

Pharmaceutical industry and chemical biology are dominated by organic chemistry with inorganic compounds playing only a minor role. This is well illustrated by a review of FDA approved drugs during 2007 in which not a single compound contains a metal atom, with most compounds being reversible enzyme inhibitors.[1] However, our laboratory recently demonstrated that chemically inert metal complexes can serve as promising scaffolds for the design of enzyme inhibitors and we reported several compounds with high affinities and promising selectivity profiles for protein kinases and lipid kinases.[2–4] For example, we have recently introduced the ruthenium half-sandwich complexes HB12 and DW12 as potent protein kinase inhibitors, in particular for GSK-3 and Pim-1.[5–7] DW12 and its derivatives induce strong biological responses such as the activation of the wnt signaling pathway in mammalian cells, strong pharmacological effects during the development of frog embryos, and the efficient induction of apoptosis in some melanoma cell lines.[8,9]
Moreover, in an independent previous study we discovered by a combinatorial approach that the introduction of a D-alanine amide side chain into the η5-cyclopentadienyl moiety of HB12 increased affinity by 40-fold ((RRu)-HB1229).[11,12] Based on these results, we were curious to investigate by how much we could further improve potency if we would combine these beneficial modifications at the cyclopentadienyl and pyridocarbazole moiety in one molecule. Accordingly, we synthesized the individual stereoisomers of NP549 (see supporting information for synthetic details) and found (RRu)-NP549 to be an extremely potent inhibitor for GSK-3β with an IC50 of 40 pM at 100 μM ATP.[13,14] Since this IC50 was measured in presence of the lowest possible GSK-3β concentration of 100 pM, this value reflects an upper limit. Considering that GSK-3β displays a Km for ATP of 15 μM, the binding constant can be estimated to Ki ≤ 5 pM by applying the Cheng-Prusoff equation.[15] With this, (RRu)-NP549 is one of the highest affinity ligands for a protein kinase known to date.[16]
In order to investigate the binding mode of this class of organoruthenium complexes to GSK-3β, we crystallized full-length human GSK-3β, soaked it with a solution of enantiomerically pure (RRu)-NP549 and solved to a resolution of 2.4 Å (Table 1). The global structure reveals the typical two-lobe protein kinase architecture, connected by a hinge region, with the catalytic domain positioned in a deep intervening cleft and (RRu)-NP549 occupying the ATP-binding site, similar to the binding of staurosporine and synthetic organic inhibitors (Figure 2).[17]
Table 1.
Crystallographic data and refinement statistics.
| Parameter[a] | |
|---|---|
| Space group | P212121 |
| Cell dimensions [Å] | a = 83.04, b = 86.11, c = 177.39 |
| Resolution [Å] | 2.4 |
| Total observations (unique, redundancy) | 209116 (52103, 4) |
| Completeness (outer shell) | 97.4 (98.3) |
| Rmerge (outer shell) [%] | 10.5 (71.2) |
| I/σ (outer shell) | 14.4 (2.1) |
| Rwork (Rfree) [%] | 19.0 (22.7) |
| Hetero groups | (RRu)-NP549 |
| Rmsd bond length | 0.016 |
| Rmsd bond angle | 1.548 |
| Ramachandran (allowed/ generally allowed/disallowed) | 91.1/8.6/0.3 |
Rmsd = root-mean-square deviation.
Figure 2.
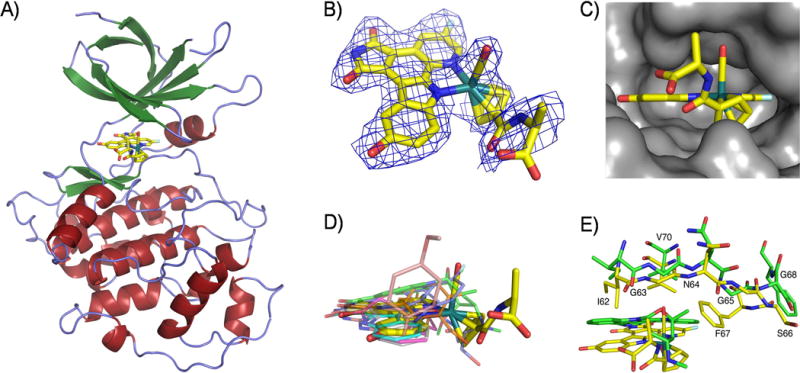
Crystal structure of GSK-3β with the ruthenium compound (RRu)-NP549 bound to the ATP-binding site. A) Overview of the complete structure. B) Electron density of the ruthenium complex contoured at 1σ. C) Fit of (RRu)-NP549 into the active site of GSK-3β with emphasis on the hydrophobic pocket for the CO ligand. D) Superimposed binding positions of (RRu)-NP549 and other small molecules (PDB codes 1UV5, 1Q3D, 1Q3W, 1Q4L, 1Q5K, 1Q41, and 1ROE) within the ATP-site of GSK-3β. E) Relative binding positions of (RRu)-NP549 and staurosporine (PDB code 1Q3D) within the ATP-site of GSK-3β.
(RRu)-NP549 forms a number of hydrogen bonds within the ATP-binding site of GSK-3β (Figure 3). The maleimide moiety and the indole OH-group establish together three important hydrogen bonds to the backbone of the hinge region: one between the imide NH group and the backbone carbonyl oxygen of Asp133, a second between one of the imide carbonyl groups and the backbone NH of Val135 and the third between the backbone carbonyl oxygen of Val135 and the indole OH. The second carbonyl group of the maleimide moiety forms a water-mediated contact to Asp200. An additional hydrogen bond is established with the amide carbonyl group at the cyclopentadienyl moiety which is in a water-mediated contact to Thr138. The carboxylate group does not form any particular hydrogen bond but is nicely placed close to a positively charged patch formed from Arg141 and Arg144 and thus contributing to electrostatic attraction. Furthermore, the fluoride atom is at a close distance to the amino group of Lys85 (3.1 Å) which suggests a weak F…H-N hydrogen bond.
Figure 3.

Interactions of (RRu)-NP549 within the ATP-binding site of GSK-3β. A) Hydrogen bonding interactions. B) The most important hydrophobic interactions. C) Highlighting the close contact of the CO ligand of (RRu)-NP549 with Gly63 and the small hydrophobic pocket for the CO. D) Proximity of the carboxylate to Arg141 and Arg144, Lys85 to the fluorine substituent, and interaction of Gln185 with the cyclopentadienyl ring and the adjacent amide carbonyl group.
(RRu)-NP549 is involved in extensive van der Waals contacts with GSK-3β. A hydrophobic pocket for the pyridocarbazole moiety is built by side chains from more than 10 amino acids, in particular Phe67, Val70, Ala83, Val110, Leu132, Tyr134, Val135, Leu188, and Cys199. Phe67 also packs against the CO ligand and one edge of the cyclopentadienyl moiety, whereas Gln185 interacts with one edge and the face of the cyclopentadienyl ring and the adjacent amide carbonyl group. Finally, the methyl group of the cyclopentadienyl amide side chain forms a hydrophobic contact with the CH2-group of Gly63 within the glycine-rich loop.
Most interestingly, the CO ligand comes in particularly close contact to Gly63, with a distance to the methylene group of only 3.1 Å. This is below the van der Waals distance and suggests dipolar interactions.[18] We have observed this close contact to the glycine-rich loop also in crystal structures of related organometallic compounds with the protein kinase Pim-1.[7,10] In addition, Gly63, together with the sidechains of Ile62, Val70, and Phe67 create a small hydrophobic pocket in which the CO ligand is buried (Figure 3C). It is noteworthy that replacing the CO by any other monodentate ligand reduces the binding affinity significantly.[19] For example, exchanging the CO group in HB12 against PF3 (CS44) increases the IC50 by around 25-fold, presumably because the PF3 ligand is too big for this pocket, whereas replacing the (η5-C5H5)RuCO moiety in HB12 by the highly similar (η6-C6H6)RuCN fragment (NP930) leads to a diminished affinity by 75-fold (Scheme 1). Such a dramatic effect by replacing a CO ligand with a cyanide we have observed before in a related octahedral scaffold.[19] Although isoelectronic, coordinated CO is hydrophobic,[20,21] whereas coordinated cyanide tends to form hydrogen bonds with its nitrogen lone pair and will therefore not have any desire to bind into the hydrophobic pocket build by the glycine-rich loop.[22,23] These examples demonstrate the importance of the CO group and, in fact, we have yet to find a highly potent and selective ruthenium complex for GSK-3 that lacks this apparently crucial CO ligand.
Scheme 1.
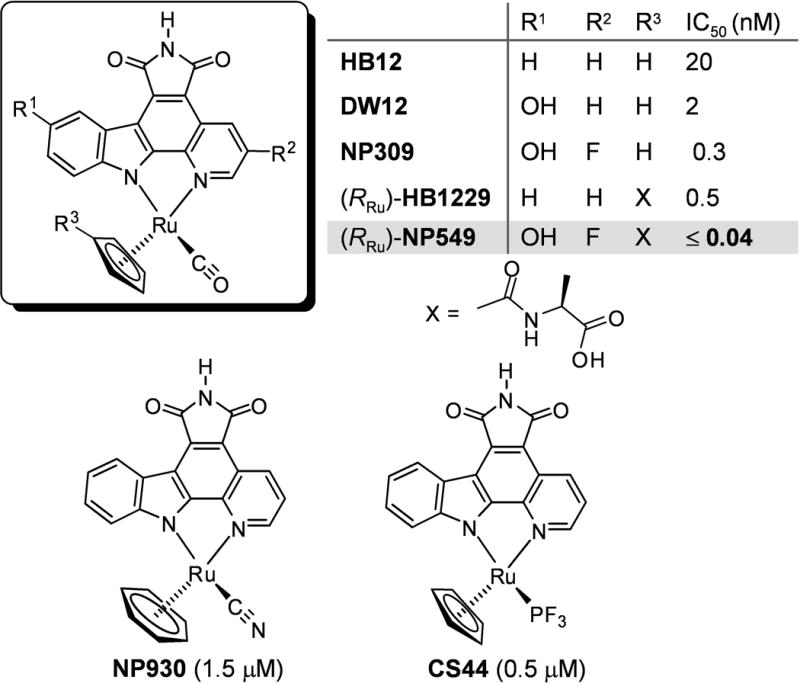
Ruthenium complex HB12 as a lead scaffold for the design of highly potent GSK-3 inhibitors. NP930 and CS44 are only weak inhibitors for GSK-3. IC50 values were measured at 100 μM ATP. Compounds are racemic if not indicated otherwise.
Finally, we compared the relative binding position of (RRu)-NP549 with cocrystal structures of small organic molecules bound to GSK-3β. A superimposition of all available structures demonstrates that (RRu)-NP549 occupies the same area of the ATP-binding site. However, it seems that the position of the CO ligand together with the perpendicular orientation to the pyridocarbazole heterocycle is a unique feature of (RRu)-NP549 which allows Val70 to reach down to the pyridocarbazole moiety, thus maximizing the hydrophobic interactions with the pyridocarbazole moiety and creating the hydrophobic pocket for the CO ligand. Although the pyrane oxygen atom of staurosporine occupies a similar position in the active site compared to the CO oxygen of the ruthenium complex, the glycine-rich loop is in a significantly more open position as displayed in Figure 2E and does not allow the same closure of the active site with its optimized contacts.
In conclusion, we here reported an extremely high affinity GSK-3 inhibitor and its binding to the ATP-binding site of GSK-3β. Overall, (RRu)-NP549 perfectly complements the shape of the ATP-binding site and forms three direct hydrogen bonds, two water mediated hydrogen bonds, one fluorine-mediated hydrogen bond, undergoes electrostatic contacts between the carboxylate tail and two arginines, and is involved in van der Waals interactions with more than 10 amino acids. Furthermore, the CO ligand stacks against the glycine-rich loop and is buried in a small pocket which appears to be crucial for affinity and selectivity for GSK-3β. With a Ki value of around 5 pM or less, (RRu)-NP549 is one of the most potent protein kinase inhibitors reported to date and by almost 4 orders of magnitude more potent than the related natural product staurosporine (IC50 = 180 nM at 100 μM ATP), demonstrating that this organoruthenium structure is a priviledged scaffold for the design of GSK-3 inhibitors.
Experimental Section
Cloning, expression, purification and crystallization of GSK-3β
GSK-3β was cloned into pET151 vector by using Champion™ pET Directional TOPO Expression kit (Invitrogen). The plasmid was expressed in Rosetta2DE3 cells as an N-terminal cleavable His6-tag fusion protein with TEV cleavage site. The cells were grown at 37 °C until they reached O.D. = 0.4 at which time the temperature was decreased to 17 °C. Once they reached O.D. = 0.6, protein expression was induced with 1 mM IPTG overnight. The next morning, the cells were centrifuged, resuspended in lysis buffer (50 mM HEPES pH 7.2, 50 mM NaCl, 5% glycerol) and supplemented with Complete Protease Inhibitor Cocktail tablets (Roche). The cells were then lysed by sonication at 4 °C. The supernatant was collected and applied to ion exchange chromatography (SP-Sepharose, GE Biosciences). The protein was bound onto the column in 50 mM HEPES pH 7.2 and eluted with 50 mM HEPES pH 7.2, 1 M NaCl. The eluted fractions were applied to a Talon affinity column equilibrated with 50 mM HEPES pH 7.5, 300 mM NaCl. The column was washed with 50 mM HEPES pH 7.5, 300 mM NaCl, 20 mM imidazole and the protein was eluted with 50 mM HEPES pH 7.5, 300 mM NaCl, 250 mM imidazole. GSK-3β was treated overnight with λ-phosphatase and TEV-protease to remove phosphate and His-tag, respectively. Further purification was achieved with a MONO-S column (GE Biosciences). The protein was loaded onto the MONO-S column in 50 mM HEPES pH 7.2, 10% glycerol, 1 mM DTT and eluted with a linear gradient of 0–1 M NaCl in 50 mM HEPES pH 7.2, 10% glycerol, 1 mM DTT to ensure the separation of phosphorylated and non-phosphorylated species. Non-phosphorylated GSK-3β was subjected to gel filtration chromatography (Superdex 26/60, GE Biosciences) in 50mM HEPES, 1 mM DTT, 2 mM MgCl2, 500 mM NaCl, pH 7.2 and concentrated to 3–4 mg/mL for crystallization purposes. Crystals of the apo-protein were grown at 4 °C in 4 μL hanging drops where 2 μL of protein solution was mixed with 2 μL of the precipitant solution. Crystals were observed in 100 mM Tris pH 7.2, 20% PEG 6000 or 12.5% PEG 8000 within 3 days. The crystal quality was improved by overlaying 400 μL of oil (50% silicon oil in paraffin oil) onto 600 μL of reservoir solution. Apo form crystals were soaked with the ruthenium compound overnight at 4 °C in 100 mM Tris pH 7.2, 20% PEG 6000, 1 mM (RRu)-NP549, 10% DMSO and 1% glycerol. The next morning the crystals were cryoprotected in 25% glycerol and flash frozen in liquid nitrogen.
Data collection and structure determination
Cryoprotected crystals diffracted up to 2.4 Å on A1 beam line at Cornell High Energy Synchrotron Source (Ithaca, NY). After indexing and merging the data using HKL2000, the structure was solved by molecular replacement using a crystal structure of GSK-3β (PDB code 1J1B) as a search model calculated with Phaser. Refinement and manual rebuilding of the model were performed using REFMAC5 and Coot, respectively. Data collection and current refinement statistics are listed in Table 1. Coordinates of the structure have been deposited in the Protein Data Bank (PDB code 2JLD).
Measurement of IC50-values with GSK-3β
GSK-3β and phospho-glycogen synthase peptide-2 (PGSP-2) were purchased from Upstate Biosciences (USA). Various concentrations of inhibitors were incubated at room temperature in 20 mM MOPS, 30 mM MgCl2, 0.8 μg/μl BSA, 5% DMSO (resulting from the inhibitor stock solution), pH 7.0, in presence of 20 μM PGSP-2, and GSK-3β (200 pM for NP930, HB12, and DW12; 100 pM for HB1229, NP309, and NP549). After 15 minutes, the reaction was initiated by addition of 100 μM ATP including approximately 0.2 μCi/μl [γ-32P]ATP. Reactions were performed in a total volume of 25 μl. After 60 minutes, the reaction was terminated by spotting 17.5 μl of the reaction mixture on a circular P81-phosphocellulose paper (diameter 2.1 cm, Whatman) followed by washing four times (five minutes each wash) with 0.75% phosphoric acid and once with acetone. The dried P81-papers were transferred to a scintillation vial and 4 ml of scintillation cocktail were added. The counts per minute (CPM) were determined with a Beckmann 6000 scintillation counter and IC50 values were defined to be the concentration of inhibitor at which the CPM was 50% of the control sample, corrected by the background.
Supplementary Material
{kind=link}
Figure 1.
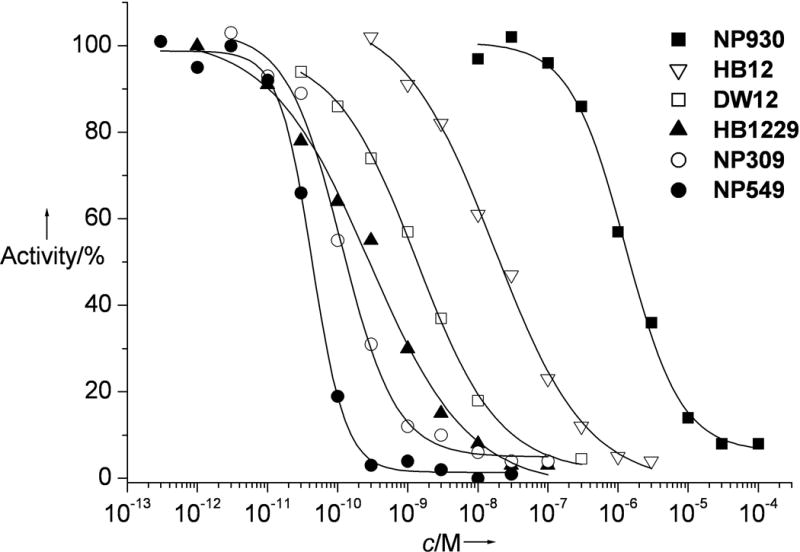
IC50 curves with GSK-3β obtained by phosphorylation of phospho-glycogen synthase peptide-2 with [γ-32P]ATP at 100 μM ATP. HB1229 and NP549 were used as the (RRu)-isomers.
Acknowledgments
We thank the US National Institutes of Health for support (GM071695).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Hughes B. Nature Rev Drug Discov. 2008;7:107–109. doi: 10.1038/nrd2514. [DOI] [PubMed] [Google Scholar]
- 2.Meggers E. Curr Opin Chem Biol. 2007;11:287–292. doi: 10.1016/j.cbpa.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Meggers E, Atilla-Gokcumen GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, Williams DS. Synlett. 2007;8:1177–1189. [Google Scholar]
- 4.Xie P, Williams DS, Atilla-Gokcumen GE, Milk L, Xiao M, Smalley KSM, Herlyn M, Meggers E, Marmorstein R. ACS Chem Biol. 2008;3:305–316. doi: 10.1021/cb800039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bregman H, Williams DS, Atilla GE, Carroll PJ, Meggers E. J Am Chem Soc. 2004;126:13594–13595. doi: 10.1021/ja046049c. [DOI] [PubMed] [Google Scholar]
- 6.Williams DS, Atilla GE, Bregman H, Arzoumanian A, Klein PS, Meggers E. Angew Chem Int Ed. 2005;44:1984–1987. doi: 10.1002/anie.200462501. [DOI] [PubMed] [Google Scholar]
- 7.Debreczeni JÉ, Bullock AN, Atilla GE, Williams DS, Bregman H, Knapp S, Meggers E. Angew Chem Int Ed. 2006;45:1580–1585. doi: 10.1002/anie.200503468. [DOI] [PubMed] [Google Scholar]
- 8.Atilla-Gokcumen GE, Williams DS, Bregman H, Pagano N, Meggers E. ChemBioChem. 2006;7:1443–1450. doi: 10.1002/cbic.200600117. [DOI] [PubMed] [Google Scholar]
- 9.Smalley KSM, Contractor R, Haass NK, Kulp AN, Atilla-Gokcumen GE, Williams DS, Bregman H, Flaherty KT, Soengas MS, Meggers E, Herlyn M. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 10.Pagano N, Maksimoska J, Bregman H, Williams DS, Webster RD, Xue F, Meggers E. Org Biomol Chem. 2007;5:1218–1227. doi: 10.1039/b700433h. [DOI] [PubMed] [Google Scholar]
- 11.Bregman H, Meggers E. Org Lett. 2006;8:5465–5468. doi: 10.1021/ol0620646. [DOI] [PubMed] [Google Scholar]
- 12.The absolute configuration at the ruthenium has been assigned according to the priority order of the ligands being η5-C5H5 > pyridine [N(C, C, C)] > indole [N(C, C, lone pair)] > CO.
- 13.The diastereomer (SRu)-NP549 is a weaker inhibitor for GSK-3β with an IC50 value of 220 pM at 100 μM ATP.
- 14.The absolute configuration at the ruthenium center was assigned from the crystal structure of (RRu)-NP549 with GSK-3β.
- 15.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 16.for reversible low picomolar protein kinase inhibitors, see:; a) Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson JM, Leopold WR, Connors RW, Bridges AJ. Science. 1994;265:1093–1095. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]; b.) Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA. J Med Chem. 1996;39:267–276. doi: 10.1021/jm9503613. [DOI] [PubMed] [Google Scholar]; c) Bridges AJ. Chem Rev. 2001;101:2541–2571. doi: 10.1021/cr000250y. [DOI] [PubMed] [Google Scholar]
- 17.For cocrystal structures of organic compounds with GSK-3β, see:; a) Bertrand JA, Thieffine S, Vulpetti A, Cristiani C, Valsasina B, Knapp S, Kalisz HM, Flocco M. J Mol Biol. 2003;333:393–407. doi: 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]; b) Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]; c) Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. J Biol Chem. 2003;278:45937–45945. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]; d) Zhang HC, Bonaga LV, Ye H, Derian CK, Damiano BP, Maryanoff BE. Bioorg Med Chem Lett. 2007;17:2863–2868. doi: 10.1016/j.bmcl.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 18.For orthogonal multipolar interactions in crystal structures, see:; Paulini R, Müller K, Diederich F. Angew Chem Int Ed. 2005;44:1788–1805. doi: 10.1002/anie.200462213. [DOI] [PubMed] [Google Scholar]
- 19.See also:; Bregman H, Carroll PJ, Meggers E. J Am Chem Soc. 2006;128:877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 20.Ott I, Kircher B, Dembinski R, Gust R. Expert Opin Ther Pat. 2008;18:327–337. [Google Scholar]
- 21.For hydrophobic interactions of CO bound to a heme-containing transcriptional activator, see:; Youn H, Kerby RL, Roberts GP. J Biol Chem. 2003;278:2333–2340. doi: 10.1074/jbc.M210825200. [DOI] [PubMed] [Google Scholar]
- 22.This is comparable to the nonproteic cyanide and CO ligands bound to iron in the active site of hydrogenases. Whereas the cyanides are involved in specific hydrogen bonding interactions, the CO ligands are surrounded by hydrophobic residues. See, for example:; Frey M. ChemBioChem. 2002;3:153–160. doi: 10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 23.For the protonation of coordinated cyanides in the presence of a CO ligand, see for example:; Lai CH, Lee WZ, Miller ML, Reibenspies JH, Darensbourg DJ, Darensbourg MY. J Am Chem Soc. 1998;120:10103–10114. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.