

Abstract
This study analyzes whether the release of nitric oxide (NO) and thromboxane A2 (TXA2) depends on the time lapsed since gonadal function is lost, and their correlation with the proliferation of vascular smooth muscle cells (VSMC) mediated by the epidermal growth factor receptor (EGFR). For this purpose, aortic and mesenteric artery segments from control and 6-weeks or 5-months orchidectomized rats were used to measure NO and TXA2 release. The results showed that the basal and acetylcholine (ACh)-induced NO release were decreased 6 weeks post-orchidectomy both in aorta and mesenteric artery, but were recovered 5 months thereafter up to levels similar to those found in arteries from control rats. The basal and ACh-induced TXA2 release increased in aorta and mesenteric artery 6 weeks post-orchidectomy, and was maintained at high levels 5 months thereafter. Since we previously observed that orchidectomy, which decreased testosterone level, enlarged the muscular layer of mesenteric arteries, the effect of testosterone on VSMC proliferation was analyzed. The results showed that treatment of cultured VSMC with testosterone downregulated mitogenic signaling pathways initiated by the ligand-dependent activation of the EGFR. In contrast, the EGFR pathways were constitutively active in mesenteric arteries of long-term orchidectomized rats. Thus, the exposure of mesenteric arteries from control rats to epidermal growth factor (EGF) induced the activation of EGFR signaling pathways. However, the addition of EGF to arteries from orchidectomized rats failed to induce a further activation of these pathways. In conclusion, this study shows that the release of NO depends on the time lapsed since the gonadal function is lost, while the release of TXA2 is already increased after short periods post-orchidectomy. The alterations in these signaling molecules could contribute to the constitutive activation of the EGFR and its downstream signaling pathways after long period post-orchidectomy enhancing the proliferation of the vascular muscular layer.
Introduction
The vascular tone is regulated by several mechanisms that implicate the participation of hormonal, neuronal and endothelial factors [1]. It has been established that sex hormones are able to modify the production of different vasoactive factors released from the vessel wall. Among them, nitric oxide (NO), prostanoids and reactive oxygen species play pivotal roles regulating the vascular tone through their vasoactive properties as well as regulating cell proliferation [2]–[4]. An altered production of these factors could modify the regulation of the vascular tone leading to the development of different vascular pathologies.
Clinical studies have shown a correlation between hypotestosteronemia and incidence of cardiovascular diseases [5] and mortality risk [6]. These issues, as well as different mechanisms of action by which testosterone causes vasodilation were reviewed by Jones [7], In this regard, previous studies from our group have demonstrated an increase in the production of superoxide anion [8], prostanoids, such as thromboxane A2 (TXA2) [9], [10] and prostaglandin E2 (PGE2) [11] five months post-orchidectomy. Concerning the effect of androgens on endothelial NO release, most of studies were carried out in endothelial cells culture showing an increased release [12]–[14]. However, when the effect of androgenic derivatives was studied in orchidectomized animals, the vasodilator action of NO rather than its release was analyzed, and contradictory results were often obtained [15]–[17]. Thus, the involvement [16] or lack of involvement [17] of NO in testosterone-induced relaxation has been reported. Moreover, androgen-induced relaxation has been reported to be mediated by endothelium-independent mechanisms [18]. This variety of results could depend on the tissue, the concentration, administration-time, and the molecular structure of the androgenic derivatives used.
Concerning the effects of sex hormones deprivation on vascular function, we previously demonstrated in mesenteric artery of orchidectomized rats that the increased activity of protein kinase C (PKC) positively regulated eNOS activity [19], preventing a decrease in the release of endothelial NO. In aorta from rats, we reported that the effect of ovariectomy on NO release depended on the time lapsed since the loss of the gonads [20]. This implies that different compensatory mechanisms are likely to be at work during prolonged periods of time after gonadectomy preventing vascular failure, as already suggested [21]. In view of these data, our first objective in the present study was to determine whether there were differences in the release of NO and TXA2 in aorta and mesenteric arteries of rats subjected to short (6 weeks) and long (5 months) periods post-orchidectomy.
We have also found in a previous study an enlargement of the media muscular layer of mesenteric arteries from orchidectomized rats [8], suggesting that testosterone deprivation could enhance the proliferation of VSMC. The ability of TXA2 [22], PGE2 [23], superoxide anion [24] and PKC [25] to induce epidermal growth factor receptor (EGFR) transactivation have been reported, suggesting that this receptor could be implicated in the hyperproliferation of the mesenteric muscular layer from rats after 5-months post-orchidectomy. The EGFR is a receptor tyrosine kinase (RTK), which upon ligand-dependent dimerization leads to the activation of several intracellular signaling pathways, including the mitogen-activated protein kinase/extracellular-regulated kinases 1/2 (MAPK-ERK1/2) and the phosphatidylinositol 3-kinase (PI3K)/Akt pathways that control cell survival and cell growth [26]. In view of these data, our second objective was to analyze whether the loss of gonadal function for long periods enhances the ligand-dependent activation of the EGFR and its downstream signaling pathways in mesenteric arteries, and whether this can be correlated with the effect of testosterone on VSMC.
Materials and Methods
Animal protocols
Male Sprague-Dawley rats (6 months-old) were housed in the Animal Facility of the Universidad Autónoma de Madrid (registration number EX-021U) in accordance with directives 609/86 CEE and RD 233/88 of the Ministerio de Agricultura, Pesca y Alimentación of Spain. The animals were subjected to 12 hours of light/dark cycles and standard feeding with fodder and water ad libitum. In a first group, orchidectomy was performed 4 weeks after birth and 5 months later the animals were sacrificed. In a second group, the orchidectomy was performed 4.5 months after birth and 6 weeks later the animals were sacrificed. A control group not subjected to orchidectomy was also included. Surgery was performed under anesthesia by isoflurane inhalation. Absence of retraction reflex in the hind-legs after mechanical stimulation and regular respiratory rhythm were tested to determine the adequacy of anesthesia. For analgesia, rats were treated with 0.30 mg/Kg SC meloxicam (Metacam from Boehringer-Ingelheim) immediately after surgery and with 50 mg/Kg ibuprofen for 4 days. Systolic blood pressure was indirectly measured in animals 2–3 days before sacrifice by the tail-cuff method [21] using a Leica Digital Pressure Meter LE5000 (Barcelona, Spain). Extraction of blood samples (by cardiac puncture) for testosterone levels determination and body weight measurement were done the day of the experiment before the animals were sacrificed by CO2 inhalation. The aorta and the superior mesenteric artery were carefully dissected and removed, cleaned of connective tissue, cut into 4 mm long segments and placed in Krebs-Henseleit solution (KHS) at 4°C containing (in mM): NaCl 115, CaCl2 2.5, KCl 4.6, KH2PO4 1.2, MgSO4 1.2, NaHCO3 25, glucose 11.1, Na2-EDTA 0.03 (pH 7.4). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the USA National Institutes of Health (NIH publication No. 85.23 revised 1985). This study was approved by the Ethical Committee of the Universidad Autónoma de Madrid.
Testosterone levels
The testosterone level in the serum was determined using an enzyme immunoassay kit (Cayman Chemical; Ref. No. 582701). The assays were performed according to the manufacturer's instructions.
Cells culture
Vascular smooth muscle cells (SV40LT-VSMC) were obtained from the American Type Culture Collection (ATCC) (Lot No. 3350860) and grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 40 µg/ml gentamicine and 2 mM l-glutamine in F75 flasks at 37°C in a humidified atmosphere containing 5% CO2. Cells were seeded (450,000 cells/well) in P6 plates in the same medium, and maintained overnight in the absence of FBS and in the absence and presence of testosterone (10 nM) before performing the experiments.
NO release
Endothelium-intact arterial segments from control and orchidectomized rats were subjected to a resting tension of 9.8 and 4.9 mN in aorta and mesenteric artery, respectively. After an equilibration period of 60 min in a buffer containing (in mM): NaCl 119, N-(2-hydroxyethyl)piperazine-N-2-ethane-sulfonic acid (HEPES) 20, CaCl2 1.2, KCl 4.6, MgSO4 1, NaHCO3 5, glucose 5.5, KH2PO4 0.4, Na2H2PO4 0.15 (pH 7.4), the presence of vascular endothelium was tested by the ability of ACh (10 µM) to relax segments pre-contracted with noradrenaline (NA, 0.1 and 1 µM in aortic and mesenteric segments, respectively). Only arterial segments in which the ACh-induced relaxation was higher than 75% of the previous contraction were used. After, the arterial segments were rinsed several times and recovered the basal tone, arteries were incubated with the fluorescent probe 4,5-diaminofluorescein (DAF-2) (0.5 µM) for 45 min, as previously described [8]. Then, the medium was collected to measure the basal NO release. Once the organ bath was refilled, arteries were pre-contracted with NA for 2 min (0.1 µM and 1 µM in aorta and mesenteric segments, respectively), and then followed by the addition of cumulative ACh concentrations (0.1 nM–10 µM) applied at 1 min intervals to induce relaxation and to measure NO release. The fluorescence emitted by DAF-2 was measured in a spectrofluorimeter (LS50 Perkin Elmer instruments, FL WINLAB Software) using an excitation wavelength of 495 nm and an emission wavelength of 515 nm. When required assays were performed in the presence of the NO synthetase (NOS) inhibitor l-N G-nitroarginine methyl ester (l-NAME), in which the ACh-induced fluorescence was abolished [27]. Blanks were collected from segment-free medium in order to subtract the background fluorescence. The amount of NO released was expressed as arbitrary units/mg tissue.
Release of TXA2
The production of TXA2 in vivo is typically monitored by measuring its stable metabolite TXB2 using an enzyme immunoassay kit [11]. Endothelium-intact aortic and mesenteric arterial segments from control and orchidectomized rats were subjected to an equilibration period of 30 min in KHS at 37°C followed by 2 wash periods of 10 min using 0.2 ml of the same medium and collecting samples to measure the basal release. Once fresh KHS was replaced, arteries were exposed to NA for 2 min (0.1 µM and 1 µM in aorta and mesenteric segments, respectively), and then cumulative ACh concentrations (0.1 nM–10 µM) were applied at 1 min interval. The medium was collected, and stored at −80°C until use. To analyze the effect of testosterone on the release of TXA2, vascular smooth muscle cells (VSMC) were seeded (450,000 cells/well) in P6 plates and grown at 37°C. Thereafter, plates were maintained overnight in the same conditions but in the absence of FBS and in the absence and presence of testosterone (10 nM). Media were collected and stored at −80°C until use. The TXB2 assay was carried out according to the manufacturer's instructions. Results were expressed as pg TXA2/mg tissue or as pg TXA2/ml of medium for arteries or VSMC, respectively.
Preparation of cell and tissue extracts
VSMC were seeded (450,000 cells/well) as previously mentioned in P6 plates and grown at 37°C. Plates were maintained overnight in the absence of FBS and in the absence and presence of testosterone (10 nM). Time-course stimulation (0–30 min) with EGF (10 nM) was carried out and the reaction was stopped with 10% (w/v) trichloroacetic acid. Artery segments were incubated in the absence and presence of EGF (10 nM) during 2 minutes. The reaction was stopped with liquid nitrogen and the arteries were stored at −80°C until used. Arterial segments were homogenated at 4°C in 150 µl of RIPA buffer containing 50 mM Tris-HCl (pH 8), 150 mM NaCl, 1% (w/v) deoxycholic acid, 1% (v/v) NP-40, 1% (v/v) SDS, 100 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail (Calbiochem; Ref. No. 539134) supplemented with freshly prepared 1 mM phenylmethylsulfonyl fluoride. Samples were centrifuged at 16,000 g during 30 min at 4°C and the supernatant was collected to quantify protein concentration by the bicinchoninic acid assay using the BCA™ Protein Assay Kit (Pierce).
Western blotting analysis
Electrophoresis Laemmli's loading buffer was added to each sample and heated 5 min at 100°C. Proteins were separated by SDS-PAGE in linear gradient (5–20%) gels, and transferred to polyvinylidene difluoride (PVDF) membranes (BioTRACE™). Membranes were stained with 0.1% (w/v) Fast Green in 50% (v/v) methanol and 10% (v/v) acetic acid to ascertain equal loading. Membrane segments were blocked with 5% (w/v) fat-free powdered milk or 5% (w/v) bovine serum albumin following the instructions of the antibodies' manufactures in 10 mM Tris-HCl (pH 7.4), 150 mM NaCl and 0.1% (v/v) Tween-20 (TBS-T), and incubated overnight with the following primary antibodies (Cell Signalling Technology) at a 1∶2000 dilution: monoclonal anti-phospho-tyrosine (4G10) and anti-phospho-ERK1/2 (Thr202/Tyr204); and polyclonal anti-Akt (pan), anti-phospho-Akt (S473) and anti-ERK1/2. Membranes segments were washed with TBS-T and incubated with the corresponding anti-IgG horseradish peroxidase-conjugated antibody (Invitrogene) at a 1∶5000 dilution following commercial recommendations. Finally, membranes were developed with the ECL™ Western Blotting detection kit (GE Health Care) exposing X-ray films for appropriate periods of time. The intensity of the bands was quantified with a computer-assisted scanning densitometer using the NIH Image 1.60 program.
Statistical Analysis
All data are presented as the mean ± SEM. The experiments on NO and TXA2 release, testosterone level, body weight and blood pressure were analyzed using an unpaired Student's t-test (GraphPad Prism software), and the activation of the EGFR and signaling pathways in VSMC in the absence and presence of testosterone by the two-way analysis of variance (ANOVA). A p<0.05 was considered significant.
Reagents
The reagents used were: NA, ACh, L-NAME and DAF-2 were obtained from Sigma-Aldrich, and human recombinant EGF was from PeproTech EC. Reagents were prepared in distilled water except NA that was dissolved in a solution of 0.9% (w/v) NaCl and 0.01% (w/v) ascorbic acid, and EGF that was dissolved in 25 mM HEPES-NaOH (pH 7.4). Reagent stock solutions were kept at −20°C and appropriate dilutions were made in KHS or HEPES-buffer on the day of the experiment.
Results
Effect of short and long periods post-orchidectomy on serum testosterone levels, body weight and blood pressure
Table 1 shows that the levels of testosterone in serum samples drastically decreased in rats 6 weeks post-orchidectomy. These levels were maintained low in 5-months post-orchidectomized animals. The body weight was not modified in 6-weeks post-orchidectomized rats, but in orchidectomized rats after 5 months it was slightly decreased. Orchidectomy did not significantly modify the systolic blood pressure in either group (Table 1). We have observed, however, that in old orchidectomized rats the systolic blood pressure was increased (control young, 141.5±4.1 mmHg; control aged, 149.2±3.8 mmHg; orchidectomized aged, 159.5±2.9 mmHg, p<0.05).
Table 1. Time-dependent effect of orchidectomy (Orch) on the serum testosterone level, blood pressure and body weight in male rats.
| Control (n = 7) | 6 weeks-Orch (n = 6) | 5 months-Orch (n = 8) | |
| Testosterone (pg/mL) | 2368±323 | 235±45* | 220±52* |
| Systolic blood pressure (mmHg) | 143±5.6 | 147.5±7.2 | 145±6.2 |
| Body weight (g) | 473±9.4 | 480±4.7 | 432±6.8+ |
The number of animal used are indicated.
*p<0.0001 vs control rats.
p<0.05 vs control rats.
Effect of short and long periods post-orchidectomy on NO and TXA2 release in aorta and mesenteric arteries
ACh enhanced the basal release of NO in aortic segments from control and orchidectomized rats. However, both basal and ACh-induced release of NO decreased in 6 weeks post-orchidectomized rats. These values were restored to similar levels found in aorta from control animals 5 months after orchidectomy (Fig. 1A).
Figure 1. Time-dependent effect of orchidectomy on the release of NO from rat aorta and mesenteric artery.

The plots present the basal- and ACh-induced NO release in aorta (A) and mesenteric (B) segments from control, 6 weeks and 5 months post-orchidectomized rats. Results (mean ± SEM) are expressed as arbitrary units (AU)/mg of tissue. * p<0.05, compared with its respective basal condition; # p<0.05 compared with basal NO release in control animals; + p<0.05, ++ p<0.001 compared with ACh-induced NO release in control animals. The number of animals used were: control, 6; 6 week post-orchidectomy, 4; 5 months post-orchidectomy, 7.
Similarly, in mesenteric artery, the basal NO release was strongly decreased in 6-weeks post-orchidectomized rats, and ACh did not induce a significant further release. As observed in aortic segments, the basal and ACh-induced NO release in mesenteric artery was restored to control levels in 5-months post-orchidectomized rats (Fig. 1B).
In aortic segments from control and orchidectomized rats, ACh enhanced the release of TXA2 [11]. Both basal and ACh-induced TXA2 release increased 6 weeks post-orchidectomy. This increase was not further modified in 5 months post-orchidectomized rats (Fig. 2A). Similar results to those described in aorta were found in mesenteric arteries at 6 weeks and 5 months post-orchidectomy (Fig. 2B).
Figure 2. Time-dependent effect of orchidectomy on the release of TXA2 from rat aorta and mesenteric artery.

The plots present the basal- and ACh-induced TXA2 release in aorta (A) and mesenteric (B) segments from control, 6 weeks and 5 months post-orchidectomized rats. Results (mean ± SEM) are expressed as pg/mL/mg tissue. Number of animals: 4–7. * p<0.05, ** p<0.001 compared with its respective basal condition; # p<0.01 compared with basal TXA2 release in control animals; + p<0.01 compared with ACh-induced TXA2 release in control animals. The number of animals used were: control, 6; 6 week post-orchidectomy, 4; 5 months post-orchidectomy, 7.
The levels of TXA2 were also analyzed in the medium conditioned by VSMC incubated in the absence and presence of testosterone (10 nM). Our results show that after overnight incubation of VSMC with testosterone the release of TXA2 was decreased (control, 8.17±0.9 pg/ml; testosterone-treated, 5.4±0.3 pg/ml; n = 6, p<0.05).
Effect of testosterone on the activation of EGFR signaling pathways in VSMC
The ligand-dependent activation of the EGFR and their downstream signaling kinases Akt and ERK1/2 were analyzed in homogenates of cultured VSMC incubated overnight in the absence and presence of 10 nM testosterone. Testosterone treatment slightly decreased the ligand-dependent phosphorylation (activation) of the EGFR after time course stimulation with 10 nM EGF (Figs. 3A and 3B). Moreover, the phosphorylation of a ≈115 kDa phospho-(Tyr)-protein (p115) (Figs. 3A and 3C), Akt (Figs. 3A and 3D) and ERK1/2 (Figs. 3A and 3E) were also significantly reduced. The EGFR was progressively dephosphorylated after addition of its ligand, and this was attributed to the combined action of phosphatases and the internalization and proteolytic processing of the receptor, as it is apparent by the progressive decrease of the total EGFR signal (Figs. 3A). Immunoprecipitation experiments showed that the ≈115 kDa phospho-(Tyr)-protein was not the catalytic subunit of PI3K (data not shown).
Figure 3. Effect of testosterone treatment on ligand-dependent activation of the EGFR and downstream signaling pathways in smooth muscle vascular cells.

Serum starved SMVC were incubated with 10% (w/v) trichloroacetic acid. The samples were processed by Western blots using the indicated antibodies to determine the phosphorylation levels of the EGFR, p115, Akt, and ERK1/2, and the total levels of EGFR Akt, ERK1/2 and GAPDH as described in Materials and Methods. The total EGFR level decreased after EGF stimulation due to the expected proteolytic processing of the receptor after ligand-dependent internalization. The total Akt, ERK1/2 and GAPDH levels were used as loading controls. The intensity of the bands was measured densitometrically and the signal of the different phosphoproteins was corrected using appropriate loading controls. The photograph (A) shows typical Western blots of the proteins. The top and bottom arrows point to the phosphorylated EGFR and p115, respectively. The plots (B–E) present the mean ± SEM phosphorylation of the EGFR (n = 4) (B), p115 (n = 5) (C), Akt (n = 6) (D), and ERK1/2 (n = 6) (E) from a set of experiments similar to those shown in A.
Effect of long period post-orchidectomy on the activation of EGFR signaling pathways in mesenteric artery
We also determined the activation of the EGFR and the downstream Akt and MAPK(ERK1/2) signaling pathways in homogenates of superior mesenteric artery from control and orchidectomized rats 5 months after surgery. The stimulation of mesenteric artery from control rats with 10 nM EGF for 2 min produced the phosphorylation (activation) of a faint 170 kDa band corresponding to the EGFR. This was accompanied by a significant increased phosphorylation of the ≈115 kDa phospho-(Tyr)-protein (p115), which is likely to correspond to a substrate of the EGFR, and the phosphorylation (activation) of Akt and ERK1/2 in arteries from control rats (Fig. 4). However, in arteries from orchidectomized rats the EGFR, p115, Akt and ERK1/2 were already activated in basal conditions and did not further increase their phosphorylation upon addition of EGF (Fig. 4).
Figure 4. Effect of orchidectomy on the ligand-dependent activation of the EGFR and downstream signaling pathways in the mesenteric artery.
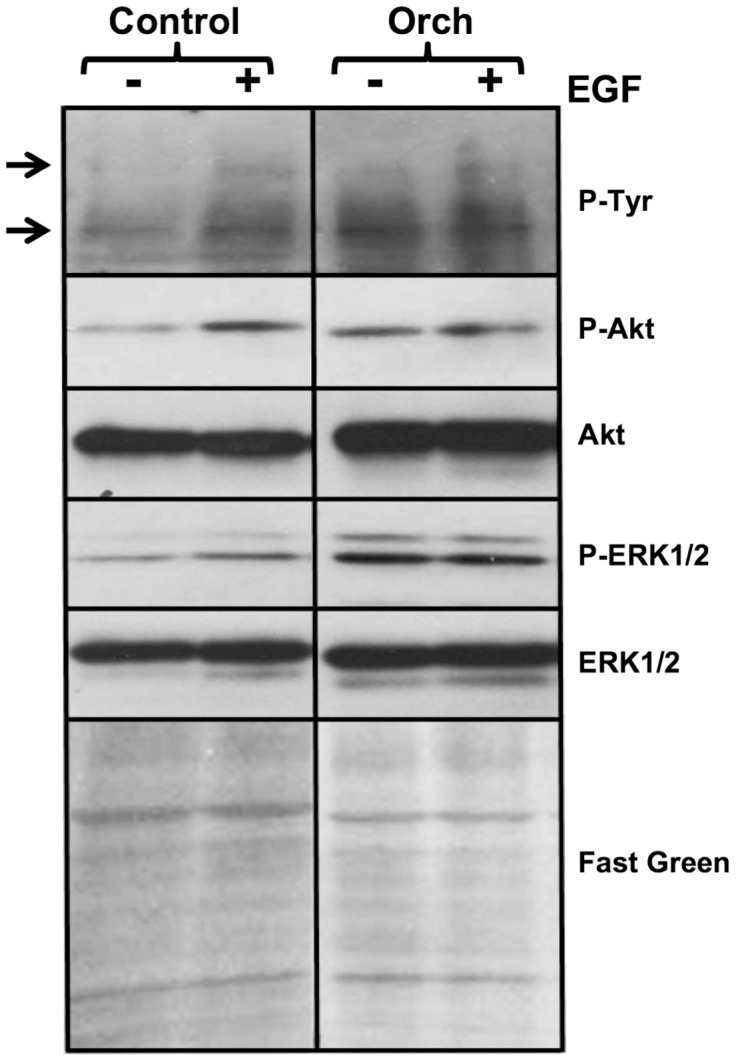
Segments of the superior mesenteric artery from control and 5 months post-orchidectomized rats were incubated in the absence (−) and presence (+) of 10 nM EGF during 2 min, and the reaction was thereafter arrested freezing the samples in liquid nitrogen as described in Materials and Methods. The frozen samples were stored and thereafter homogenized and processed by Western blots using the indicated antibodies. The photograph shows the phosphorylation levels of the EGFR, p115, Akt and ERK1/2, and the total levels of Akt and ERK1/2 and segments of PVDF membranes stained with Fast Green used as additional loading control. The top and bottom arrows point to the phosphorylated EGFR and p115, respectively. The figure shows a typical experiment from control (n = 4) and orchidectomized (n = 4) rats.
Discussion
The pivotal role of NO and prostanoids in the regulation of the vascular tone has been established [3], [4], [28]. Likewise, the beneficial effect of androgens on the vascular function of males is widely recognized [5], [29]–[31]. The effects of androgenic derivatives on NO [12], [13] and prostanoids [32], [33] signalling pathways in different tissues have been described. Concerning NO release, both genomic [12] and non-genomic [13],[34] actions of dehydroepiandrosterone haven been reported. Testosterone has been also shown to induce NO release via activation of the PI3K/Akt cascade [14]. The majority of studies performed in arteries have been focused on analysing NO function instead of NO release, and discrepant results have been described. Thus, it has been reported that testosterone can increase [1], [16] and decrease [15], [17] the endothelium-dependent relaxation. However, endothelium-independent relaxation induced by testosterone and its 5-reduced metabolites has been also reported [18]. Likewise, it has been described that androgens induce vasorelaxation by activating K channels by increasing K+ efflux and by inhibiting calcium channels causing hyperpolarization [30]. These discrepancies could be explained depending on the tissue or animal model, the concentration, administration-time and molecular structure of the androgenic derivatives used. In addition, it is important to note that most of the published studies analyzed the effect of specific androgenic derivatives in orchidectomized animals. However, a more integrative approach could be more informative, especially taking into account that in 5 months-orchidectomized rats different signaling pathways are simultaneously working to try to maintain the vascular function [8], [19], [27], [35].
Taking into account that in aorta from female rats the basal and ACh-induced NO release was decreased 6 weeks post-ovariectomy, but recovered up to levels similar to those found in control rats 5 months thereafter [20], we analyzed whether the vessels from male rats showed a similar pattern. The results shown in the present study confirm that this is indeed the case, as both aorta and mesenteric arteries presented lower basal and ACh-induced NO release in 6 weeks post-orchidectomized rats. It is important to note that the mesenteric artery showed greater reduction in NO release than the aorta, since ACh failed to induce significant NO release in the former. This result is in agreement with that reported in mesenteric arteries of female rats, in which the involvement of endothelial NO was abolished four weeks post-ovariectomy [28]. This suggests that the effects due to the loss of gonadal function also depend on the specific vascular bed.
In addition to NO, prostanoids exert important regulatory effects on the vascular function. It has been reported that TXA2 is one of the most important vasoconstrictor prostanoids produced by the vascular wall that may participate in vascular dysfunction associated with cardiovascular risk factors [36]–[38]. We have already demonstrated that the release of TXA2 was increased 5 months post-orchidectomy in mesenteric artery [9], [10], [19], probably as consequence of increased oxidative stress observed in these experimental conditions. To ascertain whether the release of TXA2 also depends on the time lapsed since the gonadal function is lost, its release was also analyzed in vessels from 6 weeks post-orchidectomized rats. The results showed that after short periods of gonadal function loss the production of TXA2 was already increased up to similar levels of those observed in vessels from 5 months post-orchidectomized rats. This increased release of TXA2 over time, could be involved in the maintenance of NO release during a prolonged period of time after gonadectomy, as observed in aorta from male (unpublished results) and female rats [21].
Since the reduction of testosterone in the serum was already evident in 6 weeks post-orchidectomized rats and maintained 5 months post-orchidectomy, the effect of testosterone on TXA2 release in VSMC was analyzed. The results show that the incubation of VSMC with testosterone decreased the release of TXA2, in accordance with data previously reported [32]. Overall, these results indicate that testosterone is involved in the effects observed, although the participation of gonadal factors or hormones other than testosterone cannot be discarded.
It is well known that NO and TXA2 are able to regulate platelet aggregation and vasomotor response [38]. Moreover, it has been described that NO and TXA2 are important regulators of endothelial cell migration, angiogenesis and cell proliferation [39]–[42]. The participation of the EGFR in cell proliferation has been widely documented [43], [44], since its activation initiates intracellular signaling pathways with cell proliferation effects in which the kinases ERK1/2 and Akt are involved [45]. On the other hand, it has been reported that the TXA2 receptor transactivates the EGFR by a mechanism implicating Src-mediated phosphorylation of the receptor [25], and that NO negatively regulates the EGFR inducing the reversible S-nitrosylation of the receptor [46], [47]. Since we have found in a previous study an enlargement of the media muscular layer of mesenteric arteries from orchidectomized rats [8], we correlated the changes in NO and TXA2 levels with cell proliferation by analyzing the activation of EGFR-induced signaling pathway. Our results demonstrate that testosterone downregulates mitogenic signaling pathways initiated by the ligand-dependent activation of the EGFR in SMVC. Conversely, a decrease in testosterone after orchidectomy results in the basal activation of these EGFR pathways. The exposure of mesenteric arteries from control rats to EGF for 2 min induced the activation of the EGFR signaling pathways. However, in arteries from orchidectomized rats the addition of EGF did not induce a further increase in these signaling pathways. Overall, these results suggest that a decrease in the level of circulating testosterone may lead to increased proliferation of cells in the vascular wall, as previously observed in the muscular layer [8]. Likewise, the results obtained could account for the inhibitory effect of testosterone on vascular remodeling described in resistance mesenteric arteries [48]. Although the exact mechanism by which decreased level of testosterone upregulates EGFR signaling was not studied in the present work, the overproduction of TXA2, PGE2 and superoxide anion in arteries from orchidectomized rats could be involved, since transactivation of the EGFR by these signaling molecules has been described [25], [38], [49], [50], initiating the proliferation signal via the MAPK and Akt pathways. In this context, it has been reported that hypertension increased the transactivation of RTKs induced by proinflammatory mediators [51]. In the present study we reported the activation of the EGFR in mesenteric arteries after long period post-orchidectomy, and the animals did not develop hypertension, probably due to the existence of compensatory mechanisms. We believe that maintaining gonadal function is essential to prevent the development of hypertension, as only old orchidectomized animals develop hypertension. Taken together, these results could contribute to the understanding of the signaling pathways implicated in pathophysiological situations in which gonadal function is impaired. (i.e.: aging, hypogonadism, and pharmacological treatment of prostate cancer).
In summary, this study shows that the release of NO depends on the time lapsed since the gonadal function is lost, while the release of TXA2 is already increased after short period post-orchidectomy, and that this correlates with the possible consequences of the modifications along the time of these two crucial factors in regulating the vascular tone and the proliferation of vascular cells. In addition, this study describes for the first time the increased activation of the EGFR and its downstream signaling pathways in rat mesenteric arteries after long period post-orchidectomy, that maintained for prolonged periods of time could contribute to the development of hypertension.
Acknowledgments
We thank the veterinarian Dr. Ma del Carmen Fernández-Criado and Mr. David Muñoz for the care of animals.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
This work was supported by grants to M.F. from the Fondo de Investigaciones Sanitarias (PI1100406) and Fondo Europeo de Desarrollo Regional, and grants to A.V. from the Secretaría de Estado de Investigación, Desarrollo e Innovación (SAF2011-23494), the Consejería de Educación de la Comunidad de Madrid (S2011/BMD-2349), and the European Commission (contract PITN-GA-2011-289033). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Orshal JM, Khalil RA (2004) Gender, sex hormones and vascular tone. Am J Physiol 286: R233–R249. [DOI] [PubMed] [Google Scholar]
- 2. Wang Y, Ji L, Jiang R, Zheng L, Liu D (2014) Oxidized high-density lipoprotein induces the proliferation and migration of vascular smooth muscle cells by promoting the production of ROS. J Atheroscler Thromb 21: 204–216. [DOI] [PubMed] [Google Scholar]
- 3. Félétou M, Huang Y, Vanhoutte PM (2010) Vasoconstrictor prostanoids. Pflugers Arch 459: 941–950. [DOI] [PubMed] [Google Scholar]
- 4. Atochin DN, Huang PL (2010) Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch 46: 965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao J, Zou H, Zhu BP, Wang H, Li J, et al. (2010) Sex hormones and androgen receptor: risk factors of coronary heart disease in elderly men. Chin Med Sci J 25: 44–49. [DOI] [PubMed] [Google Scholar]
- 6. Güder G, Frantz S, Bauersachs J, Allolio B, Ertl G, et al. (2010) Low circulating androgens and mortality risk in heart failure. Heart 96: 504–509. [DOI] [PubMed] [Google Scholar]
- 7. Jones TH (2010) Testosterone deficiency: A risk factor for cardiovascular disease? Trends Endocr Rev 24: 313–340. [DOI] [PubMed] [Google Scholar]
- 8. Martín MC, Balfagón G, Minoves N, Blanco-Rivero J, Ferrer M (2005) Androgen deprivation increases neuronal nitric oxide metabolism and its vasodilator effect in rat mesenteric arteries. Nitric Oxide 12: 163–176. [DOI] [PubMed] [Google Scholar]
- 9. del Campo L, Sagredo A, Aras-López R, Balfagón G, Ferrer M (2008) Orchidectomy increases the formation of non-endothelial thromboxane A2 and modulates its role in the electrical field stimulation-induced response in rat mesenteric artery. J Endocrinol 197: 371–379. [DOI] [PubMed] [Google Scholar]
- 10. Blanco-Rivero J, Balfagón G, Ferrer M (2005) Orchidectomy modulates α2-adrenoceptor reactivity in rat mesenteric artery through increased thromboxane A2 formation. J Vasc Res 43: 101–108. [DOI] [PubMed] [Google Scholar]
- 11. Martorell A, Blanco-Rivero J, Aras-López R, Sagredo A, Balfagón G, et al. (2008) Orchidectomy increases the formation of prostanoids and modulates their role in the acetylcholine-induced relaxation in the rat aorta. Cardiovasc Res 77: 590–599. [DOI] [PubMed] [Google Scholar]
- 12. Simoncini T, Mannella P, Fornari L, Varone G, Caruso A, et al. (2003) Dehydroepiandrosterone modulates endothelial nitric oxide synthesis via direct genomic and nongenomic mechanisms. Endocrinology 144: 3449–3455. [DOI] [PubMed] [Google Scholar]
- 13. Liu D, Dillon JS (2004) Dehydroepiandrosterone stimulates nitric oxide release in vascular endothelial cells: evidence for a cell surface receptor. Steroids 69: 279–289. [DOI] [PubMed] [Google Scholar]
- 14. Yu J, Akishita M, Eto M, Koizumi H, Hashimoto R, et al. (2012) Src kinase-mediates androgen receptor-dependent non-genomic activation of signaling cascade leading to endothelial nitric oxide synthase. Biochem Biophys Res Commun 424: 538–543. [DOI] [PubMed] [Google Scholar]
- 15. Ba ZF, Wang P, Koo DJ, Ornan DA, Bland KI, et al. (2001) Attenuation of vascular endothelial dysfunction by testosterone receptor blockade after trauma and hemorrhagic shock. Arch Surg 136: 1158–1163. [DOI] [PubMed] [Google Scholar]
- 16. Wynne FL, Khalil RA (2003) Testosterone and coronary vascular tone: implications in coronary artery disease. J Endocrinol Invest 26: 181–186. [DOI] [PubMed] [Google Scholar]
- 17. Gonzales RJ, Krause DN (2004) Duckles SP (2004) Testosterone suppresses endothelium-dependent dilation of rat middle cerebral arteries. Am J Physiol 286: H552–H560. [DOI] [PubMed] [Google Scholar]
- 18. Perusquía M, Espinoza J, Montaño LM, Stallone A (2012) Regional differences in the vasorelaxing effects of testosterone and its 5-reduced metabolites in the canine vasculature. Vasc Pharmacol 56: 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blanco-Rivero J, Sagredo A, Balfagón G, Ferrer M (2007) Protein kinase C activation increases endothelial nitric oxide release in mesenteric arteries from orchidectomized rats. J Endocrinol 192: 189–197. [DOI] [PubMed] [Google Scholar]
- 20.Sagredo A, del Campo L, Ferrer M (2011) Time-dependent effect of ovariectomy on nitric oxide release and function in rat aorta. Joint Meeting of the European Society for Microcirculation (ESM) and the German Society of Microcirculation and Vascular Biology (GFMVB). Munich, 2011.
- 21. Martorell A, Sagredo A, Aras-López R, Balfagón G, Ferrer M (2009) Ovariectomy increases the formation of prostanoids and modulates their role in acetylcholine-induced relaxation and nitric oxide release in the rat aorta. Cardiovasc Res 84: 300–308. [DOI] [PubMed] [Google Scholar]
- 22. Gallet C, Blaie S, Lévy-Toledano S, Habib A (2003) Epidermal-growth-factor receptor and metalloproteinases mediate thromboxane A2-dependent extracellular-signal-regulated kinase activation. Biochem J 371: 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fernández-Martínez AB, Lucio Cazaña FJ (2013) Epidermal growth factor receptor transactivation by intracellular prostaglandin E2-activated prostaglandin E2 receptors. Role in retinoic acid receptor-β up-regulation. Biochim Biophys Acta 1833: 2029–2038. [DOI] [PubMed] [Google Scholar]
- 24. Oeckler RA, Kaminski PM, Wolin MS (2003) Stretch enhances contraction of bovine coronary arteries via an NAD(P)H oxidase-mediated activation of the extracellular signal-regulated kinase mitogen-activated protein kinase cascade. Circulation Res 92: 23–31. [DOI] [PubMed] [Google Scholar]
- 25. Gao Y, Tang S, Zhou S, Ware JA (2001) The thromboxane A2 receptor activates mitogen-activated protein kinase via protein kinase C-dependent Gi coupling and Src-dependent phosphorylation of the epidermal growth factor receptor. J Pharmacol Exp Ther 296: 426–433. [PubMed] [Google Scholar]
- 26. Aksamitiene E, Kiyatkin A, Kholodenko BN (2012) Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans 40: 139–146. [DOI] [PubMed] [Google Scholar]
- 27. Blanco-Rivero J, Sagredo A, Balfagón G, Ferrer M (2006) Orchidectomy increases expression and activity of Cu/Zn-superoxide dismutase, while decreasing endothelial nitric oxide bioavailability. J Endocrinol 190: 771–778. [DOI] [PubMed] [Google Scholar]
- 28. Ferrer M, Osol G (1998) Estrogen replacement modulates resistance artery smooth muscle and endothelial α2-adrenoceptor reactivity. Endothelium 6: 133–141. [DOI] [PubMed] [Google Scholar]
- 29. Liu PY, Death AK, Handelsman DJ (2003) Androgens and cardiovascular disease. Endocr Rev 24: 313–340. [DOI] [PubMed] [Google Scholar]
- 30. Perusquia M, Stallone JN (2010) Do androgens play a beneficial role in the regulation of vascular tone? non genomic vascular effects of testosterone metabolites. Am J Physiol 298: H1301–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cutini PH, Campelo AE, Agriello E, Sandoval MJ, Rauschemberger MB, et al. (2012) The role of sex steroids on cellular events involved in vascular disease. J Steroid Biochem Mol Biol 132: 322–330. [DOI] [PubMed] [Google Scholar]
- 32. Myers SI, Turnage RH, Bartula L, Kalley B, Meng Y (1996) Estrogen increases male rat aortic endothelial cell PGI2 release. Prostaglandins Leukot Essent Fatty Acids 54: 403–409. [DOI] [PubMed] [Google Scholar]
- 33. Gonzales RJ, Ghaffari AA, Duckles SP, Krause DN (2005) Testosterone treatment increases thromboxane function in rat cerebral arteries. Am J Physiol 286: 552–560. [DOI] [PubMed] [Google Scholar]
- 34. Formoso G, Chen H, Kim JA, Montagnani M, Consoli A, et al. (2006) Dehydroepiandrosterone mimics acute actions of insulin to stimulate production of both nitric oxide and endothelin 1 via distinct phosphatidylinositol 3-kinase- and mitogen-activated protein kinase-dependent pathways in vascular endothelium. Mol Endocrinol 20: 1153–1163. [DOI] [PubMed] [Google Scholar]
- 35. Blanco-Rivero J, Balfagón G, Ferrer M (2006) Male castration increases neuronal nitric oxide synthase activity in the rat mesenteric artery through protein kinase C activation. J Vasc Res 42: 526–534. [DOI] [PubMed] [Google Scholar]
- 36. Shimokawa H (1998) Endothelial dysfunction in hypertension. J Atheroscler Thromb 4: 118–127. [DOI] [PubMed] [Google Scholar]
- 37. Matz RL, de Sotomayor MA, Schott C, Stoclet JC, Andriantsitohaina R (2000) Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. Br J Pharmacol 131: 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sellers MM, Stallone JM (2008) Sympathy for the devil: the role of TXA2 in regulation of vascular tone and blood pressure. Am J Physiol Heart Circ Physiol 294: 1978–1986. [DOI] [PubMed] [Google Scholar]
- 39. Nie D, Lamberti M, Zacharek A, Li L, Szekeres K, et al. (2000) Thromboxane A2 regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun 267: 245–251. [DOI] [PubMed] [Google Scholar]
- 40. Morinelli TA, Zhang LM, Newman WH, Meier KE (1994) Thromboxane A2/prostaglandin H2-stimulated mitogenesis of coronary artery smooth muscle cells involves activation of mitogen-activated protein kinase and S6 kinase. J Biol Chem 269: 5693–5698. [PubMed] [Google Scholar]
- 41. Villalobo A (2006) Nitric oxide and cell proliferation. FEBS J 273: 2329–2344. [DOI] [PubMed] [Google Scholar]
- 42. Villalobo A (2007) Enhanced cell proliferation induced by nitric oxide. Dynamic Cell Biol 1: 60–64. [Google Scholar]
- 43. Jorissen RN, Walker F, Pouliot N, Garret TPJ, Ward CW, et al. (2003) Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 284: 31–53. [DOI] [PubMed] [Google Scholar]
- 44. Citri A, Yarden Y (2006) EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 7: 505–515. [DOI] [PubMed] [Google Scholar]
- 45. Schaeffer HJ, Weber MJ (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19: 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Estrada C, Gómez C, Martín-Nieto J, de Frutos T, Jiménez A, et al. (1997) Nitric oxide reversibly inhibits the epidermal growth factor receptor tyrosine kinase. Biochem J 326: 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murillo-Carretero M, Torroglosa A, Castro C, Villalobo A, Estrada C (2009) S-Nitrosylation of the epidermal growth factor receptor: a regulatory mechanism of receptor tyrosine kinase activity. Free Radical Biol Med 46: 471–479. [DOI] [PubMed] [Google Scholar]
- 48. del Campo L, Guvenc TB, Ferrer M, van Bavel E, Bakker NTPE (2013) Testosterone and β-oestradiol prevent inward remodeling of rat small mesenteric arteries: role of NO and transglutaminase. Clin Sci 124: 719–728. [DOI] [PubMed] [Google Scholar]
- 49. Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP, Versteeg HH (2004) Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Cell Biol 36: 1187–1205. [DOI] [PubMed] [Google Scholar]
- 50. Alfranca A, Iñiguez MA, Fresno M, Redondo JM (2006) Prostanoid signal transduction and gene expression in the endothelium: role in cardiovascular diseases. Cardiovasc Res 70: 446–456. [DOI] [PubMed] [Google Scholar]
- 51. Yogi A, Callera GE, Aranha AB, Antunes TT, Graham D, et al. (2011) Sphingosine-1-phosphate-induced inflammation involves receptor tyrosin kinase transactivation in vascular cells. Upregulation in hypertension. Hypertension 57: 809–818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.