

Abstract
In the presence of ERβ, trans-hydroxytamoxifen (TOT) protects cells against 17β-estradiol(E2)-induced oxidative DNA damage (ODD) and this correlates with increased expression of the antioxidative enzyme quinone reductase (QR). Here, we investigate the molecular mechanism responsible for ERβ-mediated protection against ODD. We observe constitutive interaction between ERβ and the novelprote in hPMC2. Using a combination of breast epithelial cell lines that are either positive or negative for ERα, we demonstrate TOT-dependent recruitment of both ERβ and hPMC2 to the EpRE (electrophile response element)-regulated antioxidative enzyme QR. We further demonstrate TOT-dependent corecruitment of the coactivators Nrf2, PARP-1 (poly (ADP-ribose) polymerase 1) and topoisomerase IIβ, both in the presence and absence of ERα. However, absence of either ERβ or hPMC2 results in nonrecruitment of PARP-1 and topoisomerase IIβ, loss of antioxidative enzyme induction and attenuated protection against ODD by TOT even in the presence of Nrf2 and ERα. These findings indicate minor role for Nrf2 and ERα in TOT-dependent antioxidative gene regulation. However, downregulation of PARP-1 attenuates TOT-dependent antioxidative gene induction. We conclude that ERβ and hPMC2 are required for TOT-dependent recruitment of coactivators such as PARP-1 to the EpRE resulting in the induction of antioxidative enzymes and subsequent protection against ODD.
Keywords: PARP-1, ERβ-dependent transcription, anti-oxidative enzymes, tamoxifen, breast cancer
Introduction
Prolonged exposure to estrogen is strongly associated with increased risk for developing breast cancer (Bolton and Thatcher, 2008). Metabolism of estrogens in breast epithelial cells generates highly reactive catechol estrogen quinones (CE-Q) that form mutagenic DNA adducts. Further, the interconversion of quinone– semiquinone forms generates reactive oxygen species, which induce oxidative stress inside the cells (Cavalieri et al., 2006). The sustained oxidative stress, together with the mutagenic potential of CE-Qs contributes to the initiation and progression of breast cancer (Liehr, 2000). This hypothesis is strengthened by our observation that downregulation of the antioxidative enzyme quinone reductase (QR), coupled with exposure to 17β-estradiol (E2) leads to the increased levels of CE-Q and transformation of nontumorigenic breast epithelial cells (Montano et al., 2007). QR catalyzes the reduction of estradiol-3,4-quinone, thus preventing the generation of reactive oxygen species by quinone–semiquinone interconversion (Gaikwad et al., 2007). Also, a strong correlation exists between breast cancer incidence and polymorphisms in several antioxidative genes including NQO1, indicating an important role for antioxidative enzymes in breast cancer prevention and treatment (Menzel et al., 2004; Oestergaard et al., 2006; Sakoda et al., 2007; Udler et al., 2007).
We observed that QR expression is increased in mammary glands of rats treated with tamoxifen and this increased expression correlates with a decrease in E2-induced ODD (oxidative DNA damage) levels (Montano et al., 2007). Also, treatment of human breast epithelial cells with trans-hydroxytamoxifen (TOT) prevents E2-induced increase in CE-Q levels (Montano et al., 2007). This ODD protective ability of tamoxifen makes it an attractive candidate for chemoprevention of breast cancer. In fact, the recently concluded Study of Tamoxifen and Raloxifene (STAR) and International Breast Cancer Intervention Study (IBIS) breast cancer trials highlight the advantage of using tamoxifen and raloxifene as chemopreventive agents (Fabian, 2007; Jordan, 2007).
TOT treatment increases the activity of reporter genes regulated by an EpRE (electrophile response element) sequence (Montano et al., 2004). EpRE is a cis-regulatory element present in the upstream of many antioxidative gene promoters including GCSH, GSTP1 and NQO1 (Rushmore et al., 1991; Tew, 1994; Prestera and Talalay, 1995; Strange et al., 2001). GSTpi (glutathione-S-transferase pi) detoxifies CE-Qs by conjugation with the cellular antioxidant glutathione, while GCSh (γ glutamylcysteine synthase heavy subunit) catalyzes the rate-limiting step in the de novo synthesis of glutathione (Liehr, 2000). EpRE-regulated reporter genes are activated by TOT-ERβ (estrogen receptor β), but not significantly by TOT-ERα (estrogen receptor α), and the ODD protective ability of TOT is not observed in the absence of ERβ (Bianco et al., 2003; Montano et al., 2004). These data indicate a key for TOT-ERβ in mediating oxidative stress response through the regulation of EpRE promoters.
The role of tamoxifen in regulating ERα-dependent transcription is well characterized, but mechanism of ERβ-mediated transcription in breast cancer cells is not well defined. Even less is known about the pathway responsible for TOT-ERβ-mediated upregulation of EpRE-regulated genes. Hence, our goal in the current study was to analyze TOT-mediated events at the EpRE locus and thereby define the transcription coactivator complex responsible for ERβ-dependent upregulation of EpRE genes.
Results
The ERβ-interacting protein, hPMC2 is important for tamoxifen-mediated protection against estrogen-induced oxidative DNA damage
TOT-induced transcription of EpRE genes is further enhanced by the ERβ-interacting protein hPMC2 (prevention of mitotic catastrophe; Montano et al., 2000), suggesting a role for hPMC2 in TOT-mediated protection against ODD. So, we transiently downregulated the expressions of both hPMC2 and QR (Figure 1a) by retroviral infection, and immunostained for 8-OHdG to analyze the relative roles of hPMC2 and QR in mediating TOT-dependent protection against E2-induded ODD in response to TOT.
Figure 1.

hPMC2 interacts directly with ERβ and is involved in mediating tamoxifen-dependent decrease in ODD levels. (a) Western blotting analysis of QR and hPMC2 expression levels to confirm transient knockdown in MCF7 cells. Cytokeratin 18 and GAPDH were used as loading controls, con (control shRNA). (b) MCF7 cells transiently infected with retroviral particles to knockdown QR and hPMC2 expression were treated for 24 h with 0.01% ethanol, 50 nM E2 or 100 nM TOT or a combination of E2 and TOT as indicated. Cells were immunostained with an antibody against 8-OHdG and the intensity of the staining quantitated in each case. The bars represent the average of three independent experiments and the standard error is given by the error bars. *Represents P<0.001 vs the E2-treated samples. (c) Serum-starved MCF7 cells were treated with either 0.01% ethanol (con), 10 nM E2 or 10 nM TOT for 3 h. Lysates were immunoprecipitated using antibodies against ERβ or hPMC2 and analyzed for coimmunoprecipitating proteins by western blotting. Normal rabbit immunoglobilin was used as a specificity control. Input lanes represent 25% of the total protein. ChIP, chromatin immunoprecipitation; ER, estrogen receptor; ODD, oxidative DNA damage; QR, quinone reductase; TOT, trans-hydroxytamoxifen.
Cells expressing knockdown levels of either hPMC2 or QR displayed higher basal levels of ODD compared with control-infected cells, while E2 treatment resulted in greater than twofold increase in the ODD levels of all three cell lines (hPMC2 shRNA, QR shRNA, control shRNA)(Figure 1b). Treatment with TOT alone revealed 8-OHdG levels lesser than or similar to that of the ethanol-treated samples, indicating that TOT by itself does not contribute to ODD. Knockdown of QR did not result in a complete loss of the protective ability of TOT, in confirmation with our previous observations (Bianco et al., 2003). This finding suggests the contribution of other antioxidative enzymes (GSTpi, GCSh and so on) in mediating the ODD protective ability of TOT. However, in hPMC2 knockdown cells 8-OHdG levels were similar to that of the E2-treated samples even with the TOT treatment, indicating a requirement for hPMC2 in TOT–ERβ-mediated protection against E2-induced ODD.
In vitro studies indicate the selective interaction of hPMC2 with ERβ. (Montano et al., 2000). Immunoprecipitation of MCF7 lysates for unliganded, E2-liganded and TOT-liganded ERβ resulted in coimmunoprecipitation of hPMC2 in all three cases. Conversely, hPMC2 immunoprecipitation pulled down ERβ both in the presence and absence of E2 or TOT, indicating constitutive interaction between ERβ and hPMC2 (Figure 1c). Taken together, our data indicate a novel role for hPMC2 in TOT-mediated protection against ODD by selective interaction with ERβ to induce antioxidative gene expression.
Tamoxifen treatment results in the recruitment of ERβ, hPMC2 and transcriptional coactivators to the EpRE
ERβ and hPMC2 interact in vivo and hPMC2 together with ERβ binds to the EpRE sequence in vitro (Montano et al., 2000). These data suggest potential recruitment of ERβ and hPMC2 to the EpRE regions in vivo. To test the TOT-dependent recruitment of these proteins, we used chromatin immunoprecipitation (ChIP) to analyze the EpRE region of the NQO1 gene (Figure 2a). We treated the cells for 3 h with the indicated ligands, as that was the earliest time point at which we observed transcriptional induction (data not shown). To identify other factors involved in ERβ-mediated induction of EpRE, we first studied the recruitment of known ERα-associated transcriptional coactivators: SRC-1 (steroid receptor coactivator-1), PARP-1 (poly (ADP-ribose) polymerase 1) and topoisomerase IIβ (Shang and Brown, 2002; Ju et al., 2006). We also tested for the recruitment of the EpRE-binding transcriptional activator Nrf2 (NF-E2-related factor-2) (Venugopal and Jaiswal, 1996).
Figure 2.

Tamoxifen-dependent recruitment of coactivators to the EpRE sequence of NQO1 (a) Representation of NQO1 gene locus with the thick lines representing regions analyzed in subsequent PCR experiments. (b) Serum-starved MCF7 cells were treated with either 0.01% ethanol (con), 10 nM E2 or 10 nM TOT for 3 h and processed for ChIP analysis. Immunoprecipitated DNA was PCR analyzed to determine the recruitment patterns of the indicated proteins. Input (Inp) represents 2% of total DNA. (c) Average recruitment levels of the indicated factors in TOT-treated samples relative to that of the ethanol-treated samples. Error bars represent standard errors of three or more independent experiments. (d) ChIP analysis of TOT-treated cells for recruitment of the indicated factors using primers upstream and downstream of the EpRE region (e) MCF7 cells were treated with 10 nM TOT for 3 h and processed lysates were subjected to ChIP using an antibody against hPMC2 (rabbit IgG was used as a specificity control). hPMC2- precipitated chromatin was diluted 1:20 and reimmunoprecipitated using the indicated antibodies. The precipitates were then used to isolate DNA and subjected to PCR analysis at the EpRE locus. The results in each case are representatives of two or more independent experiments. ChIP, chromatin immunoprecipitation; EpRE, electrophile response element; IgG, immunoglobulin G; TOT, trans-hydroxytamoxifen.
We observed weak recruitment of PARP-1 and Nrf2 in control-treated samples but a strong recruitment of all the factors tested in the TOT-treated cells indicating a predominantly TOT-dependent recruitment of not only ERβ, hPMC2 and Nrf2, but also ERα-associated coactivators (Figures 2b and c). Analysis of TOT-treated samples revealed little or no recruitment of any of the proteins tested either at ~800 bp upstream of the EpRE or at the NQO1 promoter region located ~400 bp downstream to the EpRE (Figure 2d), suggesting a localized and selective recruitment to the EpRE region.
An initial ChIP of the TOT-treated samples with an antibody to hPMC2, followed by reimmunoprecipitation of the chromatin using antibodies to the indicated proteins confirmed mutual recruitment of ERβ, hPMC2 Nrf2, ERα and ERα-associated coactivators to the EpRE (Figure 2e). Taken together, the data indicate that TOT–ERβ together with hPMC2, recruits an ERα-like activation complex localized to the EpRE region, resulting in transcriptional induction.
ERβ and hPMC2 are required for effective inhibition of estrogen-induced oxidative DNA damage by tamoxifen
To examine the ERα-independent role of ERβ and hPMC2 in TOT-mediated induction of EpRE and in protection against E2-induced ODD, we used the ER negative, nontumorigenic breast epithelial cell line, MCF10A (Montano et al., 2007). The lack of ERα expression enabled us to examine the ability of ERβ and hPMC2 to recruit ER-associated coactivators and for inducing antioxidative genes in the absence of ERα. We established two MCF10A cell lines, both of which constitutively express Flag-ERβ (FL-ERβ), and one of them expresses stably knocked down levels of hPMC2 (hPMC2mi; Figure 3a).
Figure 3.

Both ERβ and hPMC2 are required for tamoxifen-mediated increase in antioxidative enzyme expression and protection against ODD. (a) Western blot analysis of MCF7, MCF10A parental cells (10A(P)), MCF10A stably expressing Flag-ERβ (FL-ERβ) and MCF10A FL-ERβ-positive cells that express stably knockeddown levels of hPMC2 (hPMC2mi). An anti-Flag antibody was used to detect FL-ERβ and GAPDH is used as a loading control. (b) Cells belonging to the indicated cell lines were treated with either 0.01% ethanol or 10 nM TOT for 3 h. Lysates were analyzed for expression levels of GCSh, QR and GSTpi by western blotting. The signals were normalized to their respective GAPDH loading controls and the fold change relative to the ethanol-treated group was calculated as described in the Materials and methods. The bars indicate the average of three independent experiments and the standard error is given by the error bars. (c and d) Cells from the indicated cell lines were treated with either 0.01% ethanol (con), 50 nM E2 or, a combination of 50 nM E2 and 10 nM TOT for 24 h and (c) immunostained for 8-OHdG as before. The bars represent average of four independent experiments and the error bars represent the standard error. (d) The culture media was subjected to LC–MS analysis to detect the indicated metabolite levels. The bars represent the average fold change in metabolite levels relative to the ethanol-treated group and error bars represent standard error from three independent experiments. ER, estrogen receptor; LC–MS, liquid chromatography/mass spectrometry; ODD, oxidative DNA damage; QR, quinone reductase; TOT, trans-hydroxytamoxifen.
No significant change was observed in expression levels of QR, GCSh and GSTpi upon TOT treatment of MCF10A(P) cells (Figure 3b). In contrast, a twofold to threefold increase in the expression of all three enzymes in FL-ERβ stables was observed, indicating that the transcriptional induction by TOT requires ERβ. Also, no significant changes were observed in antioxidative enzyme expression levels upon TOT treatment of hPMC2mi cells, strongly suggesting a requirement for hPMC2 in this process (Figure 3b). Similar results were obtained when the cell lines were tested for the ability of TOT to protect against E2-induced ODD (Figure 3c). The protective ability of TOT was severely attenuated in the absence of either ERβ or hPMC2.
We also measured the effect of E2 treatment on the levels of catechol estrogens and their quinone conjugates in the presence and absence of TOT (Figure 3d). We measured the levels of 4-hydroxyestradiol–quinone conjugate (4-con) as this metabolite is predominantly responsible for the formation of mutagenic DNA adducts (Li et al., 2004). Following E2-treatment, we observed comparable levels of 4-con induction in MCF10A(P) and FL-ERβ cells, while cells downregulated for hPMC2 exhibited lower levels of 4-con. This could be either due to a slower conversion of 4-OHE to 4-con or increased metabolism of 4-con to form DNA adducts in the absence of hPMC2. The similar levels of E2-induced ODD across all three cell lines (Figure 3c), suggest that the lower levels of 4-con observed in E2-treated hPMC2mi cells may not be reflective of lower levels of 4-con formation.
As a comparison, we also measured the levels of 2-hydroxyestradiol quinone conjugates (2-con). The FL-ERβ cells revealed significantly higher levels of 2-con metabolite in response to E2 treatment in comparison with the other two cell lines tested. Since the levels of 2-OHE are comparable across the cell lines, the higher levels of 2-con more likely reflects slower catabolism of the 2-con metabolite as a result of ERβ expression.
More importantly, TOT treatment reduced both 4- and 2-hydroxyestradiol–quinone conjugates levels by fourfold to sixfold in FL-ERβ cells, while very little or no reduction were observed in the absence of ERβ (MCF10A(P) cells), or hPMC2 (FL-ERβ/hPMC2mi cells). This indicated that TOT-dependent decrease in the levels of E2-induced quinone conjugates is dependent on the presence of ERβ and hPMC2 and not on the levels of the conjugates themselves. Further, 2-OHE2 and 4-OHE2 (2-OHE and 4-OHE) levels were similar both in the presence and absence of TOT in all three cell lines, indicating that the reduction in the levels of 2-con and 4-con is not simply a result of lower levels of 2-OHE2 and 4-OHE2 in response to TOT treatment.
Collectively, these results demonstrate a strong link between the expression levels of antioxidative enzymes and levels of quinone conjugates and underscore the key roles played by ERβ and hPMC2 in mediating oxidative stress responses.
Tamoxifen-mediated recruitment of the coactivators PARP-1, topoisomerase IIβ and SRC-1 to the EpRE, is dependent on both ERβ and hPMC2
TOT treatment resulted in the recruitment of an ERα-like activation complex at the EpRE that included both ERα and ERβ (Figure 2). However, our previous studies indicate that TOT–ERα alone does not significantly induce antioxidative gene expression but TOT–ERβ induces the expression even in the absence of ERα (Montano et al., 2004). Hence, we analyzed the E2- and TOT-dependent recruitment of the ERβ, hPMC2, Nrf2 and ERα-associated coactivators to the EpRE region, in MCF10A FL-ERβ cells (Figures 4a and b). We observed TOT-dependent recruitment of Nrf2, ERβ, hPMC2, PARP-1, topoisomerase IIβ and SRC-1, indicating that the absence of ERα does not significantly affect the recruitment of coactivators. However, induction levels of antioxidative genes by TOT in MCF7 and MCF10A FL-ERβ cells are comparable (Supplementary Figure 1D and Figure 3b), suggesting that ERα recruitment is unlikely to be a major contributor to ERβ-dependent induction of EpRE promoters. In the absence of either ERβ or hPMC2, we observed little or no recruitment of the factors tested, except for Nrf2. This indicates a dependence on both ERβ and hPMC2 for TOT-mediated recruitment of the coactivator complex at the EpRE.
Figure 4.
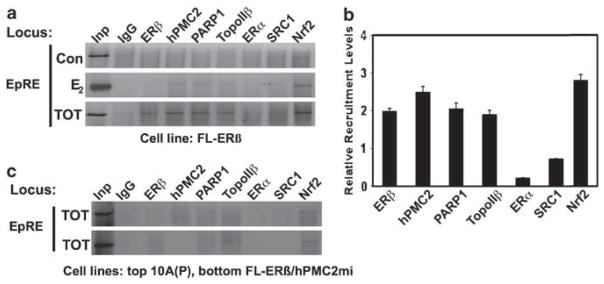
Tamoxifen-dependent recruitment of coactivators to the EpRE sequence of NQO1 requires both ERβ and hPMC2. (a) The indicated cell lines were treated with either 0.01% ethanol (con), 10 nM E2, or TOT for 3 h. Cells were processed for ChIP analysis and immunoprecipitated DNA was analyzed by PCR to determine the recruitment of the indicated proteins at the EpRE region. (b) Average recruitment levels of the indicated factors in TOT-treated samples relative to that of the ethanol-treated cells. Error bars represent the standard errors of two or more independent experiments. (c) ChIP analysis of TOT-treated samples for the recruitment of the indicated factors in the absence of ERα, ERβ and hPMC2. ChIP, chromatin immunoprecipitation; EpRE, electrophile response element; ER, estrogen receptor; TOT, trans-hydroxytamoxifen.
The above results taken together confirms our observations in MCF7 cells (Figure 2) that ERβ and hPMC2 are capable of assembling an E2-liganded ERα-like transcription activation complex at the EpRE in response to TOT. More importantly, this result is not cell line-specific and demonstrates that ERβ can assemble a functional ERα-like transcriptional complex in the absence of ERα.
PARP-1 is involved in tamoxifen-mediated increase of antioxidative enzyme expression
PARP-1 and topoisomerase IIβ together are required for E2-dependent activation by ERα and only PARP-1 is recruited by TOT-ERα resulting in the repression (Ju et al., 2006). However, we observe a TOT-dependent corecruitment of PARP-1 and topoisomerase IIβ along with hPMC2 and ERβ (Figures 2 and 4). To investigate the functional relevance of PARP-1 recruitment in the induction of EpRE-regulated genes, we transiently downregulated PARP-1 expression by siRNA transfection in MCF10A FL-ERβ cells (Figure 5a). The downregulation of PARP-1 resulted in >50% decrease in the ability of TOT to induce the antioxidative enzymes GCSh, QR and GSTpi (Figure 5b). Similar results were obtained by four different siRNA oligos, targeted against different regions of PARP-1 mRNA, indicating that the result is unlikely to be an off target-effect (data not shown). The basal expression levels of GCSh, QR and GSTpi were comparable in both siRNA-transfected and control cells (Supplementary Figure 3), indicating that the enzyme expression levels in TOT-treated samples were not the consequence of a general decrease in protein expression. This observation, along with TOT-dependent recruitment to the EpRE, suggests an important role for PARP-1 in ERβ-mediated induction of antioxidative genes.
Figure 5.

Knockdown of PARP-1 attenuates tamoxifen-dependent increase in the expression of antioxidative enzymes. FL-ERβ cells were treated for 3 h with 0.01% ethanol (c) or 10nM TOT (T) after 72 h posttransfection. (a) Western blot analysis of knockdown in PARP-1 expression. si1, si2, si3 and si4 are siRNA oligos targeted against different regions of PARP-1 mRNA. Numbers at the bottom represent the percentage of knockdown in siRNA-transfected cells as compared to their corresponding lanes in the control–transfected samples. GAPDH was used as a loading control. (b) Expression levels of GCSh, QR and GAPDH were analyzed and normalized to that of GAPDH in each case. The bars represent the fold change in the expression levels of the indicated enzymes as compared to their corresponding ethanol-treated samples. Error bars indicate standard deviation of four independent experiments. EpRE, electrophile response element; FL-ERβ, Flag-ERβ; PARP-1; poly (ADP-ribose) polymerase 1; QR, quinone reductase; TOT, trans-hydroxytamoxifen.
Discussion
Our studies demonstrate a TOT-dependent, localized co-recruitment of the transcription factors ERβ, hPMC2, PARP-1, topoisomerase IIβ, SRC-1, ERα and Nrf2 to the EpRE. Recruitment of the entire complement of coactivators is observed both in the presence and absence of ERα. More importantly, except for Nrf2, recruitment of all other coactivators is dependent on the presence of both ERβ and hPMC2. Together, our data indicate TOT-dependent targeting of ERβ and hPMC2 to the EpRE, with subsequent recruitment of other transcriptional regulators to form a coactivator complex resulting in transcriptional induction. The key roles played by ERβ and hPMC2 are highlighted by our observations that the lack of either of these factors results in an almost complete loss of (1) TOT-induced antioxidative gene expression, (2) TOT-dependent decrease in the levels of catechol estrogen quinones and (3) protection against E2-induced ODD by TOT. Transcriptional induction by ERβ and hPMC2 at least in part is dependent upon PARP-1 recruitment, as downregulation of PARP-1 expression results in attenuated induction of antioxidative genes by TOT. However, our observations do not support a significant role for ERα and Nrf2 in TOT–ERβ-mediated transcription at the EpRE.
We verified induced transcription of the EpRE-regulated genes NQO1, GCSH and GSTP1 in MCF7 breast epithelial cells (Supplementary Figure 1). This induction exhibited a strong correlation with the ability of TOT to protect against E2-induced ODD, as evidenced by the fact that the absence of either ERβ or hPMC2 results in the loss of both TOT-dependent gene induction and also protection against E2-induced ODD after TOT treatment (Figures 1b and 3). The EpRE-transcription factor Nrf2 is required for basal transcription of EpRE genes and induces EpRE promoters in response to high levels of oxidative stress (Ramos-Gomez et al., 2001). Accordingly, we observed constitutive localization of Nrf2 at the EpRE (Figures 2a, b, 4a and b) However, in the absence of either ERβ or hPMC2, Nrf2 was insufficient by itself to induce antioxidative genes, or protect against E2-induced ODD (Figures 1b and 3), indicating that it does not contribute significantly to TOT–ERβ-mediated regulation of EpRE. A similar observation is true for ERα, even though ERα is recruited to the EpRE in a TOT-dependent manner. FL-ERβ cells lacking ERα expression revealed effective TOT-dependent antioxidative enzyme induction and were protected against E2-induced ODD by TOT (Figure 3). This observation is also supported by our previous findings where TOT– ERα did not significantly induce the transcription of EpRE-regulated reporter genes (Montano et al., 1998). TOT treatment alone was insufficient to prevent MCF10A cells from acquiring tumorigenicity upon exposure to E2 (Montano et al., 2007). Also, exogenous expression of ERβ, not ERα, inhibited the tumorigenic potential of E2 in the presence of TOT (Montano et al., 2007).
Analysis of the mechanistic basis of ERβ and hPMC2 functions yielded a constitutive interaction between ERβ and hPMC2 (Figure 1c). We observe simultaneous recruitment of both ERβ and hPMC2 to the EpRE when both of them are expressed, but little or no recruitment of either protein in the absence of the other (Figures 2e and 4c). These results suggest the recruitment of an ERβ–hPMC2 complex to the EpRE. ERβ– hPMC2 interaction is constitutive (Figure 1c), while recruitment to the EpRE is TOT-dependent (Figures 2a and 4a). An explanation is that while the presence or absence of a ligand does not affect the interaction between ERβ and hPMC2, it could affect the conformation of the ERβ–hPMC2 complex. This could in turn result in differential recruitment of ERβ and hPMC2 to target sequences in response to different ligands. This explanation seems more likely when we consider the recruitment of ERβ and hPMC2 to the ERE region of the pS2 gene. ChIP analysis in MCF10A FL-ERβ cells revealed recruitment of ERβ and hPMC2 to the ERE under both E2 and TOT treatments (Supplementary Figure 2), in contrast to the predominantly TOT-dependent recruitment observed at EpRE sequences.
Transcriptional induction at the EpRE by the TOT– ERβ–hPMC2 pathway involves a coactivator complex very similar to that of E2–ERα (Figures 2a and b), but independent of ERα recruitment (Figures 4a and b). An explanation is that even though both ERα and ERβ are recruited to the EpRE, only TOT–ERβ recruitment results in transcriptional induction. In fact, studies on ligand-dependent recruitment to both classical and nonclassical ER response genes indicate that the ability of either ERα or ERβ to activate transcription is not solely dependent on their recruitment to DNA, but also depend on both the ligand and the promoter context (Paech et al., 1997; Routledge et al., 2000; Wong et al., 2001; Matthews and Gustafsson, 2003; Chang et al., 2006; Matthews et al., 2006). TOT-dependent corecruitment of ERα and ERβ to the EpRE (Figure 2e), suggests the possibility of an ERα–ERβ heterodimer. It has been shown that ERα and ERβ form functional heterodimers and that the heterodimer form predominates when both isoforms are expressed (Pettersson et al., 1997). Alternately, the presence of ERα could be a consequence of protein–protein interactions with the coactivators recruited to the EpRE.
We observe increased recruitment of Nrf2 in response to TOT (Figures 2a and 4a). This can be explained by considering both in vitro (Montano et al., 2004) and in vivo (Figure 2e) data that indicate corecruitment of Nrf2 with ERβ and hPMC2. Such an interaction can potentially result in more stable binding of Nrf2–EpRE or indirect recruitment of Nrf2 by the ERβ-coactivator complex. Even though Nrf2 is required for transcription of EpRE-regulated genes, TOT treatment neither increases antioxidative enzyme levels nor inhibits E2-induced ODD in the absence of ERβ or hPMC2 (Figure 3). This indicates that the ODD protective effect of TOT is primarily mediated by ERβ and hPMC2, as Nrf2 alone is insufficient to induce antioxidative gene expression in response to TOT.
Finally, both PARP-1 and topoisomerase IIβ are corecruited to the EpRE in response to TOT (Figures 2a, e and 4a). It has been proposed that E2– ERα recruitment switches promoter occupancy from PARP-1 to PARP-1/topoisomerase IIβ complex, resulting in gene activation (Lis and Kraus, 2006). Such a scenario would explain the presence of PARP-1 at the estrogen receptor response element (ERE) region in response to TOT treatment (Supplementary Figure 2). However, at the EpRE region, we do not observe PARP-1/topoisomeasre IIβ recruitment in the absence of either ERβ or hPMC2 (Figure 4c). It is possible that PARP-1 is recruited subsequent to ERβ and hPMC2, but plays a role in recruiting other coactivators by protein–protein interaction. Such a role for PARP-1 is shown to be necessary for transcription from RARβ2 promoters (Pavri et al., 2005). Even though PARP-1 is localized to the promoters of many actively transcribing genes, knockdown of PARP-1 does not affect the transcription of all the genes (Krishnakumar et al., 2008), indicating that mere recruitment does not indicate a functional role. In this regard, our observation that the downregulation of PARP-1 attenuates TOT-dependent induction of EpRE promoters coupled with TOT-dependent PARP-1/topoisomerase IIβ recruitment to the EpRE underscores the contribution of PARP-1 in mediating oxidative stress responses through the TOT– ERβ pathway.
In conclusion, our data provide a mechanistic basis for the protective effect of TOT against E2-induced DNA damage. We show that the protective effect of TOT is primarily mediated by ERβ and hPMC2. This is significant given that ERβ expression is increasingly being recognized as a positive prognostic factor for tamoxifen treatment, especially in ERα-negative breast cancers (Gruvberger-Saal et al., 2007; Speirs and Walker, 2007). Our studies imply an important role for hPMC2 in mediating chemoprevention against breast cancer. Not much is known about the biological role of this protein apart from its ability to increase transcription from EpRE promoters in response to tamoxifen. The C-terminus of hPMC2 encodes a putative exonuclease domain (ExoIII), while in vitro data suggest that that interaction with ERβ is mediated through the N-terminal (unpublished data). We can speculate that the transcriptional regulation by ERβ and hPMC2 could potentially involve double-strand DNA breaks induced by the exonuclease activity and subsequent recruitment of multifunctional proteins such as PARP-1. However, further studies are required to identify the biological functions of this protein. Nevertheless, our characterization of the mechanism of transcriptional regulation by antiestrogen-ERβ and identification of key players in this pathway will help us devise new strategies to prevent breast cancer progression.
Materials and methods
Plasmids
The construction of pCMV-–Flag–ERβ has been described previously (Montano et al., 1998). pSuper-QRshRNA and pSuper-hPMC2shRNA were constructed by annealing to its complement, an oligonucleotide that specifies a 19 nucleotides sequence derived from the NQO1 and hPMC2 genes, respectively, and separated by a short spacer from the reverse complement of the same 19 nucleotide sequence. Oligonucleotides were ligated to the pSUPER vector (Brummelkamp et al., 2002). To make pcDNA-hPMC2 569miR, the oligos encoding the miRNA sequences were annealed and cloned into the pcDNA 6.2 GW/EmGFP vector (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. The miRNA sequences used to construct the plasmids are listed in Supplementary Table 1.
Tissue culture and retroviral transfection
Breast epithelial cells (MCF7 and MCF10A) and PA317 amphotropic packaging cells were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained according to their recommended protocols. Retroviruses were made by transfecting PA317 cells with the pSuper plasmid, pSuper-hPMC2shRNA or pSuper-QRshRNA. MCF7 cells were infected with retrovirus as described previously (Bianco et al., 2003). For all experiments, breast epithelial cells were depleted of estrogen by growth in improved minimal essential media minus phenol red containing 5% charcoal–dextran treated calf serum for 5 days before ligand treatment. Twenty-four hours postinfection cells were treated with the indicated ligands for 24 h, and processed either for western blot analysis or immunostaining.
RNA extraction and semiquantitative reverse transcriptase–PCR
RNA was extracted from MCF7 cells and total mRNA was analyzed by reverse transcriptase–PCR as described in the Supplementary Methods. The primer sequences used are listed in Supplementary Table 2.
Generation of stable MCF10A cell lines
MCF10A FL-ERβ cell line was generated by transfecting MCF10A human breast epithelial cells with pCMV–Flag–ERβ using FuGene HD Transfection Reagent (Roche, Indianapolis, IN, USA). Cells were selected on growth media containing 500 μg/ml G418 and resistant colonies were isolated and expanded. Positive colonies were identified by western blot analysis of FL-ERβ expression.
To generate MCF10A Fl-ERβ/hPMC2mi cell line, MCF10A FL-ERβ cells were transfected with pcDNA-hPMC2 569miR, and stable transfectants cells were selected by growing cells in the presence of 5μg/ml blasticidin. The cells were allowed to reach 70% confluency and were then flow sorted for green fluorescent protein (GFP) expression. Knockdown of hPMC2 expression was confirmed by western blot analysis.
Immunostaining for 8-OHdG
MCF7 and MCF10A cells grown on coverslips were processed further for immunostaining with anti-8-OHdG antibody as described in the Supplementary Methods.
Endogenous. immunoprecipitation and western blot analysis
Cell lysates were subjected to immunoprecipitation and the eluates were analyzed by western blotting as described in the Supplementary Methods.
Chromatin immunoprecipitation
Cells fixed with 1% formaldehyde were processed further for immunoprecipitation of the chromatin using various antibodies as described in the Supplementary Methods. The primer sequences used are listed in Supplementary Table 2 and the list of antibodies in Supplementary Table 3.
Liquid chromatography/mass spectrometry analysis of E2 metabolites
Cells were treated with either 50 nM E2 alone or together with 10 nM TOT for 24 h. The media was collected and subjected to liquid chromatography/mass spectrometry analysis to detect levels of the indicated metabolites as described by (Montano et al., 2007). The amount of each metabolite was normalized to the total number of cells and the resulting values were used to calculate the fold change. Fold change in metabolite levels for each cell line was calculated as the ratio of the normalized value from ligand-treated sample to that of its corresponding ethanol-treated cell line.
Transient siRNA transfection
Cells were maintained on stripped serum media 2 days before transfection. Trypsinized cells were then independently transfected with four different double-stranded siRNA oligos from Qiagen (Valencia, CA, USA) targeted along the PARP-1 mRNA. The transfections were carried out using siPORT amine reagent from Ambion (Austin, TX, USA) as per the reverse transfection protocol provided by the manufacturer. After 72 h, both transfected and nontransfected cells were treated with 0.01% ethanol (control) or 10 nM TOT for 3 h. Cells were harvested in phosphate-buffered saline (PBS) and whole cell lysates were analyzed by western blotting to determine the protein expression levels.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants (CA92440 and CA130066) to MMM.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Bianco NR, Perry G, Smith MA, Templeton DJ, Montano MM. Functional implications of antiestrogen induction of quinone reductase: inhibition of estrogen-induced DNA damage. Mol Endocrinol. 2003;17:1344–1355. doi: 10.1210/me.2002-0382. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbcan.2006.03.001. e-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- Fabian C. Tamoxifen or raloxifene in postmenopausal women for prevention of breast cancer: a tale of two choices–counterpoint. Cancer Epidemiol Biomarkers Prev. 2007;16:2210–2212. doi: 10.1158/1055-9965.EPI-06-1065. [DOI] [PubMed] [Google Scholar]
- Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Rad Biol Med. 2007;43:1289–1298. doi: 10.1016/j.freeradbiomed.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruvberger-Saal SK, Bendahl PO, Saal LH, Laakso M, Hegardt C, Eden P, et al. Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin Cancer Res. 2007;13:1987–1994. doi: 10.1158/1078-0432.CCR-06-1823. [DOI] [PubMed] [Google Scholar]
- Jordan VC. Tamoxifen or raloxifene for breast cancer chemoprevention: a tale of two choices–point. Cancer Epidemiol Biomarkers Prev. 2007;16:2207–2209. doi: 10.1158/1055-9965.EPI-07-0629. [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science. 2008;319:819–821. doi: 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- Li KM, Todorovic R, Devanesan P, Higginbotham S, Kofeler H, Ramanathan R, et al. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis. 2004;25:289–297. doi: 10.1093/carcin/bgg191. [DOI] [PubMed] [Google Scholar]
- Liehr JG. Is estradiol a genotoxic mutagenic carcinogen? Endocr Rev. 2000;21:40–54. doi: 10.1210/edrv.21.1.0386. [DOI] [PubMed] [Google Scholar]
- Lis JT, Kraus WL. Promoter cleavage: a topoIIbeta and PARP-1 collaboration. Cell. 2006;125:1225–1227. doi: 10.1016/j.cell.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv. 2003;3:281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- Matthews J, Wihlen B, Tujague M, Wan J, Strom A, Gustafsson JA. Estrogen receptor (ER) beta modulates ERalpha-mediated transcriptional activation by altering the recruitment of c-Fos and c-Jun to estrogen-responsive promoters. Mol Endocrinol. 2006;20:534–543. doi: 10.1210/me.2005-0140. [DOI] [PubMed] [Google Scholar]
- Menzel HJ, Sarmanova J, Soucek P, Berberich R, Grunewald K, Haun M, et al. Association of NQO1 polymorphism with spontaneous breast cancer in two independent populations. Br J Cancer. 2004;90:1989–1994. doi: 10.1038/sj.bjc.6601779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano MM, Chaplin L, Deng H, Mesia-Vela S, Gaikwad N, Zahid M, et al. Protective roles of quinone reductase and antiestrogens in estrogen-induced mammary cell tumorigenesis. Oncogene. 2007;26:3587–3590. doi: 10.1038/sj.onc.1210144. [DOI] [PubMed] [Google Scholar]
- Montano MM, Deng H, Liu M, Sun X, Singal R. Transcriptional regulation by the estrogen receptor of antioxidative stress enzymes and its functional implications. Oncogene. 2004;23:2442–2453. doi: 10.1038/sj.onc.1207358. [DOI] [PubMed] [Google Scholar]
- Montano MM, Jaiswal AK, Katzenellenbogen BS. Transcriptional regulation of the human quinone reductase gene by antiestrogen-liganded estrogen receptor-alpha and estrogen receptor- beta. J Biol Chem. 1998;273:25443–25449. doi: 10.1074/jbc.273.39.25443. [DOI] [PubMed] [Google Scholar]
- Montano MM, Wittmann BM, Bianco NR. Identification and characterization of a novel factor that regulates quinone reductase gene transcriptional activity. J Biol Chem. 2000;275:34306–34313. doi: 10.1074/jbc.M003880200. [DOI] [PubMed] [Google Scholar]
- Oestergaard MZ, Tyrer J, Cebrian A, Shah M, Dunning AM, Ponder BA, et al. Interactions between genes involved in the antioxidant defence system and breast cancer risk. Br J Cancer. 2006;95:525–531. doi: 10.1038/sj.bjc.6603272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- Pavri R, Lewis B, Kim TK, Dilworth FJ, Erdjument-Bromage H, Tempst P, et al. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell. 2005;18:83–96. doi: 10.1016/j.molcel.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Pettersson K, Grandien K, Kuiper GG, Gustafsson JA. Mouse estrogen receptor beta forms estrogen response element-binding heterodimers with estrogen receptor alpha. Mol Endocrinol. 1997;11:1486–1496. doi: 10.1210/mend.11.10.9989. [DOI] [PubMed] [Google Scholar]
- Prestera T, Talalay P. Electrophile and antioxidant regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA. 1995;92:8965–8969. doi: 10.1073/pnas.92.19.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routledge EJ, White R, Parker MG, Sumpter JP. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) alpha and ER beta. J Biol Chem. 2000;275:35986–35993. doi: 10.1074/jbc.M006777200. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- Sakoda LC, Blackston CR, Xue K, Doherty JA, Ray RM, Lin MG, et al. Glutathione S-transferase M1 and P1 polymorphisms and risk of breast cancer and fibrocystic breast conditions in Chinese women. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9633-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Speirs V, Walker RA. New perspectives into the biological and clinical relevance of oestrogen receptors in the human breast. J Pathol. 2007;211:499–506. doi: 10.1002/path.2130. [DOI] [PubMed] [Google Scholar]
- Strange RC, Spiteri MA, Ramachandran S, Fryer AA. Glutathione-S-transferase family of enzymes. Mutat Res. 2001;482:21–26. doi: 10.1016/s0027-5107(01)00206-8. [DOI] [PubMed] [Google Scholar]
- Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54:4313–4320. [PubMed] [Google Scholar]
- Udler M, Maia AT, Cebrian A, Brown C, Greenberg D, Shah M, et al. Common germline genetic variation in antioxidant defense genes and survival after diagnosis of breast cancer. J Clin Oncol. 2007;25:3015–3023. doi: 10.1200/JCO.2006.10.0099. [DOI] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P) H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CW, Komm B, Cheskis BJ. Structure-function evaluation of ER alpha and beta interplay with SRC family coactivators. ER selective ligands Biochemistry. 2001;40:6756–6765. doi: 10.1021/bi010379h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.