin-1-yl)ethoxy]benzaldehyde (42)
To a suspension of potassium carbonate (0.21 g, 1.5 mmol) and potassium iodide (0.18 g, 1.1 mmol) in acetonitrile were added compound 41 (0.18 g, 1.0 mmol) and N-methylpiperazine (0.15 g, 1.5 mmol) under refluxing condition. The reaction mass was refluxed for 6 h and worked up by diluting with a small quantity of water. The diluted mass was extracted thrice with ethyl acetate (3 × 20 mL). The combined organic layers were dried over magnesium sulfate and concentrated under vacuum to obtain a crude mass. The crude product was finally purified by preparative TLC (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield compound 42 in the form of a pale yellow oil (0.17 g, yield 70%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.41 (s, 3H), 2.63–2.83 (m, 8H), 2.86 (t, J = 5.3 Hz, 2H), 4.17 (t, J = 5.9 Hz, 2H), 6.98 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H), 9.86 (s, 1H).
Synthesis of 4-(3-Chloropropoxy)benzaldehyde (43)
43 was synthesized in the same manner as that for compound 37 by using equimolar proportions of 4-hydroxybenzaldehyde (2.00 g, 16.4 mmol) and bromochloropropane (2.62 g, 16.4 mmol) using potassium carbonate (3.40 g, 24.6 mmol) as a base. The mixture was refluxed for 3 h. The crude mass obtained upon workup was purified by column chromatography employing a mobile phase of n-hexane/ethyl acetate 90:10 to yield yellow oil which eventually turned solid upon prolonged standing (2.00 g, yield 61%): mp 28–31 °C (lit. 29 °C);581H NMR (400 MHz, CDCl3, TMS) δ 2.40 (t, J = 7.4 Hz, 2H), 3.59 (t, J = 4.7 Hz, 2H), 3.99 (t, J = 6.1 Hz, 2H), 6.88 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 8.8 Hz, 2H), 9.74 (s, 1H).
Synthesis of 4-(3-Morpholin-4-ylpropoxy)benzaldehyde (44)
44 was synthesized in the same manner as that for compound 37 by using equimolar proportions of compound 43 (2.00 g, 10.1 mmol) and morpholine (0.90 g, 10.1 mmol) utilizing potassium carbonate (2.10 g, 15.15 mmol) as a base. The mixture was refluxed for 6 h and the product was purified by column chromatography (employing a mobile phase of ethyl acetate/methanol/2 M methanolic ammonia 90:10:2) to yield 44 as dark brown viscous oil (2.10 g, yield 82%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.99 (q, J = 6.5 Hz, 2H), 2.37–2.50 (m, 4H), 2.52 (t, J = 7.3 Hz, 2H), 3.62–3.78 (s, 4H), 4.10 (t, J = 6.3 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 9.85 (s, 1H).
Synthesis of 2-Chloro-1-morpholin-4-ylethanone (45)
45 was synthesized by performing diminutive changes in the reported procedure.43,44 A solution of morpholine (0.87 g, 10.0 mmol) in DCM was added dropwise to an ice cold vigorously stirred solution of equimolar proportions of chloroacetyl chloride (1.13 g, 10.0 mmol) and triethylamine (1.50 g, 15.0 mmol) in DCM. The mixture was brought to room temperature after 30 min and allowed to stir for additional 3 h. After 3 h, the reaction mass was evaporated under reduced pressure and the resulting crude product was purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2), yielding the desired product 45 as dark brown oil (1.45 g, yield 89%): bp 292–296 °C (lit. 294 °C);441H NMR (400 MHz, CDCl3, TMS) δ 3.25–3.37 (m, 4H), 3.42 (t, J = 4.8 Hz, 2H), 3.45 (t, J = 4.8 Hz, 2H), 3.89 (s, 2H).
Synthesis of 4-(2-Morpholin-4-yl-2-oxoethoxy)benzaldehyde (46)
46 was synthesized in the same manner as that for compound 37 by using equimolar proportions of 4-hydroxybenzaldehyde (2.00 g, 16.4 mmol) and 45 (2.67 g, 16.4 mmol) utilizing potassium carbonate (3.40 g, 24.6 mmol) as a base. The reaction mixture was refluxed for 6 h and the product was purified by column chromatography (employing a mobile phase of ethyl acetate/methanol/2 M methanolic ammonia 90:10:2) to yield 46 as dark brown oil (3.10 g, yield 76%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.40–3.52 (m, 4H), 3.53–3.68 (m, 4H), 5.01 (s, 2H), 7.11 (d, J = 8.6 Hz, 2H), 7.86 (d, J = 8.6 Hz, 2H), 9.87 (s, 1H).
Synthesis of 4-Oxiranylmethoxybenzaldehyde (47)
47 was synthesized in the same manner as that for compound 37 by using equimolar proportions of 4-hydroxybenzaldehyde (0.50 g, 4.09 mmol) and epibromhydrin (0.56, 4.09 mmol) utilizing potassium carbonate (0.85 g, 6.14 mmol) as a base. The reaction mass was refluxed for 6 h and the product was purified by column chromatography (utilizing a mobile phase of n-hexane/ethyl acetate 70:30) to yield compound 47 as pale brown oil (0.44 g, yield 60%): bp >300 °C (lit. 327 °C);591H NMR (400 MHz, CDCl3, TMS) δ 2.58–2.63 (m, 1H), 2.74 (t, J = 4.6 Hz, 1H), 3.17–3.25 (m, 1H), 3.78 (dd, J = 11.2, 6.2 Hz, 1H), 4.20 (dd, J = 11.2, 3.2 Hz, 1H), 6.84 (d, J = 8.5 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 9.69 (s, 1H).
Synthesis of 4-(2-Hydroxy-3-morpholin-4-ylpropoxy)benzaldehyde (48)
This compound was synthesized by the procedure used for compound 37, by alkylating morpholine (0.3 g, 3.44 mmol) with intermediate 47 (0.61 g, 3.44 mmol) utilizing potassium carbonate (0.70 g, 5.16 mmol) as a base. The reaction mass was refluxed for 6 h. It was then filtered and concentrated under vacuum. The resulting crude product was purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield compound 48 as a pale viscous brown oil (0.53 g, yield 58%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.42–2.75 (m, 6H), 3.61–3.87 (m, 5H), 4.03–4.23 (m, 3H), 7.03 (d, J = 8.7 Hz, 2H), 7.82 (d, J = 8.7 Hz, 2H), 9.86 (s, 1H).
Synthesis of 4-{2-[4-(4-Fluorophenyl)piperazin-1-yl]ethoxy}benzaldehyde (49)
49 was synthesized in the same manner as that for compound 37 by using equimolar proportions of compound 41 (0.46 g, 2.50 mmol) and 4-fluorophenylpiperazine (0.45 g, 2.50 mmol) utilizing potassium carbonate (0.52 g, 3.75 mmol) as a base. The reaction was refluxed for 48 h and the product was purified by column chromatography (utilizing a mobile phase of n-hexane/ethyl acetate 70:30) to obtain compound 49 as a pale brown oil (0.50 g, yield 61%): 1H NMR (400 MHz, CDCl3, TMS) δ 2.53–2.76 (m, 4H), 2.79 (t, J = 5.4 Hz, 2H), 2.98–3.21 (m, 4H), 4.11 (t, J = 5.4 Hz, 2H), 6.71–6.82 (m, 2H), 6.87 (t, J = 8.7 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 9.77 (s, 1H).
General Procedure for Synthesis of Compounds 50–73
To a well stirred suspension having equimolar proportions of compound 36 and the corresponding benzaldehyde (commercially available or synthesized) in toluene (3–10 mL) was added ammonium acetate (1–1.5 equiv). The reaction mass was refluxed until completion (1–24 h) and intermittently monitored using TLC. Upon completion of the reaction, the toluene was evaporated under vacuum and the ensuing mass was purified by either column chromatography or preparative TLC to obtain the desired compound. The isolated compound was washed with water and dried under vacuum to yield the title compounds 50–73.
Synthesis of (Z)-2-Benzylidene-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (50)
The title compound was prepared according to general procedure using 36 (0.07 g, 0.39 mmol), ammonium acetate (0.03 g, 0.39 mmol), and benzaldehyde (0.04 g, 0.39 mmol), and the mixture was refluxed for 2 h. It was purified by column chromatography (ethyl acetate/methanol 95:5) and obtained as a pale yellow solid (0.07 g, yield 70%): mp 212–214 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 7.05 (s, 1H), 7.24 (t, J = 7.3 Hz, 1H,), 7.40 (t, J = 7.7 Hz, 1H,), 7.50 (m, 3H), 7.87 (s, 1H), 7.95 (m, 2H), 8.06 (m, 2H). Anal. Calcd for C16H11NO3·1H2O: C, 67.84; H, 4.63; N, 4.94. Found: C, 67.56; H, 4.51; N, 4.80.
Synthesis of (Z)-3-Oxo-2(3′-phenoxybenzylidene)-2,3-dihydrobenzofuran-7-carboxamide (51)
The title compound was obtained following the general procedure using 36 (0.1 g, 0.56 mmol), ammonium acetate (0.06 g, 0.78 mmol), and 3-phenoxybenzaldehyde (0.11 g, 0.56 mmol). The reaction mass was refluxed for 24 h and purified by column chromatography (ethyl acetate/methanol 95:5) to yield pale yellow solid (0.13 g, yield 65%): mp 195–198 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 7.05 (s,1H), 7.06–7.10 (m, 3H), 7.17 (t, J = 7.4 Hz, 1H), 7.37–7.44 (m, 3H), 7.50 (t, J = 8.1 Hz, 1H), 7.64 (s, 1H), 7.79 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.91 (s, 1H), 7.93 (dd, J = 7.6, 1.4 Hz, 1H), 8.04 (dd, J = 7.6, 1.3 Hz, 1H). Anal. Calcd for C22H15NO4·1/2H2O: C, 72.12; H, 4.23; N, 3.92. Found: C, 71.98; H, 4.76; N, 3.49.
Synthesis of (Z)-2-(4′-Chlorobenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (52)
The title compound was synthesized as per the general procedure using 36 (0.1 g, 0.56 mmol), ammonium acetate (0.05 g, 0.6 mmol), and 4-chlorobenzaldehyde (0.08 g, 0.56 mmol), and the mixture was refluxed for 4 h. It was purified by column chromatography (ethyl acetate/methanol 95:5) to yield a pale yellow solid (0.11 g, yield 62%): mp 290–293 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 8.06 (d, J = 7.1 Hz, 3H), 7.94 (d, J = 7.1 Hz, 2H), 7.57 (d, J = 8.5 Hz, 2H), 7.39 (t, J = 7.6 Hz, 1H), 7.07 (s, 1H), 7.82 (s, 1H). Anal. Calcd for C16H10ClNO3: C, 64.12; H, 3.36; N, 4.67. Found: C, 64.56; H, 3.43; N, 4.30.
Synthesis of (Z)-2-(4′-Hydroxy-3′-methoxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (53)
The title compound was prepared according to the general procedure using 36 (0.1 g, 0.56 mmol), ammonium acetate (0.06 g, 0.78 mmol), and 4-hydroxy-3-methoxybenzaldehyde (0.09 g, 0.56 mmol), and the mixture was refluxed for an hour. The product obtained was purified by column chromatography (ethyl acetate/methanol 95:5) to yield bright yellow solid (0.14 g, yield 80%): mp 268–270 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.87 (s, 3H), 6.88 (d, J = 7.9 Hz, 1H,), 6.98 (s, 1H), 7.34–7.41 (m, 2H), 7.85 (s, 1H), 7.91 (d, J = 7.2 Hz, 1H,), 7.96 (s, 2H), 8.05 (d, J = 7.4 Hz, 1H,), 9.94 (s, 1H). Anal. Calcd for C17H13NO5·2/3H2O: C, 63.16; H, 4.47; N, 4.33. Found: C, 63.26; H, 4.77; N, 4.41.
Synthesis of (Z)-2-(4′-Methoxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (54)
The title compound was synthesized by following the general procedure using 36 (0.15 g, 0.84 mmol), ammonium acetate (0.07 g, 0.84 mmol), and 4-methoxybenzaldehyde (0.11 g, 0.84 mmol). The reaction mass was refluxed for 1.5 h and purified by column chromatography (ethyl acetate/methanol 95:5) to yield a bright yellow solid (0.20 g, yield 80%): mp 248–250 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.84 (s, 3H), 7.04 (s, 1H), 7.07 (d, J = 7.6 Hz, 2H), 7.37 (t, J = 7.5 Hz, 1H), 7.82 (s, 1H), 7.87–7.99 (m, 2H), 8.03 (d, J = 7.2 Hz, 3H). Anal. Calcd for C17H13NO4·1/2H2O: C, 67.10; H, 4.64; N, 4.60. Found: C, 67.29; H, 4.45; N, 4.54.
Synthesis of (Z)-2-(2′-Hydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (55)
The compound was obtained by following the general procedure using 36 (0.13 g, 0.73 mmol), ammonium acetate (0.06 g, 0.78 mmol), and 2-hydroxybenzaldehyde (0.09 g, 0.73 mmol). The mixture was refluxed for 6 h and purified by column chromatography (ethyl acetate/methanol 95:5) to yield a pale yellow solid (0.08 g, yield 56%): mp 239–241 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.92 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 7.28–7.35 (m, 2H), 7.38 (t, J = 7.5 Hz, 1H), 7.84–7.90 (m, 2H), 7.92 (d, J = 7.8 Hz, 1H), 8.04 (d, J = 7.5 Hz, 1H), 8.20 (d, J = 7.5 Hz, 1H), 10.58 (s, 1H). Anal. Calcd for C16H11NO4·1/2H2O: C, 66.20; H, 4.17; N, 4.83. Found: C, 66.29; H, 4.43; N, 4.52.
Synthesis of (Z)-2-(3′-Hydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (56)
The title compound was prepared according to the general procedure using 36 (0.10 g, 0.56 mmol), ammonium acetate (0.05 g, 0.60 mmol), and 3-hydroxybenzaldehyde (0.07 g, 0.56 mmol), and the mixture was refluxed for 4 h. It was purified by column chromatography (ethyl acetate/methanol 95:5) to obtain a pale yellow solid (0.09 g, yield 61%): mp 238–240 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.90 (d, J = 8.0 Hz, 1H), 6.94 (s, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.35–7.42 (m, 2H), 7.51 (d, J = 7.6 Hz, 1H), 7.83–7.90 (m, 2H), 7.93 (d, J = 7.6 Hz, 1H), 8.05 (d, J = 7.3 Hz, 1H), 9.75 (s, 1H). Anal. Calcd for C16H11NO4: C, 64.21; H, 4.38; N, 4.68. Found: C, 64.10; H, 4.43; N, 4.78.
Synthesis of (Z)-2-(4′-Hydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (57)
The target compound was prepared by following the general procedure using 36 (0.10 g, 0.56 mmol), ammonium acetate (0.05 g, 0.60 mmol), and 4-hydroxybenzaldehyde (0.07 g, 0.56 mmol). The reaction mass was refluxed for 4 h and purified by column chromatography (ethyl acetate/methanol 95:5) to obtain a pale yellow solid (0.10 g, yield 67%): mp 298–300 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.89 (d, J = 8.6 Hz, 2H), 6.99 (s, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.85 (s, 1H), 7.88–7.94 (m, 4H), 8.02 (d, J = 7.6 Hz, 1H), 10.35 (s, 1H). Anal. Calcd for C16H11NO4: C, 64.21; H, 4.38; N, 4.68. Found: C, 64.65; H, 4.43; N, 4.90.
Synthesis of (Z)-2-(3′,4′-Dihydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (58)
The title compound was synthesized according to the general procedure using 36 (0.07 g, 0.39 mmol), ammonium acetate (0.05 g, 0.60 mmol), and 3,4-dihydroxybenzaldehyde (0.05 g, 0.39 mmol), and the mixture was refluxed for 4 h. The reaction mixture was purified by column chromatography (ethyl acetate/methanol 95:5) to obtain a dark brown solid (0.17 g, yield 56%): mp 257–259 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.81–6.94 (m, 2H), 7.30–7.49 (m, 2H), 7.77–7.88 (m, 2H), 7.90 (d, J = 7.5 Hz, 1H), 8.02 (d, J = 7.3 Hz, 1H), 9.23 (bs, 1H), 9.92 (bs, 1H). Anal. Calcd for C16H11NO5·1H2O: C, 61.15; H, 3.83; N, 4.46. Found: C, 61.52; H, 3.91; N, 5.14.
Synthesis of (Z)-2-(2′,4′-Dihydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (59)
The target compound was prepared according to the general procedure using 36 (0.07 g, 0.39 mmol), ammonium acetate (0.03 g, 0.39 mmol), and 2,4-dihydroxybenzaldehyde (0.05 g, 0.39 mmol). The reaction lasted for almost 10 h and the mixture was purified by column chromatography (ethyl acetate/methanol 95:5) to obtain a brick red solid (0.19 g, yield 65%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.81–6.94 (m, 2H), 7.30–7.49 (m, 2H), 7.77–7.88 (m, 2H), 7.90 (d, J = 7.5 Hz, 1H), 8.02 (d, J = 7.3 Hz, 1H), 9.23 (bs, 1H), 9.92 (bs, 1H). Anal. Calcd for C16H11NO5: C, 64.65; H, 3.73; N, 4.71. Found: C, 64.76; H, 3.98; N, 4.60.
Synthesis of (Z)-2-(3′,5′-Dihydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (60)
The title compound was synthesized according to the general procedure using 36 (0.07 g, 0.39 mmol), ammonium acetate (0.03 g, 0.39 mmol), and 3,5-dihydroxybenzaldehyde (0.05 g, 0.39 mmol). The mixture was refluxed for 18 h and the title compound was purified by column chromatography (ethyl acetate/methanol 95:5) to obtain a light brown solid (0.19 g, yield 65%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.39 (s, 1H), 6.81 (s, 1H), 6.87 (s, 2H), 7.38 (t, J = 7.4 Hz, 1H), 7.76 (s, 1H), 7.87 (s, 1H), 7.92 (d, J = 6.8 Hz, 1H), 8.05 (d, J = 7.6 Hz, 1H), 9.54 (s, 2H). Anal. Calcd for C16H11NO5: C, 64.65; H, 3.73; N, 4.71. Found: C, 64.52; H, 3.43; N, 5.14.
Synthesis of (Z)-2-(2′,5′-Dihydroxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (61)
The target compound was synthesized following the general procedure using 36 (0.07 g, 0.39 mmol), ammonium acetate (0.03 g, 0.39 mmol), and 2,5-dihydroxybenzaldehyde (0.05 g, 0.39 mmol), and the mixture was refluxed for 21 h. The title compound was purified by column chromatography (ethyl acetate/methanol 95:5) to obtain a dark brown solid (0.16 g, yield 55%): mp >300 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 6.78 (s, 1H), 6.80 (s, 1H), 7.25 (s, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.54 (s, 1H), 7.78 (s, 1H), 7.87 (s, 1H), 7.92 (d, J = 7.5 Hz, 1H), 8.05 (d, J = 7.5 Hz, 1H), 9.04 (s, 1H), 9.85 (s, 1H). Anal. Calcd for C16H11NO5: C, 64.65; H, 3.73; N, 4.71. Found: C, 64.90; H, 4.03; N, 4.59.
Synthesis of (Z)-2-[3′-(2-Morpholin-4-ylethoxy)benzylidene]-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (62)
The title compound was synthesized according to the general procedure using 36 (0.07 g, 0.39 mmol), ammonium acetate (0.03 g, 0.39 mmol), and compound 37 (0.09 g, 0.39 mmol), and the mixture was refluxed for 6 h and purified by flash chromatography (employing a mobile phase of ethyl acetate/methanol/2 M methanolic ammonia 90:10:2) to yield a bright yellow solid (0.11 g, yield 68%): mp 189–193 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.55–2.62 (m, 4H), 2.75 (t, J = 5.6 Hz, 2H), 3.59 (t, J = 4.0 Hz, 4H), 4.18 (t, J = 5.6 Hz, 2H), 7.02 (s, 1H), 7.06 (d, J = 8.4 Hz, 1H), 7.40 (t, J = 7.8 Hz, 2H), 7.59 (d, J = 7.5 Hz, 1H), 7.72 (s, 1H), 7.86 (s, 1H), 7.93 (d, J = 8.3 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 8.07 (d, J = 7.4 Hz, 1H). Anal. Calcd for C22H22N2O5·3/4H2O: C, 64.77; H, 5.81; N, 6.87: found: C, 65.12; H, 5.74; N, 6.90.
Synthesis of (Z)-3-Oxo-2-[3′-(2-piperidine-1-ylethoxy)benzylidene]-2,3-dihydrobenzofuran-7-carboxamide (63)
The target compound was prepared by following the general procedure using 36 (0.09 g, 0.50 mmol), ammonium acetate (0.04 g, 0.50 mmol), and compound 38 (0.12 g, 0.50 mmol). The mixture was refluxed for 4 h and purified by flash chromatography (employing a mobile phase of ethyl acetate/methanol/2 M methanolic ammonia 90:10:2) to obtain bright yellow solid (0.12 g, yield 61%): mp 187–191 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.09–1.28 (m, 2H), 1.33–1.58 (m, 8H), 2.74 (t, J = 5.4 Hz, 2H), 4.18 (t, J = 5.6 Hz, 2H), 7.03 (s, 1H), 7.05 (d, J = 8.0 Hz, 1H), 7.40 (t, J = 8.0 Hz, 2H), 7.60 (d, J = 6.9 Hz, 1H), 7.71 (s, 1H), 7.92 (s,1H), 7.96 (d, J = 8.0 Hz, 2H), 8.06 (d, J = 6.9 Hz, 1H). Anal. Calcd for C23H24N2O4: C, 70.39; H, 6.16; N, 7.14. Found: C, 70.26; H, 6.39; N, 7.49.
Synthesis of (Z)-3-Oxo-2-[4′-(2-piperidine-1-ylethoxy)benzylidene]-2,3-dihydrobenzofuran-7-carboxamide (64)
The title compound was synthesized according to the general procedure using 36 (0.11 g, 0.60 mmol), ammonium acetate (0.05 g, 0.60 mmol), and 40 (0.14 g, 0.60 mmol). The reaction mixture was refluxed for 10 h and purified by flash chromatography (employing a mobile phase of ethyl acetate/methanol/2 M methanolic ammonia 90:10:5) to yield a bright yellow solid (0.13 g, yield 66%): mp 210–213 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.33–1.41 (m, 2H), 1.49 (q, J = 5.6 Hz, 4H), 2.43 (s, 4H), 2.67 (t, J = 5.4 Hz, 2H), 4.16 (t, J = 5.9 Hz, 2H), 7.03 (s, 1H), 7.06 (d, J = 8.7 Hz, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.82 (s, 1H), 7.91 (d, J = 7.3 Hz, 1H), 7.93 (s, 1H), 8.01 (d, J = 8.3 Hz, 2H), 8.04 (s, 1H). Anal. Calcd for C23H24N2O4·1/2H2O: C, 68.81; H, 6.28; N, 6.98. Found: C, 68.96; H, 6.28; N, 6.98.
Synthesis of (Z)-2-[4′-(2-Morpholin-4-ylethoxy)benzylidene]-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (65)
The title compound was prepared according to the general procedure using 36 (0.12 g, 0.70 mmol), ammonium acetate (0.06 g, 0.70 mmol), and compound 39 (0.17 g, 0.70 mmol), and the mixture was refluxed for 5 h. It was purified by flash chromatography (employing a mobile phase of ethyl acetate/methanol/2 M methanolic ammonia 90:10:2) to obtain a bright yellow solid (0.17 g, yield 64%): mp 216–220 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.46–2.48 (m, 4H), 2.71 (t, J = 5.3 Hz, 2H), 3.58 (t, J = 4.4 Hz, 4H), 4.19 (t, J = 5.5 Hz, 2H), 7.03 (s, 1H), 7.08 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.82 (s, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.94 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 8.04 (s, 1H). Anal. Calcd for C22H22N2O5·1/2H2O: C, 65.50; H, 5.75; N, 6.94: found: C, 65.73; H, 5.74; N, 6.90.
Synthesis of (Z)-2-{4′-[2-(4-Methylpiperazin-1-yl)ethoxy]benzylidene}-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (66)
The target compound was synthesized following the general procedure using 36 (0.08 g, 0.45 mmol), ammonium acetate (0.03 g, 0.45 mmol), and 42 (0.13 g, 0.45 mmol), and the mixture was refluxed for 12 h. It was purified by preparative TLC (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield compound 66 as a yellowish brown solid (0.11 g, yield 62%): mp 210–212 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.24 (s, 3H), 2.30–2.47 (m, 5H), 2.52–2.66 (m, 3H), 2.72 (t, J = 5.1 Hz, 2H), 4.17 (t, J = 5.4 Hz, 2H), 7.03 (s, 1H), 7.07 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 7.5 Hz, 1H), 7.82 (s, 1H), 7.91 (d, J = 7.9 Hz, 1H), 7.94 (s, 1H), 7.99–8.06 (m, 3H). Anal. Calcd for C23H25N3O4·1/2H2O: C, 66.40; H, 6.29; N, 10.09. Found: C, 66.76; H, 6.55; N, 9.74.
Synthesis of (Z)-2-[4′-(3-Morpholin-4-ylpropoxy)benzylidene]-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (67)
The target compound was prepared as per the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and 44 (0.55 g, 2.20 mmol). The reaction mass was refluxed for 4 h and purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield compound 67 as a bright yellow solid (0.61 g, yield 68%): mp 186–190 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.91 (q, J = 7.0 Hz, 2H), 2.32–2.40 (m, 4H), 2.43 (t, J = 7.4 Hz, 2H), 3.58 (t, J = 4.3 Hz, 4H), 4.12 (t, J = 6.0 Hz, 2H), 7.04 (s, 1H), 7.06 (d, J = 8.0 Hz, 2H), 7.38 (t, J = 7.6 Hz, 1H), 7.83 (s, 1H), 7.92 (d, J = 8.2 Hz, 1H), 7.95 (s, 1H), 7.99–8.06 (m, 3H). Anal. Calcd for C23H24N2O5·1/2H2O: C, 66.17; H, 6.04; N, 6.71. Found: C, 65.91; H, 6.01; N, 6.69.
Synthesis of (Z)-2-[4′-(2-Morpholin-4-yl-2-oxoethoxy)benzylidene]-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (68)
The target compound was synthesized according to the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and compound 46 (0.55 g, 2.20 mmol), and the mixture was refluxed for 15 h and purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield compound 68 as a yellowish brown solid (0.68 g, yield 76%): mp 258–260 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.41–3.51 (m, 4H), 3.51–3.68 (m, 4H), 4.98 (s, 2H), 7.04 (s, 1H), 7.06 (d, J = 7.8 Hz, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.83 (s, 1H), 7.90–8.06 (m, 5H). Anal. Calcd for C22H20N2O6: C, 64.70; H, 4.94; N, 6.86. Found: C, 65.06; H, 4.81; N, 6.80.
Synthesis of (Z)-2-(4′-Oxiranylmethoxybenzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (69)
The title compound was obtained according to the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and 47 (0.39 g, 2.20 mmol). The reaction mixture was refluxed for 8 h and purified by column chromatography (employing a mobile phase of n-hexane/ethyl acetate 60:40) to yield compound 69 as a yellowish brown solid (0.44 g, yield 59%): mp 167–170 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.71–2.76 (m, 1H), 2.88 (t, J = 4.6 Hz, 1H), 3.36–3.39 (m, 1H), 3.94 (dd, J = 11.2, 4.2 Hz, 1H), 4.46 (dd, J = 11.2, 3.2 Hz, 1H), 7.05 (s, 1H), 7.11 (d, J = 8.8 Hz, 2H), 7.39 (t, J = 8.2 Hz, 1H), 7.84 (s, 1H), 7.93 (d, J = 7.1 Hz, 2H), 7.96 (s, 1H), 8.04 (d, J = 8.2 Hz, 2H). Anal. Calcd for C19H15NO5: C, 67.65; H, 4.48; N, 4.15. Found: C, 67.41; H, 4.57; N, 4.13.
Synthesis of (Z)-2-[4′-(2-Hydroxy-3-morpholin-4-ylpropoxy)benzylidene]-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (70)
The target compound was prepared by following the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and compound 48 (0.58 g, 2.20 mmol). The reaction mass was refluxed for 18 h and purified by column chromatography (employing a mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield 70 as a yellowish brown solid (0.48 g, yield 52%): mp 210–212 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.34–2.47 (m, 6H), 3.57 (t, J = 4.3 Hz, 4H), 3.93–4.05 (m, 2H), 4.10 (d, J = 7.2 Hz, 1H), 4.97 (s, 1H), 7.04 (s, 1H), 7.08 (d, J = 8.5 Hz, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.83 (s, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.95 (s, 1H), 8.00–8.07 (m, 3H). Anal. Calcd for C23H24N2O6·1H2O: C, 62.43; H, 5.92; N, 6.33: found: C, 62.19; H, 5.74; N, 6.59.
Synthesis of (Z)-2-(4′-{2-[4-(4-Fluorophenyl)piperazin-1-yl]ethoxy}benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (71)
The title compound was prepared as per the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and 49 (0.72 g, 2.20 mmol), and the mixture was refluxed for 8 h and purified by column chromatography employing a mobile phase of n-hexane/ethyl acetate 50:50 to yield compound 71 as a bright yellow solid (0.62 g, yield 58%): mp 206–208 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.65 (t, J = 4.9 Hz, 4H), 2.79 (t, J = 5.8 Hz, 2H), 3.09 (t, J = 4.4 Hz, 4H), 4.24 (t, J = 6.0 Hz, 2H), 6.90–7.13 (m, 8H), 7.38 (t, J = 7.7 Hz, 1H), 7.83 (s, 1H), 7.92 (d, J = 7.1 Hz, 1H), 7.95 (s, 1H), 8.00–8.03 (m, 2H). Anal. Calcd for C28H26FN3O4·1H2O: C, 65.55; H, 5.40; N, 8.19. Found: C, 65.15; H, 5.62; N, 7.98.
Synthesis of (Z)-2-{4-[2-(4-Methylpiperazin-1-yl)ethanesulfonylamino]benzylidene}-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (72)
Compound 72 was prepared by following the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and compound 72d (0.68 g, 2.20 mmol). The reaction mass was refluxed for 6 h and purified by column chromatography (mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield 72 as a yellowish brown solid (0.55 g, 52%): mp 265–268 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.88 (s, 3H), 2.43–2.47 (m, 4H), 2.59–2.68 (m, 4H), 3.16 (t, J = 7.1 Hz, 2H), 4.02 (t, J = 7.7 Hz, 2H), 7.01 (s, 1H), 7.11 (d, J = 8.1 Hz, 2H), 7.37 (t, J = 8.0 Hz, 1H), 7.80–7.93 (m, 4H), 7.96–8.16 (m, 3H). Anal. Calcd for C23H26N4O5S·1.5H2O: C, 57.25; H, 5.71; N, 11.61. Found: C, 57.51; H, 5.62; N, 11.87.
Synthesis of (Z)-2-(4-(3-(4-Methylpiperazin-1-yl)propylsulfonamido)benzylidene)-3-oxo-2,3-dihydrobenzofuran-7-carboxamide (73)
The title compound was prepared by following the general procedure using 36 (0.39 g, 2.20 mmol), ammonium acetate (0.15 g, 2.20 mmol), and compound 72e (0.71 g, 2.20 mmol). The reaction mass was refluxed for 8 h and purified by column chromatography (mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield 73 as a yellowish brown solid (0.60 g, 55%): mp 287–290 °C; 1H NMR (400 MHz, DMSO-d6, TMS) δ 1.85 (s, 3H), 2.36–2.47 (m, 4H), 3.29–3.41 (m, 4H), 3.60 (q, J = 7.6 Hz, 2H), 3.84 (t, J = 7.4 Hz, 2H), 4.13 (t, J = 7.1 Hz, 2H), 7.03 (s, 1H), 7.24–7.32 (m, 2H), 7.39 (t, J = 8.2 Hz, 1H), 7.84 (s, 1H), 7.86–7.98 (m, 3H), 8.06 (d, J = 8.3 Hz, 3H). Anal. Calcd for C24H28N4O5S·1H2O: C, 57.36; H, 6.02; N, 11.15. Found: C, 57.46; H, 5.75; N, 11.46.
Synthesis of 4-Aminobenzaldehyde (72a)
72a was synthesized by following the same protocol as that of compound 6 by treating 4-nitrobenzaldehyde (1.0 g, 6.6 mmol) with tin chloride dihydrate (4.5 g, 19.8 mmol) in refluxing ethyl acetate for 4 h. The reaction was worked up as per the above-mentioned procedure (for compound 6) and the sample purified by column chromatography (n-hexane/ethyl acetate 50:50) to yield compound 72a as pale yellow solid (0.46 g, yield 58%): mp 69–72 °C (lit. 70–72 °C);601H NMR (400 MHz, DMSO-d6, TMS) δ 5.91 (s, 2H), 6.78 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 9.76 (s,1H).
Synthesis of 2-Chloro-N-(4-formylphenyl)ethanesulfonamide (72b)
72b was synthesized by following the same procedure as for compound 45 by using equimolar proportions of 72a (1.21 g, 10.0 mmol), chloroethanesulfonyl chloride (1.63 g, 10.0 mmol), and triethylamine (1.0 g, 10.0 mmol). The mixture was allowed to stir at room temperature for 6 h and the product was purified by column chromatography (n-hexane/ethyl acetate 80:20) to yield compound 72b as a pale brown solid (1.70 g, 70%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 3.35 (t, J = 7.1 Hz, 2H), 3.52 (t, J = 7.0 Hz, 2H), 7.45 (d, J = 8.8 Hz, 2H), 7.72 (s, 1H), 8.05 (d, J = 8.9 Hz, 2H), 9.95 (s, 1H).
Synthesis of 3-Chloro-N-(4-formylphenyl)propane-1-sulfonamide (72c)
The title compound was synthesized in the same way as that of compound 45 using equimolar proportions of 72a (1.21 g, 10.0 mmol), chloropropanesulfonyl chloride (1.77 g, 10.0 mmol), and triethylamine (1.0 g, 10.0 mmol). The mixture was allowed to stir at room temperature for 6 h and the product was purified by column chromatography (n-hexane/ethyl acetate 80:20) to yield compound 72c as a white solid (1.69 g, 65%): 1H NMR (400 MHz, CDCl3, TMS) δ 3.78 (q, J = 7.1 Hz, 2H), 3.95 (t, J = 7.2 Hz, 2H), 4.35 (t, J = 7.1 Hz, 2H), 7.31 (d, J = 7.5 Hz, 2H), 7.80 (s, 1H), 8.09 (d, J = 7.5 Hz, 2H), 9.90 (s, 1H).
Synthesis of N-(4-Formylphenyl)-2-(4-methylpiperazin-1-yl)ethanesulfonamide (72d)
72d was obtained in the same way as that of compound 37 using equimolar proportions of 72b (1.20 g, 5.0 mmol), N-methylpiperazine (0.5 g, 5.0 mmol), and potassium carbonate (1.0 g, 7.5 mmol) when refluxed for 6 h, and the product was purified by column chromatography (mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield 72d as a pale brown solid (0.96 g, 62%): 1H NMR (400 MHz, DMSO-d6, TMS) δ 2.10 (s, 3H), 2.67–2.89 (m, 6H), 3.15–3.21 (m, 4H), 3.46 (t, J = 7.1 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H), 7.69 (s, 1H), 7.90 (d, J = 8.9 Hz, 2H), 9.91 (s, 1H).
Synthesis of N-(4-Formylphenyl)-3-(4-methylpiperazin-1-yl)propane-1-sulfonamide (72e)
Compound 72e was obtained in the same way as that of compound 37 using equimolar proportions of 72c (2.61 g, 10.0 mmol), N-methylpiperazine (1.0 g, 10.0 mmol), and potassium carbonate (2.1 g, 15.0 mmol) when refluxed for 6 h, and the product was purified by column chromatography (mobile phase of DCM/methanol/2 M methanolic ammonia 90:10:2) to yield 72e as a pale brown solid (1.75 g, 54%): 1H NMR (400 MHz, CDCl3, TMS) δ 1.73–1.95 (m, 4H), 2.06–2.40 (m, 7H), 3.62 (q, J = 7.2 Hz, 2H), 3.85 (t, J = 7.5 Hz, 2H), 4.12 (t, 7.3 Hz, 2H), 7.33 (d, J = 7.5 Hz, 2H), 7.69 (s, 1H), 7.92 (d, J = 7.5 Hz, 2H), 9.90 (s, 1H).
Biological Methods. PARP-1 Inhibition Assay
Inhibitory activity of the synthesized PARP-1 inhibitors was determined using a commercially available 96-well assay kit (catalog no. 4677-096-K; universal colorimetric PARP assay kit from Trevigen, Inc., Gaithersburg, MD) according to the instructions provided by the manufacturer (with diminutive modifications) and our prior reports.21,22 Briefly, the stock solutions of the various test compounds were made in dimethylsulfoxide (DMSO) and then serially diluted with distilled water to the required concentrations. In the beginning the strip wells were rehydrated by adding 50 μL of 1× PARP buffer into each well followed by incubation for 30 min. After 30 min, the microplate strip plate was tapped a few times upside down onto the paper towels. Care was taken to avoid the splashing of the sample back into wells. The paper towels were discarded after each tapping to ensure thorough drying before proceeding with the assay. In the first phase of the assay, the ribosylation reaction was initiated into the wells by adding 10 μL of the inhibitor solution, 15 μL of diluted PARP-1 enzyme (providing 0.5 unit/well), and 25 μL of PARP cocktail (containing of biotinylated NAD+, activated DNA in Tris-Cl, pH 8.0, and EDTA). They were serially added into each well (PARP buffer was added instead of the inhibitor solution and enzyme solution in the case of the blank reading, while PARP buffer was added instead of the inhibitor solution in the case of the negative control). 3-Aminobenzamide (3-AB), veliparib, and olaparib were used as positive controls. The strip wells were then incubated at room temperature for 60 min and then washed twice with 200 μL of a 0.1% Triton solution in phosphate buffered saline (PBS: Na2HPO4, NaH2PO4, and NaCl) and twice with 200 μL of PBS. The wells were dried thoroughly as mentioned before and then a 50 μL of diluted Strep-HRP (containing a mixture of 1× Strep diluent and Strep-HRP enzyme in distilled water, respectively) was added to each well, and the strips were further incubated at room temperature for 60 min to detect the degree of ribosylation. After the wells were washed twice with 200 μL of 0.1% Triton solution in PBS and 200 μL of PBS, the wells were dried and 50 μL of TACS-Sapphire colorimetric substrate was added to each well and allowed to stand in the dark for 10–15 min. The intensity of the blue color that developed in each well was measured on a microplate reader at 630 nm. Reaction was stopped by the addition of 50 μL of 5% phosphoric acid to each well, and the absorbance was measured at 450 nm. All samples were tested in triplicate. Compounds that inhibited PARP-1 activity by ≥50% at a concentration of 25 μM (in the case of DHBF-7-carboxamide derivatives) and 10 μM (in the case of DHBF-3-one-7-carboxamide derivatives) were further tested to obtain IC50 values. To determine the IC50 value for each inhibitor, dose–response curves were generated with five to six different concentrations, averaging absorbance values of each inhibitor concentration (values correlated after deducting the blank reading). The data were plotted against the log of the concentration of each respective inhibitor (semilog plot), and the IC50 value for each plot was obtained using regression wizard from Sigma Plot, version 10.0 (San Jose, CA). Data presented are the results of at least two independent experiments done in triplicate. The results of these studies are presented as the mean ± standard deviation (SD).
Cell Lines
Chicken DT40 B cell lines used in this study were obtained from the laboratory of Dr. Takeda; Laboratory of Radiation Genetics, Graduate School of Medicine, Kyoto University (Kyoto, Japan).
Measurement of Cellular Sensitivity to Drugs
To measure drug cytotoxicity, cells were continuously exposed to various drug concentrations for 72 h in triplicate. Two-hundred DT40 cells were seeded into 384-well white plate (no. 6007680 PerkinElmer Life Sciences) in 40 μL of medium per well. Cell viability was determined using the ATPlite one-step kit (PerkinElmer). A 20 μL ATPlite solution was added to each well. After 5 min, luminescence was measured by EnVision 2104 multilabel reader (PerkinElmer). The ATP level in untreated cells was defined as 100%. Percent viability of treated cells was defined as (ATP level of treated cell/ATP level of untreated cells) × 100. Further information on this assay is described in ref (61).
Molecular Modeling
Computational work was carried out on a Dell Precision 490 dual processor workstation with the Linux operating system (Ubuntu 12.04 LTS). The initial protein structure of PARP-1 bound with benzoxazinone inhibitor (PDB code 4L6S)32 was refined by default parameters in Protein Preparation Wizard implemented in Maestro, version 9.5, and the Impact program, version 6.0 (Schrödinger, LLC, New York, NY, 2013), in which the protonation states of the ionizable residues were treated to their dominant ionic forms at physiological pH. Refined PARP structure was further used to generate a defined grid centered on a bound benzoxazinone inhibitor. Proposed and synthesized inhibitors were docked using the bound inhibitor grid. The inhibitory structures were built using the build tool in Maestro, version 9.5, and energy-minimized by the Macromodel program, version 10.1 (Schrödinger, LLC., New York, NY, 2013), using the steepest descent followed by truncated Newton conjugate gradient protocol. LigPrep tool, version 2.7, was used to obtain the low-energy 3D structures of all inhibitors. Default settings were used, but the “Generate Tautomers” option was not selected. Resultant inhibitor structures were docked within the active site of PARP-1 using the “Extra Precision” (XP) mode of Glide docking program, version 6.0, and the default parameters. The images were generated using PYMOL, version 1.6.
Cloning and Protein Production
The Zn1/Zn3 (a fusion of residues 1–96 and residues 207–366) and the WGR-CAT (residues 518–1014) fragments of PARP-1 used for crystallography were expressed from the pET24 expression vector with a C-terminal hexahistidine tag as previously described.62 PARP-1 proteins were expressed in Escherichia coli and purified as previously described using a three-column purification protocol.63
Crystallization of the PARP-1/DNA Complex
Crystals of the activated PARP-1 complex were grown as previously published with minor modification.23 In brief, crystals were grown in sitting drop vapor diffusion at 20 °C in 3.0 μL droplets that combined a 2.1 μL mixture of protein (300 μM Zn1/Zn3 and WGR-CAT in 25 mM Hepes, pH 8.0, 150 mM NaCl, 1 mM EDTA, and 0.1 mM TCEP) with 475 μM 26-bp palindromic DNA duplex (5′ GCCTACCGGTTCGCGAACCGGTAGGC 3′) and 0.9 μL of well solution (6.8% PEG 3350, 2% ethylene glycol, and 100 mM Hepes, pH 7.5). Crystals were grown to their largest size after approximately 3–4 weeks, upon which they were collected and soaked with inhibitors by hanging drop diffusion at 22 °C in 7 μL droplets consisting of 5 mM inhibitor in well solution (6.5% PEG 3350, 1.9% ethylene glycol, 5% DMSO, 75 mM NaCl, 0.5 mM EDTA, 0.05 mM TCEP, and 95 mM Hepes, pH 7.5). After 1–7 days of soaking, crystals were quickly transferred to a cryo solution (3% PEG 3350, 25% ethylene glycol, 25 mM NaCl, 0.05 mM TCEP, 25 mM Hepes, pH 7.5, and 5 mM inhibitor in 5% DMSO) prior to flash-cooling in liquid nitrogen. X-ray diffraction data were collected at beamline X29A of the National Synchrotron Light Source (NSLS, Brookhaven National Laboratory), and processed using Xia2 (Supporting Information Table S1).64
Structure Determination and Refinement
The structure of activated PARP-1 (PDB code 4DQY)23 was used as a starting model. The weighted Fo – Fc electron density maps calculated after rigid body refinement in REFMAC5 from the CCP4 suite resulted in significant difference density in one catalytic domain of the two present in the asymmetric unit, which was used to position the inhibitor. As with the original structure, the second copy of the PARP-1 complex is not as well-defined, presumably because of flexibility. Thus, the bound compounds were only modeled in one copy of the catalytic domain. The inhibitors were placed according to their benzamide portion of DHBF scaffold, which has a well-established binding site. The constructed model was refined in REFMAC5 using restrained and TLS refinement.65 The refined models exhibit good geometry and agree well with the original structure 4DQY, and the bound compounds are well represented in the final electron density maps (Figure 3). Coordinates and structure factors have been deposited in the Protein Data Bank (see Table S1 for the PDB entries).
Absolute Configuration Determination for 12a
The absolute structure of (R)-(−)-12a with (S)-(−)-α-methylbenzylamine was confirmed based on 1215 Bijvoet pairs measured with Cu Kα radiation on a Bruker Kappa Apex-II DUO diffractometer. Refinement of the Flack parameter66 resulted in X = −0.12(15), and the Hooft parameter67 was Y = −0.09(7), corresponding to a probability of 1.000 that the configuration shown in Figure 2 is correct, in agreement with the known (S)-configuration of the amine. (CCDC 977479; full details are in Supporting Information Tables S2–S8).
Acknowledgments
This research was supported by the Department of Pharmaceutical Sciences of St. John’s University and St. John’s University Seed Grant 579-1110 to T.T.T. and by funds from the NIH (Grant R01 GM087282) to J.M.P. J.D.S. is supported by a Ruth L. Kirschstein National Research Service Award. Y.P., A.C. and J.M. were supported by the Intramural Program, Center for Cancer Research, National Cancer Institute, NIH (Grant Z01 BC006150).
Glossary
Abbreviations Used
- 3-AB
3-aminobenzamide
- ATP
adenosine triphosphate
- AUC
area under the curve
- BER
base excision repair
- BRCA
breast cancer
- CCDC
Cambridge Crystallographic Data Centre
- CCP4
Collaborative Computing Project Number 4
- DHBF
dihydrobenzofuran
- DMF
N,N-dimethylformamide
- DMSO
dimethylsulfoxide
- EDTA
ethylenediaminetetraacetic acid
- Hepes
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high performance liquid chromatography
- HR
homologous recombination
- NAD+
nicotinamide adenine dinucleotide
- NMM
N-methylmorpholine
- PAR
poly(ADP-ribose)
- PARG
poly(ADP-ribose)glycohydrolase
- PARP
poly(ADP-ribose)polymerase
- PBS
phosphate buffered saline
- PDB
Protein Data Bank
- PEG
polyethylene glycol
- PTEN
phosphatase and tensin homologue
- SAR
structure–activity relationship
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- TEMED
N,N,N′,N′-tetramethylethylenediamine
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- TLS
translation libration screw-motion
- TMS
tetramethylsilane
- XRCC
X-ray cross-complementary protein
Supporting Information Available
Summary of the cocrystal data of compounds (−)-13c, 59, and 65 (Table S1) and crystallographic data of (R)-(−)-12a (Table S2–S8). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ M.R.P. and A.B. contributed equally to this work.
⊥ All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Gibson B. A.; Kraus W. L. New insights into the molecular and cellular functions of poly(ADPribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [DOI] [PubMed] [Google Scholar]
- Luo X.; Kraus W. L. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankevicius G.; Hassler M.; Golia B.; Rybin V.; Zacharias M.; Timinszky G.; Ladurner A. G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013, 20, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O.; Hassa P. O.; Luscher B.; Schuler H.; Koch-Nolte F. Toward a unified nomenclature for mammalian ADP ribosyl transferases. Trends Biochem. Sci. 2010, 35, 208–219. [DOI] [PubMed] [Google Scholar]
- Rouleau M.; Patel A.; Hendzel M. J.; Kaufmann S. H.; Poirier G. G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D. A.; Frizzell K. M.; Zhang T.; Kraus W. L. Regulation of chromatin structure and chromatin dependent transcription by poly(ADP-ribose) polymerase-1: possible targets for drug-based therapies. Subcell. Biochem. 2007, 41, 45–69. [DOI] [PubMed] [Google Scholar]
- El-Khamisy S. F.; Masutani M.; Suzuki H.; Caldecott K. W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L.; Nakajima S.; Oohata Y.; Takao M.; Okano S.; Masutani M.; Wilson S. H.; Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano S.; Lan L.; Tomkinson A. E.; Yasui A. Translocation of XRCC-1 and DNA ligase III-alpha from centrosomes to chromosomes in response to DNA damage in mitotic human cells. Nucleic Acids Res. 2005, 33, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L.; Denegri M.; Torti M.; Poirier G. G.; Scovassi A. I. Poly(ADP-ribose) degradation by post-nuclear extracts from human cells. Biochimie 2002, 84, 1229–1235. [DOI] [PubMed] [Google Scholar]
- Mangerich A.; Burkle A. How to kill tumor cells with inhibitors of poly(ADP-ribosyl)ation. Int. J. Cancer 2011, 128, 251–265. [DOI] [PubMed] [Google Scholar]
- Penning T.; Zhu G.; Gandhi V. B.; Gong J.; Liu X.; Shi Y.; Klinghofer V.; Johnson E. F.; Donawho C. K.; Frost D. J.; Bontcheva-Diaz V.; Bouska J. J.; Osterling D. J.; Olson A. M.; Marsh K. C.; Luo Y.; Giranda V. L. Discovery of the poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J. Med. Chem. 2009, 52, 514–523. [DOI] [PubMed] [Google Scholar]
- Bryant H. E.; Schultz N.; Thomas H. D.; Parker K. M.; Flower D.; Lopez E.; Kyle S.; Meuth M.; Curtin N. J.; Helleday T. Specific killing of BRCA2-deficient tumors with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Farmer H.; McCabe N.; Lord C. J.; Tutt A. N. J.; Johnson D. A.; Richardson T. B.; Santarosa M.; Dillon K. J.; Hickson I.; Knights C.; Martin N. M. B.; Jackson S. P.; Smith G. C. M.; Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Kummar S.; Chen A.; Parchment R. E.; Kinders R. J.; Ji J.; Tomaszewski J. E.; Doroshow J. H. Advances in using PARP inhibitors to treat cancer. BMC Med. 2012, 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin N. J.; Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol. Aspects Med. 2013, 34, 1217–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papeo G.; Casale E.; Montagnoli A.; Cirla A. PARP inhibitors in cancer therapy: an update. Expert Opin. Ther. Pat. 2013, 23, 503–514. [DOI] [PubMed] [Google Scholar]
- Underhill C.; Toulmonde M.; Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann. Oncol. 2011, 22, 268–279. [DOI] [PubMed] [Google Scholar]
- Turner N.; Tutt A.; Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [DOI] [PubMed] [Google Scholar]
- Mendes-Pereira A. M.; Martin S. A.; Brough R.; McCarthy A.; Taylor J. R.; Kim J. S.; Waldman T.; Lord C. J.; Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M. R.; Pandya K. G.; Lau-Cam C. A.; Singh S.; Pino M. A.; Billack B.; Degenhardt K.; Talele T. T. Design and synthesis of N-substituted indazole-3-carboxamides as poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors. Chem. Biol. Drug Des. 2012, 79, 488–496. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. S.; Singh S.; Shah J. R.; Low W. K.; Talele T. T. Synthesis and SAR optimization of quinazoline-4(3H)-ones as poly(ADP-ribose)polymerase-1 inhibitors. Eur. J. Med. Chem. 2012, 50, 264–273. [DOI] [PubMed] [Google Scholar]
- Langelier M. F.; Plank J. L.; Roy S.; Pascal J. M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris D. V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [DOI] [PubMed] [Google Scholar]
- Steffen J. D.; Brody J. R.; Armen R. S.; Pascal J. M. Structural implications for selective targeting of PARPs. Front. Oncol. 2013, 3, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donawho C. K.; Luo Y.; Penning T. D.; Bauch J. L.; Bouska J. J.; Bontcheva-Diaz V. D.; Cox B. F.; DeWeese T. L.; Dillehay L. E.; Ferguson D. C.; Ghoreishi-Haack N. S.; Grimm D. R.; Guan R.; Han E. K.; Holley-Shanks R. R.; Hristov B.; Idler K. B.; Jarvis K.; Johnson E. F.; Kleinberg L. R.; Klinghofer V.; Lasko L. M.; Liu X.; Marsh K. C.; McGonigal T. P.; Meulbroek J. A.; Olson A. M.; Palma J. P.; Rodriguez L. E.; Shi Y.; Stavropoulos J. A.; Tsurutani A. C.; Zhu G. D.; Rosenberg S. H.; Giranda V. L.; Frost D. J. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [DOI] [PubMed] [Google Scholar]
- Jones P.; Altamura S.; Boueres J.; Ferrigno F.; Fonsi M.; Giomini C.; Lamartina S.; Monteagudo E.; Ontoria J. M.; Orsale M. V.; Palumbi M. C.; Pesci S.; Roscilli G.; Scarpelli R.; Schultz-Fademrecht C.; Toniatti C.; Rowley M. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J. Med. Chem. 2009, 52, 7170–7185. [DOI] [PubMed] [Google Scholar]
- Thomas H. D.; Calabrese C. R.; Batey M. A.; Canan S.; Hostomsky Z.; Kyle S.; Maegley K. A.; Newell D. R.; Skalitzky D.; Wang L. Z.; Webber S. E.; Curtin N. J. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 2007, 6, 945–956. [DOI] [PubMed] [Google Scholar]
- Menear K. A.; Adcock C.; Boulter R.; Cockcroft X. L.; Copsey L.; Cranston A.; Dillon K. J.; Drzewiecki J.; Garman S.; Gomez S.; Javaid H.; Kerrigan F.; Knights C.; Lau A.; Loh V. M. Jr.; Matthews I. T.; Moore S.; O’Connor M. J.; Smith G. C.; Martin N. M. 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [DOI] [PubMed] [Google Scholar]
- Penning T. D.; Zhu G.; Gandhi V. B.; Gong J.; Thomas S.; Lubisch W.; Grandel R.; Wernet W.; Park C. H.; Fry E. H.; Liu X.; Shi Y.; Klinghofer V.; Johnson E. F.; Donawho C. K.; Frost D. J.; Bontcheva-Diaz V.; Bouska J. J.; Olson A. M.; Marsh K. C.; Luo Y.; Rosenberg S. H.; Giranda V. L. Discovery and SAR of 2-(1-propylpiperidin-4-yl)-1H-benzimidazole-4-carboxamide: a potent inhibitor of poly(ADP-ribose) polymerase (PARP) for the treatment of cancer. Bioorg. Med. Chem. 2008, 16, 6965–6975. [DOI] [PubMed] [Google Scholar]
- Griffin R. J.; Calvert A. H.; Curtin N. J.; Newell D. R.; Golding B. T.. Benzoxazole-4-carboxamides and their use in inhibiting poly(ADP-ribose) polymerase activity and improving cytotoxic effectiveness of cytotoxic drugs or radiotherapy. PCT. Int. Appl. WO 9524379 A1 19950914, 1995.
- Gangloff A. R.; Brown J.; de Jong R.; Dougan D. R.; Grimshaw C. E.; Hixon M.; Jennings A.; Kamran R.; Kiryanov A.; O’Connell S.; Taylor E.; Vu P. Discovery of novel benzo[b][1,4]oxazin-3(4H)-ones as poly(ADP-ribose)polymerase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4501–4505. [DOI] [PubMed] [Google Scholar]
- Lednicer D.; Sun J. H.. Preparation of benzo[b]furan-7-carboxamides as antiemetic or antipsychotic agents. Eur. Pat. Appl. 234872, 1987.
- Hogberg T.; Paulis T.; Johansson L.; Kumar Y.; Hall H.; Ogren S. Potential antipsychotic agents. 7. Synthesis and antidopaminergic properties of the atypical highly potent (S)-5-bromo-2,3-dimethoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl]benzamide and related compounds. A comparative study. J. Med. Chem. 1990, 33, 2305–2309. [DOI] [PubMed] [Google Scholar]
- Meyers A.; Reuman M.; Gabel R. A. Nucleophilic annulations of aromatics. Novel route to benzo-fused ring systems via oxazoline activation. J. Org. Chem. 1981, 46, 783–788. [Google Scholar]
- Meshram H. M.; Premalatha K.; Rameshbabu K.; Eeshwaraiah B.; Yadav Y. S. Zirconium(IV) chloride catalyzed cyclization of ortho-allylphenols: synthesis of 2-methyl-2,3-dihydrobenzofurans. Synth. Commun. 2004, 34, 3091–3097. [Google Scholar]
- Rozenberg V.; Dubrovina N.; Sergeeva E.; Antonov D.; Belokon Y. An improved synthesis of (S)-(+)- and (R)-(−)-[2.2]paracyclophane-4-carboxylic acid. Tetrahedron: Asymmetry 1998, 9, 653–656. [Google Scholar]
- Irie R.; Noda K.; Ito Y.; Matsumoto N.; Katsuki T. Catalytic asymmetric epoxidation of unfunctionalized olefins using chiral (salen)manganese(III) complexes. Tetrahedron: Asymmetry 1991, 2, 481–494. [Google Scholar]
- He Q.; Gomma H.; Rohani S.; Zhu J.; Jennings M. Chiral discrimination in diastereomeric salts of chlorine-substituted mandelic acid and phenylethylamine. Chirality 2010, 22, 707–716. [DOI] [PubMed] [Google Scholar]
- Stein A. R.; Dawe R. D.; Sweet J. R. Preparation of chiral 1-phenylethanols and bromides. Can. J. Chem. 1985, 63, 3442–3448. [Google Scholar]
- Riether D.; Harcken C.; Razavi H.; Kuzmich D.; Gilmore T.; Bentzien J.; Pack E. J. J.; Souza D.; Nelson R. M.; Kukulka A.; Fadra T. N.; Zuvela-Jelaska L.; Pelletier J.; Dinallo R.; Panzenbeck M.; Torcellini C.; Nabozny G. H.; Thomson D. S. Nonsteroidal dissociated glucocorticoid agonists containing azaindoles as steroid A-ring mimetics. J. Med. Chem. 2010, 53, 6681–6698. [DOI] [PubMed] [Google Scholar]
- Hadj-esfandiari N.; Navidpour L.; Shadnia H.; Amini M.; Samadi N.; Faramarzi M. A.; Shafiee A. Synthesis, antibacterial activity, and quantitative structure–activity relationships of new (Z)-2-(nitroimidazolylmethylene)-3(2H)-benzofuranone derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 6354–6363. [DOI] [PubMed] [Google Scholar]
- Zlatoidsky P.; Maliar T. Synthesis and structure–activity relationship study of the new set of trypsin-like proteinase inhibitors. Eur. J. Med. Chem. 1999, 34, 1023–1034. [Google Scholar]
- Pielichowski J.; Popielarz R. Trichloroethylene in organic synthesis: II. Reaction of trichloroethylene with secondary amines. Tetrahedron 1984, 40, 2671–2675. [Google Scholar]
- Knoevenagel E. Condensation of malonic acid with aromatic aldehydes with ammonia and amines. J. Am. Chem. Soc. 1898, 31, 2596–2619. [Google Scholar]
- Scarpelli R.; Boueres J. K.; Cerretani M.; Ferrigno F.; Ontoria J. M.; Rowley M.; Schultz-Fademrecht C.; Toniatti C.; Jones P. Synthesis and biological evaluation of substituted 2-phenyl-2H-indazole-7-carboxamides as potent poly(ADP-ribose) polymerase (PARP) inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 488–492. [DOI] [PubMed] [Google Scholar]
- Ferraris D.; Ficco R. P.; Pahutski T.; Lautar S.; Huang S.; Zhang J.; Kalish V. Design and synthesis of poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors. Part 3: In vitro evaluation of 1,3,4,5-tetrahydro-benzo[c][1,6]- and [c][1,7]-naphthyridin-6-ones. Bioorg. Med. Chem. Lett. 2003, 13, 2513–2518. [DOI] [PubMed] [Google Scholar]
- Varma R.; Varma M. Alumina-mediated condensation. A simple synthesis of aurones. Tetrahedron Lett. 1992, 33, 5937–5940. [Google Scholar]
- Lea T. V.; Suhb J. H.; Kimb N.; Park H. In silico identification of poly(ADP-ribose)polymerase-1 inhibitors and their chemosensitizing effects against cisplatin-resistant human gastric cancer cells. Bioorg. Med. Chem. Lett. 2013, 23, 2642–2646. [DOI] [PubMed] [Google Scholar]
- Murai J.; Huang S. N.; Das B. B.; Renaud A.; Zhang Y.; Doroshow J. H.; Ji J.; Takeda S.; Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg E.; Karlberg T.; Kouznetsova E.; Markova N.; Macchiarulo A.; Thorsell A. G.; Pol E.; Frostell Å.; Ekblad T.; Öncü D.; Kull B.; Robertson G. M.; Pellicciari R.; Schüler H.; Weigelt J. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [DOI] [PubMed] [Google Scholar]
- Murai J.; Huang S. N.; Renaud A.; Zhang Y.; Ji J.; Takeda S.; Morris J.; Teicher B.; Doroshow J. H.; Pommier Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. L.; Lacefield W. B.. Preparation of 5-HT3 antagonist. Eur. Pat. Appl. 307172 A2 19890315, 1989.
- Hartz R. A.; Ahuja V. T.; Rafalski M.; Schmitz W. D.; Brenner A. B.; Denhart D. J.; Ditta J. L.; Deskus J. A.; Yue E. W.; Arvanitis A. G.; Lelas S.; Li Y.; Molski T. F.; Wong H.; Grace J. E.; Lentz K. A.; Li J.; Lodge N. J.; Zaczek R.; Combs A. P.; Olson R. E.; Mattson R. J.; Bronson J. J.; Macor J. E. In vitro intrinsic clearance-based optimization of N3-phenylpyrazinones as corticotropin-releasing factor-1 (CRF1) receptor antagonists. J. Med. Chem. 2009, 52, 4161–4172. [DOI] [PubMed] [Google Scholar]
- Rhoads S. J.; Raulins R.; Reynolds R. D. The para-Claisen rearrangement. I. The preparation and rearrangement of the α- and γ-ethylallyl ethers of methyl ο-cresotinate. A reinvestigation. J. Am. Chem. Soc. 1954, 76, 3456–3463. [Google Scholar]
- Nagarapu L.; Aneesa; Satyender A.; Chandana G.; Bantu R. Synthesis and antimicrobial activity of novel analogs of trifenagrel. J. Heterocycl. Chem. 2009, 46, 195–200. [Google Scholar]
- Katz L.; Karger L. S.; Schroeder W.; Cohen M. S. Hydrazine derivatives. I. Benzalthio- and bisbenzaldithiosalicoylhydrazides. J. Org. Chem. 1953, 18, 1380–1402. [Google Scholar]
- Ouach A.; Gmouh S.; Pucheault M.; Vaultier M. Onium salt supported organic synthesis in water: application to Grieco’s multicomponent reaction. Tetrahedron 2008, 64, 1962–1970. [Google Scholar]
- Ohno T.; Ogawa K.; Yano S.; Fukushima M.; Suzuki N.; Asao T. Synthesis and structure–activity relationship of 4-substituted benzoic acids and their inhibitory effect on the biosynthesis of fatty acids and sterols. Arch. Pharm. 2005, 338, 147–158. [DOI] [PubMed] [Google Scholar]
- Keelara A.; Srinivasa G. R.; Channe G. D. Palladium-catalyzed simple and efficient hydrogenative cleavage of azo compounds using recyclable polymer-supported formate. Can. J. Chem. 2005, 83, 517–520. [Google Scholar]
- Maede Y.; Shimizu H.; Fukushima T.; Kogame T.; Nakamura T.; Miki T.; Takeda S.; Pommier Y.; Murai J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 2014, 13, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen J. D.; Tholey R. M.; Langelier M. F.; Planck J. L.; Schiewer M. J.; Lal S.; Bildzukewicz M. A.; Yeo C. J.; Knudsen K. E.; Brody J. R.; Pascal J. M. Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer. Cancer Res. 2014, 74, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier M. F.; Plank J. L.; Servent K. M.; Pascal J. M. Purification of human PARP-1 and PARP-1 domains from Escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 2011, 780, 209–226. [DOI] [PubMed] [Google Scholar]
- Winter G. Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar]
- Winn M. D.; Isupov M. N.; Murshudov G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. 2001, D57, 122–133. [DOI] [PubMed] [Google Scholar]
- Flack H. D. On enantiomorph-polarity estimation. Acta Crystallogr. 1983, A39, 876–881. [Google Scholar]
- Hooft R. W.; Straver L. H.; Spek A. L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Crystallogr. 2008, 41, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.