

SUMMARY
Gut infections often lead to epithelial cell damage followed by a healing response. We examined changes in the epithelial cell cytoskeleton and the involvement of host adaptive immunity in these events using an in vivo model of parasitic infection. We found that both ezrin and villin, key components of the actin cytoskeleton comprising the brush border (BB) of intestinal epithelial cells (IECs) underwent significant post-translational changes following gut infection and during the recovery phase of gut infection. Intriguingly, using mice lacking either CD4+ or CD8+ T cell responses, we demonstrated that the mechanisms by which ezrin and villin are regulated in response to infection are different. Both ezrin and villin undergo proteolysis during the recovery phase of infection. Cleavage of ezrin requires CD4+ but not CD8+ T cells, while cleavage of villin requires both CD4+ and CD8+ T cell responses. Both proteins were also regulated by phosphorylation; reduced levels of phosphorylated ezrin and increased levels of villin phosphorylation were observed at the peak of infection and correlated with reduced BB enzyme activity. Finally, we show that infection also leads to enhanced proliferation of IECs in this model. Cytoskeletal remodeling in IECs can play critical roles in the immunopathology and healing responses observed during many infectious and non-infectious intestinal conditions. These data indicate that cellular immune responses can be significant drivers of these processes.
Keywords: Intestinal epithelial cells, cytoskeletal remodeling, ezrin, villin, gut infection, immunopathology
INTRODUCTION
The structural integrity of the intestinal epithelium is a crucial factor for proper gut function and absorption. The BB surface provides the major interface for nutrient absorption and reduction in the surface area of the microvilli is linked with reduced levels of enzymes such as sucrase and lactase that are necessary for proper digestion and absorption of sugars.1 Damaged intestinal epithelial cells could result in reduced intestinal enzymes, disposing infected individuals to malnutrition.
The structural proteins ezrin and villin are among the most abundant proteins present in the intestinal BB. Ezrin functions to cross-link the plasma membrane and the actin cytoskeleton. Evidence suggests that ezrin participates in a wide range of biological functions such as signal transduction, cell division, cell growth, cytoskeletal organization and morphogenesis.2 Ezrin also plays an important role in both microvillus formation and tight junction integrity, key elements in proper nutrient absorption.3,4 Given that ezrin has a major role in the formation of microvilli and is a linker between actin filaments and BB proteins, conformational changes in ezrin could drastically influence the integrity of microvilli.
Villin is another actin-binding protein which is present in gastrointestinal, renal and urogenital epithelial cells; villin is an extraordinarily versatile actin modifying protein in that it has the ability to both polymerize and depolymerize actin as well as sever, nucleate, bundle and cap actin filaments.5 Unlike gelsolin, another protein in the same superfamily, villin has the ability to assemble actin filaments as well.6 These studies provided a molecular mechanism for villin’s role in regulating epithelial cell plasticity related to cell injury.6 It has been previously indicated that fragments containing the COOH-terminus fragment of the protein have anti-apoptotic activity while the NH2-terminal-containing fragments of villin have pro-apoptotic functions through regulating the activation of caspase 3 and caspase 9. 6,7 It has been hypothesized that as intestinal epithelial cells migrate from the crypt to the villus tip to be shed into the lumen, villin is cleaved to generate pro-apoptotic fragments, thus enhancing and/or assisting the process of cell extrusion from the gastrointestinal epithelium.5 A correlation between villin deficiency and increased apoptosis has been noted in several inflammatory diseases of the gastrointestinal tract. For example, a decrease in the levels of villin expression in enterocytes (ECs) from patients with inflammatory bowel disease relative to healthy controls has been reported, 8 and villin-null mice have higher levels of apoptosis compared to their wild-type (WT) littermates that correlate with the severity of colitis induced by dextran sodium sulfate (DSS)-treatment.5 Additionally, differed villin expression has been implicated as a useful marker to predict the prognosis and the progress of different carcinomas.9,10
Tyrosine phosphorylation of villin regulates the villin/F-actin association, and tyrosine-phosphorylated villin molecules have lower affinity for actin filaments.11 Upon phosphorylation, villin molecules show different affinity for several ligands; 5,12 tyrosine phosphorylation of villin streamlines its ability to cut actin filaments at extremely low concentrations of Ca2+, suggesting that tyrosine phosphorylation of villin may be responsible for the efficient severing of actin filaments in vivo.13 However, tyrosine-phosphorylated villin promotes cell migration and induces cytoskeletal changes including the redistribution of F-actin to the cell perimeter and loss of stress fibers, suggesting that phosphorylated villin induced actin cytoskeletal remodeling is a crucial step in cell migration.13
Here we examined the hypothesis that these functional changes in the intestinal epithelium are due to immune mediated alterations in the epithelial cell cytoskeleton. We investigated the expression level, localization, and post-translational modifications of ezrin and villin as well as any involvement of host immune responses in these changes using a gut infection model. Also, we provide evidence suggesting roles for villin as an anti-apoptotic and homeostatic protein following gut infection. Furthermore, experiments using knockout animals demonstrated that ezrin modification was CD4+-dependent, while villin modification required both CD4+ and CD8+ T cells.
RESULTS
The actin-binding protein ezrin is cleaved following gut infection: ezrin cleavage is CD4+-dependent
Because the linker protein ezrin is intimately involved in the formation of microvilli, we investigated whether there were changes in the post-translational modification and protein expression levels of ezrin following gut infection, and any possible involvement of host immune responses. In order to test this, we infected WT mice with the GS strain of G. duodenalis as shown in Figure 1a and performed Western blot analysis of ezrin in jejunal homogenates of 5 and 18 day infected animals. Our findings demonstrate that total ezrin expression levels were not changed significantly following gut infections in WT mice. While a small amount of cleaved ezrin is apparent in uninfected (day 0) mice and at 5 days following infection, the full-length ezrin was almost completely cleaved by day 18 p.i. (Figure 1b). The cleavage process yielded a major fragment of 55-kD (ezrin p55). Since ezrin functions to cross-link actin and the plasma membrane, cleaved ezrin is therefore unable to connect these two structures.
Figure 1.

Post-translational regulation of ezrin following gut infection. The experimental protocol for infecting mice with both strains of G. duodenalis is depicted in (a). Mice were maintained on neomycin, vancomycin and ampicillin throughout the experiment to facilitate infection with either strain of parasite. WT (b), SCID (c), β2m−/− (d), and CD4−/− (e) mice were infected with the GS strain of G. duodenalis and jejunal homogenates were examined for post-translational modifications in ezrin using Western blots. GAPDH was used as a loading control. Each figure is representative of 4 mice / time point. Ezrin was also analyzed following infection of WT C57BL/6 mice with the WB strain of the parasite (f). Using paraffin-embedded tissue and IF, the localization of total ezrin (upper panel) and phosphorylated ezrin (p-ezrin, lower panel) was also determined in the intestines of mice infected with the GS strain of the parasite for 0, 5 or 18 days (g). Each panel represents 4 mice/time point.
In order to investigate the role of host immunity in mediating the cleavage of ezrin, we performed similar analyses following infection in SCID mice. We observed no increase in cleavage of ezrin during the course of infection, in spite of the heavy parasite loads found at days 5 and 18, 14 suggesting that the ezrin cleavage requires an intact host adaptive immune response (Figure 1c). Next, we sought to determine the specific cell types mediating these events. CD4−/− and β2m−/− mice were infected and analyzed for the cleavage of ezrin. We have previously shown that CD4−/− mice fail to control infections and do not exhibit reduced sucrase activity. In contrast, β2m−/− mice (lacking CD8+ T cells) control infections normally, but also have no reduction in sucrase activity.14 Western blot analyses showed that no increase in ezrin cleavage occurred in CD4−/− animals, while β2m−/− mice exhibited a cleavage pattern identical to WT littermates (Figure 1d and e). Thus, the cleavage of ezrin is regulated in a CD4+-T cell dependent manner. We also examined the cleavage of ezrin following infection with a different strain of the parasite. Ezrin was cleaved following WB infection in WT mice in a manner similar to WT mice infected with the GS strain (Figure 1f).
µ-calpain is activated in WT mice, but not in SCID mice, following gut infection
Ezrin serves as a substrate for calpain proteases, and calpain I has been shown to target ezrin specifically, yielding a specific 55-kD cleaved product.15 We therefore asked whether µ-calpain was activated following gut infection. We used a polyclonal antibody able to detect both inactive and active forms of µ-calpain. The native protein (~80-kD) is inactive and is processed to a 76-kD fragment representing the active form of µ-calpain. Western blot analysis showed that µ-calpain was activated at day 18 p.i., and densitometry analysis indicated that the active form of µ-calpain was about 3.5 times more abundant than that observed in uninfected animals (Figure 2a). These findings correlate with the cleavage of ezrin observed at day 18 p.i. In SCID mice, however, most µ-calpain was in its inactive form, consistent with lack of ezrin cleavage in these mice (Figure 2b). These data suggest that host µ-calpain was likely responsible for the post-translational cleavage of ezrin following infection.
Figure 2.

Activation of µ-calpain following infection. WT (a) and SCID (b) mice were infected with the GS strain of G. duodenalis and jejunal homogenates were tested for activated µ-calpain by Western blot using an antibody capable of detecting both active and inactive forms of µ-calpain. GAPDH was used as a loading control. Each figure is representative of 4 mice/time point.
Localization of total ezrin and phospho-ezrin is altered following gut infection
It is well-recognized that ezrin plays a critical role in organizing membrane domains such as the intestinal BB within cells.3,4 We therefore analyzed ezrin phosphorylation and localization using a monoclonal antibody against a major phosphorylated form of ezrin (Thr567) and also a polyclonal antibody against total ezrin. In uninfected animals, ezrin and phospho-ezrin are found localized to the BB of the intestinal epithelium. In contrast, at day 5 p.i. we observed significantly reduced staining for both ezrin and phospho-ezrin at the BB (Figure 1g and h). Together with the Western blot analysis indicating no decrease in total ezrin levels, the data suggest that ezrin may be distributed more diffusely throughout the cells. This would be consistent with a lack of phosphorylation at Thr567, since this correlates with an inactive conformation of ezrin. Staining of sections from 18 days p.i. indicates that most ezrin localization is restored.
Villin is proteolyzed following gut infection in WT in an immune-mediated manner
To investigate the impact of gut infection on the regulation of villin following gut infection, WT mice were analyzed at days 0, 5, and 18 p.i. by Western blotting using antibodies against epitopes in the COOH-terminal region of the protein. Results showed that villin was proteolyzed following infection at day 18 p.i., and that several COOH-terminal products (approximately 87-kD, 54-kD, and 41-kD) accumulated (Figure 3a). The total level of villin (native protein and proteolyzed products) was upregulated about 2.5 times by densitometry analysis at day 18 while total proteins levels at day 5 remained unchanged. The upregulation of villin could be explained by the existence of a compensatory mechanism in the infected intestine in immediate response to villin proteolysis at day 18.
Figure 3.

Villin proteolysis following gut infection. WT (a), SCID (b), β2m−/− (c), and CD4−/− mice (d) were infected with the GS strain of G. duodenalis. Jejunal homogenates were analyzed by Western blot for villin post-translational modifications using a polyclonal antibody against a peptide in the COOH-terminal portion of the protein. Villin proteolysis was also examined in WT mice infected with the WB strain of the parasite (e). Renal villin in WT mice infected with the GS strain was also analyzed (f). Each figure is representative of four animals / time point. GAPDH was used as a loading control.
Since post-translational changes in ezrin required intact immune responses we next examined the role of cellular immune responses in regulating the cleavage of villin. Intestinal tissues of SCID mice infected with the parasite were analyzed by Western blot at 0, 5, and 18 days p.i. Similar to ezrin cleavage, in the absence of adaptive immunity villin proteolysis was not seen (Figure 3b). Furthermore, no villin upregulation was observed. Villin proteolysis was also not observed in either CD4−/− or β2m−/− mice, and levels of villin were unchanged during the course of infection (Figure 3c and d). These findings suggest that both the upregulation and cleavage of villin require both CD4+ and CD8+ T cells. In contrast, while ezrin cleavage is a CD4+-dependent phenomenon, it does not require CD8+ T cells.
In order to determine the impact of pathogen strain on the cytoskeletal changes we observed during the infection of WT mice with the GS strain of G. duodenalis, we infected WT C57BL/6 mice with the WB strain of the parasite. Our findings showed that no apparent villin proteolysis occurred in WT mice following infection with the WB strain (Figure 3e). Finally, we also examined villin in kidneys from mice infected with GS and found that no upregulation or cleavage occurred at a remote site, indicating that villin proteolysis is restricted to the site of infection in this model (Figure 3f).
Villin is redistributed in IECs following gut infections
To analyze whether gut infection also induced villin redistribution, we performed an immunohistochemistry (IHC) analysis on formalin-fixed intestinal sections. Analyses showed that in uninfected mice ECs displayed strong apical decoration (the BB) as well as diffuse cytoplasmic labeling. In infected mice, however, villin localization was more diffuse with some accumulation near both the apical and baso-lateral plasma membranes (Figure 4). Given that villin abundance increases at day 18 p.i., it is possible that the changes in the localization of villin could be a consequence both of villin redistribution as well as increased expression.
Figure 4.

Villin localization following gut infection. WT mice were infected with the GS strain and villin distribution and localization were determined by IF in jejunal tissue at day 5 (middle panel) and day 18 (lower panel) compared with uninfected mice (upper panel). Each panel is representative of 4 animals / time point.
Tyrosine phosphorylation of villin occurs following gut infection with GS but not WB
Since we had observed changes in phospho-ezrin at 5 days p.i. in WT mice, we also examined phosphorylation of villin following infection. Tyrosine phosphorylation of villin promotes villin activity leading to increased cell migration and cell movement. It can induce cytoskeletal changes including the redistribution of F-actin to the cell perimeter and loss of stress fibers, suggesting that tyrosine phosphorylation of villin is a crucial step in promoting cell migration.12, 16 We, therefore, immunoprecipitated villin from cell lysates and analyzed tyrosine phosphorylation by Western blot. Our data indicate that tyrosine-phosphorylated villin was 4.1 times more abundant at day 5 p.i. in WT mice infected with the GS strain, compared with uninfected animals (Figure 5a). Moreover, villin phosphorylation was not seen following infection with the WB strain or after GS infection in SCID mice (Figure 5b and c).
Figure 5.
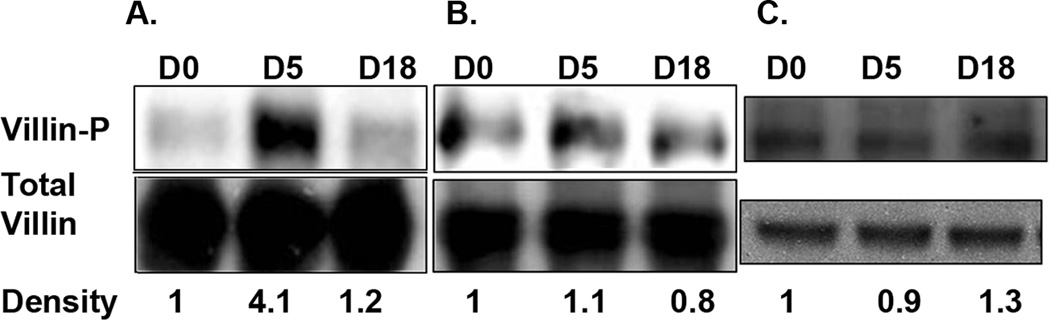
Tyrosine-phosphorylation of villin following gut infection. Villin was immunoprecipitated from jejunal homogenates of WT mice infected with the GS strain (a) or the WB strain (b). Increased tyrosine-phosphorylation of villin was not seen in SCID mice following infection with the GS strain of G. duodenalis (c). Immunoprecipitated proteins were analyzed for tyrosine phosphorylation by Western blot and levels of phosphorylated protein (upper panels) were normalized to levels of total villin (lower panels) by densitometry. Each figure represents 4 animals / time point.
COOH-terminal fragments of villin accumulate following gut infection
We next were interested in possible roles of the proteolytic fragments which accumulated. NH2-terminal fragments of villin have been reported to promote apoptosis while COOH-terminal fragments can have an anti-apoptotic role.6 We therefore asked if any cleaved products containing the NH2-terminal region of the protein accumulated by using a rabbit polyclonal antibody raised against the NH2-terminus of the protein. No NH2-terminal-containing cleaved products were detected in WT, SCID, CD4−/− or β2m−/− mice (Figure 6a-d). Together with our data that COOH-terminal fragments accumulate at day 18, these findings suggest that the proteolysis of villin following gut infections may provide anti-apoptotic signals in intestinal epithelial cells and support homeostasis in the recovering intestine.
Figure 6.

Analysis of NH2-terminal-containing villin fragments following G. duodenalis infection. WT (a), SCID (b), β2m−/− (c) and CD4−/− (d) mice were infected with the GS strain of the parasite, and jejunal homogenates were analyzed using an antibody against a peptide in the NH2-terminal portion of villin by Western blot. GAPDH was used as a loading control. Each figure is representative of four animals / time point.
Gut infection increases proliferation and migration of ECs
We hypothesized that infection might be inducing an increase in epithelial cell proliferation coupled with an accelerated rate of cell migration upward along the crypt-villus axis. This proliferation would generate a population of less mature epithelial cells which are known to express reduced levels of BB enzymes such as sucrase and lactase. We therefore asked if infection with the parasite would increase epithelial cell proliferation/migration. WT mice were infected with the GS strain of the parasite and mice were injected with 5-bromo-2'-deoxyuridine (BrdU) 2 h before euthanasia. Enumeration of cells which had incorporated BrdU in this time frame revealed a significant increase in the rate of cell proliferation on day 5 p.i. (Figure 7a). Furthermore, analysis of the distribution of these cells showed that epithelial cells in infected mice progress away from the crypts at a faster rate than in uninfected animals (Figure 7b and c). In order to evaluate the effect of the antibiotic regimen on the proliferative and migratory capacity of IECs in the absence of infection, intestinal tissue sections from uninfected time-matched controls were tested. Immunohistochemical staining of BrdU in paraffin sections showed that the antibiotic regimen alone did not alter the IECs proliferation and migration rates (data not shown).
Figure 7.
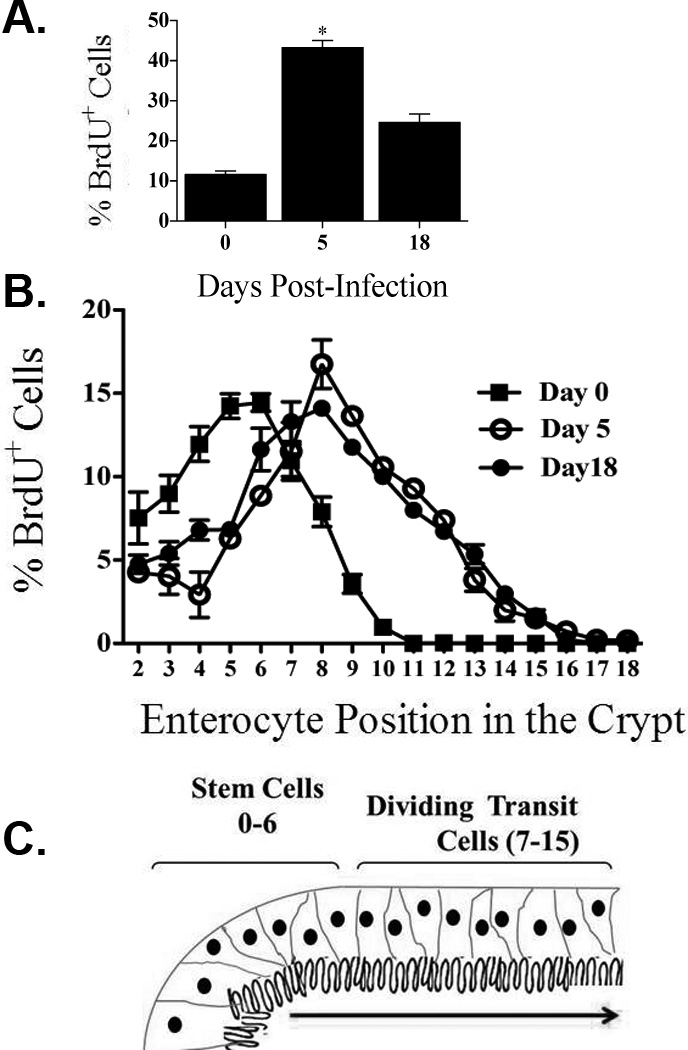
Intestinal epithelial cell proliferation and positional distribution following gut infection. WT mice were infected with the GS strain of the parasite and IEC proliferation was measured by BrdU incorporation during a two-hour pulse of BrdU. BrdU+ cells were visualized using IHC and light microscopy and the percentage of BrdU+ cells/total cells counted in each crypt that were labeled was determined (A). The positional distribution of BrdU+ cells IECs was then determined (B). A schematic diagram of an intestinal villus indicating the positioning of cells along the crypt-villus axis (C). Each figure represents 4 animals / time point. Data represent means ± SEM for four mice per group. *p < 0.05 by Mann–Whitney U test versus uninfected mice.
DISCUSSION
We have examined the mechanisms involved in pathophysiology and recovery from intestinal infection using a mouse model of infection with G. duodenalis. Specifically, we have examined the effect of infection on regulation of the IEC cytoskeleton, particularly the dynamics of the actin-binding proteins ezrin and villin. We have found that Giardia infection can induce drastic cytoskeletal changes in IECs and that these changes require a host adaptive immune response and vary based on the strain of parasite used. Both ezrin and villin were proteolyzed in WT mice at 18 days after gut infection with the GS strain of G. duodenalis but not the WB strain. Proteolysis of either protein was not observed in SCID or CD4−/− mice; cleavage of ezrin was seen following infection in β2m−/− mice. However, very low levels of villin proteolysis observed in β2m−/− mice could be explained by non-specific proteolysis occurring in the small intestines of mice examined. Activation of the protease µ-calpain was also seen on day 18 p.i. and may be involved in ezrin cleavage.15 We also showed that in WT mice localization of ezrin and villin was altered 5 days p.i. In both cases, localization at the BB was lost and these proteins appeared diffusely throughout the cytoplasm, with some localization of villin at the baso-lateral edge of the cells also observed. The NH2-terminal-containing fragments of proteolyzed villin did not accumulate in infected intestines, while fragments containing the COOH-terminal part of the protein were detected by Western blotting, suggesting an anti-apoptotic role for villin following gut infections.16 We also demonstrated an increase in tyrosine-phosphorylation of villin at day 5 p.i., in WT mice using the GS strain, but not following infection of SCID mice or using the WB strain. Similarly, we observed reduced immune-staining of phosphorylated ezrin (Thr567) at day 5 p.i. using the GS strain of G. duodenalis. Moreover, tyrosine phosphorylation of villin was seen only following infection of WT mice with the GS stain and not after infection of SCID mice or after infection with the WB strain, further supporting a role for post-translational regulation of villin in mediating pathophysiology.
Host cell cytoskeletal remodeling has been reported following several gut infections.17, 18, 19 Several entropathogens including Salmonella enterica serovar Typhimurium and Shigella flexneri alter cytoskeleton architecture at the site of bacterial attachment; changes in the actin cytoskeleton following enteric infections occur by the activation of Rho GTPases and tyrosine kinases and by directly modifying actin dynamics which facilitate bacterial invasion.20 Furthermore, some enteric pathogens manipulate the host actin cytoskeleton to enter IECs and to disseminate from cell to cell.21 Manipulation of the host cytoskeletal architecture is also seen in other intestinal pathogens; attachment of the protozoan parasite Cryptosporidium parvum triggers changes in the microvillar structures in the vicinity of the parasitophorous vacuole containing the parasite.22, 23
Villin proteolysis is also associated with infection with the causative agent of amebic colitis, Entamoeba histolytica; parasite derived cysteine proteinase activity are responsible for this cleavage since the inhibition of cysteine proteinases in trophozoites prevented villin proteolysis.24 These authors also showed that cysteine proteinases of enteric origin were likely not responsible for the proteolysis. The results of the current study, however, showed the lack of villin proteolysis in absence of host adaptive immunity in SCID mice in spite of massive infection burdens in these hosts (data not shown). Based on these observations, we concluded that a proteinase of host origin rather than a parasite-derived protease, is likely responsible for the post-translational events in villin following infection. Our data indicate that the µ-calpain protease is activated at 18 days p.i. This protease has been shown to cleave ezrin producing a 55-kD fragment similar to what we observe in these studies.15
The NH2-terminal fragment of villin have been shown to have pro-apoptotic functions, 7 and it has been hypothesized that as intestinal epithelial cells migrate from the crypt to the villus tip and are ready to be shed into the lumen, villin is cleaved to generate pro-apoptotic fragments thus enhancing and/or assisting the process of cell extrusion from the gastrointestinal epithelium.5 We found that the fragments containing the COOH-terminal portion of villin accumulated following G. duodenalis infection, but fragments containing the NH2 portion of protein did not. This suggests a role for villin as an anti-apoptotic and homeostatic protein following infection in vivo as shown before in vitro.5 Proteolysis of villin following gut infections can generate anti-apoptotic signals that counterbalance the deleterious effects of the presence of parasites and the immune responses against them, maintaining the homeostatic balance in the infected intestine.
Tyrosine kinases have been demonstrated to be important for intestinal epithelial cell migration.25 It has been shown that villin is phosphorylated in vitro by c-src kinase, 26 as well as in migrating cells in vivo.16 In the current study, we showed that in WT mice villin was tyrosine-phosphorylated at day 5 p.i. and that the levels of phosphorylated villin were approximately 4 times greater than in uninfected animals; levels of tyrosine-phosphorylated villin returned to pre-infection levels on day 18 p.i. In SCID mice, however, no changes were seen in the levels of tyrosine-phosphorylated villin during the course of infection, suggesting a role for adaptive immune responses in initiating these changes. Tyrosine phosphorylation of villin regulates the villin/F-actin association, and the tyrosine-phosphorylated villin molecules have lower affinity to bind to F-actin filaments.11 Villin has the ability to cut actin filaments at high concentrations of Ca2+ (100–200 µM) in vitro. Instead, the tyrosine-phosphorylated villin can sever actin filaments at nanomolar concentrations of Ca2+ ion, 16 suggesting that tyrosine phosphorylation of villin may be responsible for the efficient severing of action filaments in vivo.
In a few instances, it has been shown that bacterial pathogens are able to induce ezrin phosphorylation followed by proteolytic cleavage resulting in defective organ function. Helicobacter pylori CagA, for example, has been linked to src-mediated phosphorylation of ezrin, 27 inducing a conformational change that renders it susceptible to cleavage by proteases like calpains.28,29 In the current study, we found that µ-calpain is activated later following infection in a time-frame similar to the observed cleavage of ezrin. We also found that infection with G. duodenalis leads to delocalization of ezrin away from the BB. Given that ezrin is a major element of the microvilli formation and a linker between actin filaments and BB proteins, post-translational changes in ezrin could severely influence the integrity of microvilli, and this could account for abnormalities seen during gut infections.
Facilitated rates of epithelial cell turnover following intestinal infection have been reported as a defensive mechanism implemented by the host to expel intestinal helminth parasites.30 BrdU labeling studies indicate that epithelial cell proliferation and migration also increase following Giardia infection. This may result in an increase in the ratio of immature/mature IECs, and could explain the decreased intestinal enzymes observed during intestinal infections.14,31
We have recently shown that parasitic infection in mice leads to reduced levels of enzymes associated with nutrient absorption and localized to intestinal microvilli such as sucrase and lactase.14 Disaccharidase deficiency (DD) was seen in WT mice infected with the GS strain of G. duodenalis, but not the WB strain. Moreover, enzyme deficiency was not observed in severe combined immunodeficiency (SCID) mice or mice lacking either CD4+ or CD8+ T cells. Interestingly, the changes in ezrin and villin localization and phosphorylation at day 5 p.i. observed in this study, mirror the decrease of disaccharidase activity we observed in our previous study.14 However, our current findings also suggest that cleavage of ezrin and villin are not likely responsible for this pathophysiological change since cleavage of both proteins was observed only at day 18 p.i. A possible role for cleavage of ezrin and villin in restitution of the intestine and recovery of disaccharidase activity remains possible.
In conclusion, this study demonstrates that Giardia infection leads to immune activation followed by changes in the regulation of ezrin and villin. Interestingly, an increase in villin phosphorylation at day 5 p.i. in WT mice infected with the GS strain correlates kinetically and genetically with DD. Increased epithelial cell replication and migration also occur and may further contribute to the reduced levels of disaccharidase activity which are hallmarks of this disease.31,32 Cytoskeletal changes observed later following infection may facilitate cell migration and contribute to homeostasis in the recovering GI tract. This model is summarized in Figure 8. Given the importance of chronic intestinal infection in causing malnutrition and developmental defects in enormous numbers of the world’s children, efforts to understand the mechanisms leading to nutrient malabsorption and recovery of the damaged epithelium are crucial to improving global health.
Figure 8.

Model of immune-dependent changes in epithelial cell function following gut infection. Infection leads to an immune response that is required for increased villin phosphorylation at day 5. This correlates with enhanced proliferation of intestinal epithelial cells and reduced levels of disaccharidase enzymes.
METHODS
Mice
Six- to eight-week old female WT C57BL/6 mice, SCID mice, beta-2-microglobulin (β2m−/−; B6.129P2-B2mtm1Unc/J), and CD4−/− (B6.129S2-Cd4tm1Mak/J) mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Mice (n= 4/group/time point) were housed at the Georgetown University Animal Care Facility. All experiments were carried out in accordance with guidelines approved by the Georgetown University Institutional Animal Care and Use Committee in compliance with the National Institutes of Health guidelines.
Parasites, infection protocols and quantification of parasite burden
The GS/M-83-H7 (ATCC# 50581) and the WB clone-C6 (ATCC# 50803) strains of G. duodenalis were used in these experiments. Parasites were axenically grown in TYI-S-33 media supplemented with adult bovine bile, L-cysteine, ascorbic acid, and antibiotics (all from Sigma-Aldrich, St. Louis, MO). Mice were gavaged with 106 trophozoites in 100 µl PBS (pH 7.4). To equally infect mice with different strains of G. duodenalis, mice were given antibiotics in drinking water ad libitum: neomycin oral solution (1.4 mg/ml; Durvet, Blue Spring, MO), ampicillin (1 mg/ml; Sigma-Aldrich) and vancomycin (1 mg/ml; Hospira, Inc., Lake Forest, IL) for 48 h before parasite inoculation and during the entire infection period. After euthanizing mice, the first 3 cm of the small intestine (pylorus to ligament of Trietz) were discarded. The next two cm section of the duodenum were removed, opened longitudinally, and minced in 4 ml of ice-cold PBS (pH 7.4). Tissues were kept on ice for 15 minutes, and the numbers of trophozoites were counted on a hemocytometer.
Antibodies
The following antibodies were used for immunoblotting, immunopercipitation and IHC: mouse monoclonal anti-ezrin (sc-58758; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-villin (sc-28283), rabbit polyclonal against the NH2-terminal region of villin, rabbit polyclonal anti-GAPDH (sc-25778), anti-calpain-1 large subunit (no.2556; Cell Signaling), rabbit polyclonal anti-p-ezrin (Thr567; Cell Signaling), anti-phospho-tyrosine (PY-20; sc-508), peroxidase-conjugated anti-rabbit IgG and anti-mouse IgG secondary antibodies (A0545 and A4416; both from Sigma, St Louis, USA), goat anti-rabbit IgG-FITC, and goat anti-mouse IgG-TRITC (Southern Biotech, Birmingham, AL).
Immunofluorescence microscopy
Intestinal tissues for IHC were fixed in 10% buffered-formalin and subsequently embedded in paraffin as described previously. Tissue sections (5-µm thick) were deparaffinized in xylene, followed by hydration through graded ethanol solutions. In order to unmask the antigens, the slides were treated in sodium citrate buffer (pH 6.0; Vector Laboratories, Burlingame, CA) at 120°C for 20 min in a pressure cooker. Subsequently, the slides were blocked for 1 h at room temperature in a blocking solution (10% goat serum, 0.05% Tween 20 and 1% BSA in PBS). After probing with primary antibody, the sections were mounted with Vectashield mounting medium (Vector Laboratories) and a coverslip. Samples were analyzed with a Zeiss Axioplan 2 fluorescence microscope (Zeiss, Jena, Germany).
In vivo epithelial cell proliferation assay
Gender- and age-matched WT mice C57BL/6 mice (n = 4/time point) were injected intraperitoneally with 5-bromo-2'-deoxyuridine (BrdU; Sigma) dissolved in DMSO at a concentration of 50 mg/kg of body weight (1mg/mouse) 2 h before sacrifice. Control littermates (n = 4) received sham (DMSO) injections intraperitoneally. Mice were sacrificed, and tissue sections (5-µm) of paraffin-embedded duodenal, jejunal, and ileal tissues were deparaffinized and stained for the presence of BrdU-containing nuclei using a BrdU In-Situ Detection Kit according to manufacturer’s instruction (BD Pharmingen, San Diego, CA). BrdU-labeled ECs were counted at day 0, 5, and 18 p.i., and the positional distribution of IECs was determined as previously described.30 The proportion of BrdU-containing ECs was measured at high magnification under light microscopy. Intestinal epithelial cell proliferation was expressed as the percentage of BrdU-labeled ECs per 100 crypt cells, and at least 20 full-length, well-oriented duodenal crypts per animal were examined.
Western blot analysis
Fifteen cm of the small intestine were homogenized in 1 ml of tissue protein extraction reagent (T-PER; Pierce, Rockford, IL) supplemented with protease inhibitor cocktail III (Calbiochem, La Jolla, CA). Tissue lysates were centrifuged at 14000 g for 5 min, and supernatants were collected and used for further analysis. Protein concentration of each sample was determined using a Bradford assay (Bio-Rad, Hercules, CA). Equal amounts of each sample were loaded with 2× Laemmli sample buffer and were separated by gradient 4–12% SDS-PAGE (Invitrogen, Carlsbad, CA) in the MOPS buffer system (Invitrogen). Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA) by standard techniques and then probed with relevant primary antibodies. Primary antibodies were detected by using peroxidase-conjugated secondary antibodies and ECL reagents (Amersham Bioscience), and signals recorded on X-ray film (Kodak Biomax; Kodak, Rochester, NY). Protein abundance was quantified using ImageJ software (NIH); protein abundance were normalized to uninfected animals and presented relative to results obtained with the control sample set as 1.0.
Immunoprecipitation
Five hundred µg of the intestinal homogenates were pre-cleared with 20 µL of Protein A/G Plus-Agarose (sc-2003) for 1 h at 4°C. Agarose beads were removed by centrifugation, and villin polyclonal antibodies were added at a concentration of 4 µg/500 µg of homogenate and incubated with agitation for 6 h at 4°C. Subsequently, 20 µL of fresh agarose beads were added and incubated for another 2 h at 4°C. Agarose bead complexes were collected by centrifugation and washed three times with ice-cold NP40 lysis buffer (250 mM NaCl, 5 mM HEPES, 10% v/v glycerol, 0.5% NP40, 2 mM EDTA (pH 8.0) supplemented with 100 µM NaVO4 and 1 mM NaF, and protease inhibitor cocktail III). Immunocomplexes were eluted by boiling in 2× Laemmli sample buffer, resolved by gradient 4–12% SDS-PAGE (Invitrogen) in the MOPS buffer system (Invitrogen) and transferred overnight at 4°C as described earlier.
Statistical analysis
Differences between samples were evaluated using a two-tailed Mann-Whitney U test; *P values < 0.05 were considered significant.
ACKNOWLEGMENTS
This work was supported by a merit-based scholarship from Georgetown University Graduate School of Arts and Sciences (to S.S.-M.) and by National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases Grants R03-AI081033 and R15-AI094492 (to S.M.S.). The Georgetown Lombardi Shared Resources are partially supported by NIH/National Cancer Institute Grant P30-CA051008 and the Georgetown University Barrier Animal Facility by NIH/National Center for Research Resources Grant G20-RR025828. We are thankful to Professor William A. Petri, Jr., University of Virginia Health System for helpful advice and discussion. We thank Professor Anne G. Rosenwald at Georgetown University and Professor Alessio Fasano, University of Maryland, Baltimore for their useful insight. We thank members of the Singer laboratory, especially Erqiu Li and Aleksander Keselman, for assistance. Also, we thank people in the Laboratory of Mucosal Immunology at the University of California, San Diego for assistance. We thank Dr. Seema Khurana for providing the anti-NH2-terminal villin antibody.
Footnotes
DISCLOSURE
The authors declared no conflict of interest.
REFERENCES
- 1.Farthing MJ. The molecular pathogenesis of giardiasis. J. Pediatr. Gastroenterol. Nutr. 1997;24:79–88. doi: 10.1097/00005176-199701000-00018. [DOI] [PubMed] [Google Scholar]
- 2.Osawa H, Smith CA, Ra YS, Kongkham P, Rutka JT. The role of the membrane cytoskeleton cross-linker ezrin in medulloblastoma cells. Neuro. Oncol. 2009;11:381–393. doi: 10.1215/15228517-2008-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fehon RG, McClatchey AI, Bretscher A. Organizing the Cell Cortex: the role of ERM proteins. Nat. Rev. Mol. Cell. Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berryman M, Franck Z, Bretscher A. Ezrin is concentrated in the apical microvilli of a wide variety of epithelial cells whereas moesin is found primarily in endothelial cells. J. Cell. Sci. 1993;105:1025–1043. doi: 10.1242/jcs.105.4.1025. [DOI] [PubMed] [Google Scholar]
- 5.Khurana S, George SP. Regulation of cell structure and function by actin-binding proteins: villin's perspective. FEBS. Letter. 2008;582:2128–2139. doi: 10.1016/j.febslet.2008.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Srinivasan K, Siddiqui MR, George SP, Tomar A, Khurana S. A novel role for villin in intestinal epithelial cell survival and homeostasis. J. Biol. Chem. 2008;283:9454–9464. doi: 10.1074/jbc.M707962200. [DOI] [PubMed] [Google Scholar]
- 7.Tomar A, George S, Kansal P, Wang Y, Khurana S. Interaction of phospholipase C-gamma-1 with villin regulates epithelial cell migration. J. Biol. Chem. 2006;281:31972–31986. doi: 10.1074/jbc.M604323200. [DOI] [PubMed] [Google Scholar]
- 8.Kersting S, et al. Antigen transport and cytoskeletal characteristics of a distinct enterocyte population in inflammatory bowel diseases. Am. J. Pathol. 2004;165:425–437. doi: 10.1016/S0002-9440(10)63308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacchi CE, Gown AM. Distribution and pattern of expression of villin, a gastrointestinal-associated cytoskeletal protein, in human carcinomas: a study employing paraffin-embedded tissue. Lab. Invest. 1991;64:418–424. [PubMed] [Google Scholar]
- 10.Zhang PJ, Harris KR, Alobeid B, Brooks JJ. Immunoexpression of villin in neuroendocrine tumors and its diagnostic implications. Arch. Pathol. Lab. Med. 1999;123:812–816. doi: 10.5858/1999-123-0812-IOVINT. [DOI] [PubMed] [Google Scholar]
- 11.Zhai L, Zhao P, Panebra A, Guerrerio AL, Khurana S. Tyrosine phosphorylation of villin regulates the organization of the actin cytoskeleton. J. Biol. Chem. 2001;276:36163–36167. doi: 10.1074/jbc.C100418200. [DOI] [PubMed] [Google Scholar]
- 12.Tomar A, George SP, Mathew S, Khurana S. Differential effects of lysophosphatidic acid and phosphatidylinositol 4,5-bisphosphate on actin dynamics by direct association with the actin-binding protein villin. J. Biol. Chem. 2009;284:35278–35282. doi: 10.1074/jbc.C109.060830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar N, Tomar A, Parrill AL, Khurana S. Functional dissection and molecular characterization of calcium-sensitive actin-capping and actin-depolymerizing sites in villin. J. Biol. Chem. 2004;279:4536–4546. doi: 10.1074/jbc.M405424200. [DOI] [PubMed] [Google Scholar]
- 14.Solaymani-Mohammadi S, Singer SM. Host immunity and pathogen strain contribute to intestinal disaccharidase impairment following gut infection. J. Immunol. 2011;187:3769–3775. doi: 10.4049/jimmunol.1100606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao X, Thibodeau A, Forte JG. Ezrin-calpain I interactions in gastric parietal cells. Am. J. Physiol. 1993;265:36–46. doi: 10.1152/ajpcell.1993.265.1.C36. [DOI] [PubMed] [Google Scholar]
- 16.Tomar A, et al. Regulation of cell motility by tyrosine phosphorylated villin. Mol. Biol. Cell. 2004;15:4807–4817. doi: 10.1091/mbc.E04-05-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carabeo RA, Grieshaber SS, Fischer E, Hackstadt T. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect. Immun. 2002;70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker ME, et al. Toxoplasma gondii actively remodels the microtubule network in host cells. Microbes. Infect. 2008;10:1440–1449. doi: 10.1016/j.micinf.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang W, McCrae MA. The rotavirus enterotoxin (NSP4) promotes re-modeling of the intracellular microtubule network. Virus. Res. 2012;163:269–274. doi: 10.1016/j.virusres.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Dunn JD, Valdivia RH. Uncivil engineers: Chlamydia, Salmonella and Shigella alter cytoskeleton architecture to invade epithelial cells. Future. Microbiol. 2010;5:1219–1232. doi: 10.2217/fmb.10.77. [DOI] [PubMed] [Google Scholar]
- 21.Athman R, et al. Shigella flexneri infection is dependent on villin in the mouse intestine and in primary cultures of intestinal epithelial cells. Cell. Microbiol. 2005;7:1109–1116. doi: 10.1111/j.1462-5822.2005.00535.x. [DOI] [PubMed] [Google Scholar]
- 22.Elliott DA, Clark DP. Cryptosporidium parvum induces host cell actin accumulation at the host-parasite interface. Infect. Immun. 2000;68:2315–2322. doi: 10.1128/iai.68.4.2315-2322.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott DA, Coleman DJ, Lane MA, May RC, Machesky LM, Clark DP. Cryptosporidium parvum infection requires host cell actin polymerization. Infect. Immun. 2001;69:5940–5942. doi: 10.1128/IAI.69.9.5940-5942.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauwaet T, et al. Proteolysis of enteric cell villin by Entamoeba histolytica cysteine proteinases. J. Biol. Chem. 2003;278:22650–22656. doi: 10.1074/jbc.M300142200. [DOI] [PubMed] [Google Scholar]
- 25.Reiske HR, Kao SC, Cary LA, Guan JL, Lai JF, Chen HC. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J. Biol. Chem. 1999;274:12361–12366. doi: 10.1074/jbc.274.18.12361. [DOI] [PubMed] [Google Scholar]
- 26.Zhai L, Kumar N, Panebra A, Zhao P, Parrill AL, Khurana S. Regulation of actin dynamics by tyrosine phosphorylation: identification of tyrosine phosphorylation sites within the actin-severing domain of villin. Biochemistry. 2002;41:11750–11760. doi: 10.1021/bi0263762. [DOI] [PubMed] [Google Scholar]
- 27.Selbach M, Moese S, Backert S, Jungblut PR, Meyer TF. The Helicobacter pylori CagA protein induces tyrosine dephosphorylation of ezrin. Proteomics. 2004;4:2961–2968. doi: 10.1002/pmic.200400915. [DOI] [PubMed] [Google Scholar]
- 28.Zhou R, Zhu L, Kodani A, Hauser P, Yao X, Forte JG. Phosphorylation of ezrin on threonine 567 produces a change in secretory phenotype and repolarizes the gastric parietal cell. J. Cell. Sci. 2005;118:4381–4391. doi: 10.1242/jcs.02559. [DOI] [PubMed] [Google Scholar]
- 29.Zhu L, Hatakeyama J, Chen C, Shastri A, Poon K, Forte JG. Comparative study of ezrin phosphorylation among different tissues: more is good; too much is bad. Am. J. Physiol. Cell. Physiol. 2008;295:192–202. doi: 10.1152/ajpcell.00159.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cliffe LJ, Humphreys NE, Lane TE, Potten CS, Booth C, Grencis RK. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science. 2005;308:1463–1465. doi: 10.1126/science.1108661. [DOI] [PubMed] [Google Scholar]
- 31.Buret A, Hardin JA, Olson ME, Gall DG. Pathophysiology of small intestinal malabsorption in in gerbils infected with Giardia lamblia. Gastroenterology. 1992;103:506–513. doi: 10.1016/0016-5085(92)90840-u. [DOI] [PubMed] [Google Scholar]
- 32.Scott KG, Yu LC, Buret AG. Role of CD8+ and CD4+ T lymphocytes in jejunal mucosal injury during murine giardiasis. Infect. Immun. 2004;72:3536–3542. doi: 10.1128/IAI.72.6.3536-3542.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]