

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a clinicopathological change characterized by the accumulation of triglycerides in hepatocytes and has frequently been associated with obesity, type 2 diabetes mellitus, hyperlipidemia, and insulin resistance. It is an increasingly recognized condition that has become the most common liver disorder in developed countries, affecting over one-third of the population and is associated with increased cardiovascular- and liver-related mortality. NAFLD is a spectrum of disorders, beginning as simple steatosis. In about 15% of all NAFLD cases, simple steatosis can evolve into non-alcoholic steatohepatitis, a medley of inflammation, hepatocellular injury, and fibrosis, often resulting in cirrhosis and even hepatocellular cancer. However, the molecular mechanism underlying NAFLD progression is not completely understood. Its pathogenesis has often been interpreted by the “double-hit” hypothesis. The primary insult or the “first hit” includes lipid accumulation in the liver, followed by a “second hit” in which proinflammatory mediators induce inflammation, hepatocellular injury, and fibrosis. Nowadays, a more complex model suggests that fatty acids (FAs) and their metabolites may be the true lipotoxic agents that contribute to NAFLD progression; a multiple parallel hits hypothesis has also been suggested. In NAFLD patients, insulin resistance leads to hepatic steatosis via multiple mechanisms. Despite the excess hepatic accumulation of FAs in NAFLD, it has been described that not only de novo FA synthesis is increased, but FAs are also taken up from the serum. Furthermore, a decrease in mitochondrial FA oxidation and secretion of very-low-density lipoproteins has been reported. This review discusses the molecular mechanisms that underlie the pathophysiological changes of hepatic lipid metabolism that contribute to NAFLD.
Keywords: non-alcoholic fatty liver disease, molecular pathways, insulin resistance, fatty acid metabolism
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a major public health issue due to its high prevalence worldwide, and ranges widely from 11% to 46%,1–3 and has potentially serious sequelae.4 The prevalence increases to 58% in overweight individuals and can be as high as 98% in non-diabetic obese individuals.5
NAFLD is an inclusive term that takes in a spectrum of liver pathologies from simple steatosis (SS) to non-alcoholic steatohepatitis (NASH). NASH involves hepatocellular injury and inflammation of the liver.6
Whereas SS is characterized by a relatively favorable clinical course, NASH much more frequently progresses to cirrhosis and hepatocellular carcinoma.7,8 NAFLD should be suspected in individuals who are either obese, diabetic, or have metabolic syndrome.9
Moreover, NAFLD is considered a hepatic manifestation of metabolic syndrome and a risk factor for type 2 diabetes mellitus, dyslipidemia, and hypertension.10,11 The majority of patients with NAFLD are asymptomatic and the disease may be detected via routine blood tests showing elevated liver enzymes or when an ultrasound is performed for various reasons and detects liver steatosis. Secondary causes of hepatic steatosis or elevated liver enzymes, such as excess alcohol consumption, medications, toxins, lipodystrophy, autoimmune and inflammatory diseases, nutrition (malnutrition, total parenteral nutrition, severe weight loss, and refeeding syndrome), viral hepatitis, and metabolic liver disease should be excluded by reviewing the patient’s history and proper investigation.9,12
Although it is still not possible to diagnose NAFLD based solely on blood work, elevated transaminases can be used as a first step.13 An aspartate aminotransferase–alanine aminotransferase ratio <1 is also seen in NAFLD14 and supports NASH. However, it is important to note that patients with normal transaminases and liver steatosis on imaging may also have NASH.15 Ultrasonography is a noninvasive tool that is used in the detection of liver steatosis. Other imaging techniques such as computed tomography and nuclear magnetic resonance imaging can also detect liver steatosis, but neither of these more expensive techniques provide more information than ultrasonography,16,17 except for fat quantification.18 Diagnosis for NASH is confirmed when a liver biopsy shows the presence of perilobular inflammation, or the presence of hepatocyte ballooning, Mallory’s hyaline, and acidophil bodies with or without fibrosis. Noninvasive tests such as Fatty Liver Index, NAFLD fibrosis score, FibroMeter, and Fibroscan19 may suggest the presence of NASH by detecting fibrosis. Research is ongoing to assess surrogate markers for NASH such as CK18, but this remains experimental.20,21
Regarding the management of NAFLD, weight management through improvements in diet and increased physical activity can help to improve liver histology as well as delay disease progression.22–24 Lifestyle interventions may not be effective in certain cases, and thus other approaches must be considered. Pharmacological treatment has been studied in this population, specifically insulin-sensitizing agents (metformin and thiazolidinediones [TZDs]); however, there are conflicting results. Clinical studies could not demonstrate the effectiveness of metformin in the treatment of NAFLD.25 On the other hand, TZDs that are peroxisomal proliferator-activated receptor γ (PPARγ) agonists promote hepatic fatty acid (FA) oxidation and decrease hepatic lipogenesis.26,27 In NAFLD patients, TZDs have been shown to decrease hepatic fat and decrease cellular injury. However, discontinuing TZD therapy resulted in NASH recurrence and long-term use of TZDs can result in medical complications such as edema, congestive heart failure, osteoporosis, and weight gain.28,29 The use of statins in NAFLD patients with dyslipidemia can improve liver function tests,30 as well as steatosis.31 Furthermore, statins seem to be safe in NAFLD/NASH patients with dyslipidemia.32 However, there is a lack of evidence for the use of statins in the treatment of NASH patients without dyslipidemia.33 Further research is necessary to document the effect of other strategies, such as bariatric surgery, antioxidants, and fish oil in NAFLD.
Because there are currently no effective therapies for NAFLD apart from weight loss, ongoing research efforts are focused on understanding the underlying pathobiology of hepatic steatosis with the intention of identifying novel therapeutic targets. In this sense, this review analyses some of the molecular mechanisms that underlie the pathophysiological changes of hepatic lipid metabolism in NAFLD: the contribution of lipid metabolism, the influence of inflammation, and the role of lipotoxicity and cannabinoid receptors in NAFLD.
Contribution of lipid metabolism to NAFLD
The liver plays a major role in lipid metabolism, importing free FAs (FFAs) and manufacturing, storing, and exporting lipids; derangements in any of these processes can lead to the development of NAFLD.34 FAs are involved in many important cellular events, such as synthesis of cellular membranes, energy storage, and intracellular signaling pathways. However, chronically elevated FFAs can disturb diverse metabolic pathways and induce insulin resistance (IR) in many organs. Hepatic fat accumulation has been strongly associated with IR.35,36 IR in the peripheral adipose tissue enhances lipolysis and increases the delivery of adipose-derived FFAs to the liver. In particular, obesity increases tumor necrosis factor α (TNFα) production in adipocytes, facilitates adipocyte IR, and increases lipolysis rate.37 Thus, the circulating pool of FFAs is increased in obese individuals and accounts for the majority of liver lipids in NAFLD.38
Under physiological conditions, triglyceride (TG) synthesis is stimulated to dispose of the excess of FFAs. The TGs can then be stored as lipid droplets within hepatocytes or secreted into the blood as very-low-density lipoprotein (VLDL).39 Rodent studies have shown that the mechanisms leading to the excessive accumulation of hepatic TGs are associated with an increased supply of FFAs from peripheral adipose tissue to the liver and an enhanced de novo lipid synthesis via the lipogenic pathway. Conversely, liver disposal via β-oxidation and VLDL export are moderately affected.40 At the cellular level, defects in the insulin signaling pathways contribute to the increase of FFA flux in the liver, which in turn activates a series of signaling cascades and leads to the phosphorylation of several substrates.41 Despite TGs being the main lipids stored in the liver of patients with NAFLD, large epidemiological studies suggest that they might exert protective functions. TG synthesis seems to be an adaptive, beneficial response in situations where hepatocytes are exposed to potentially toxic TG metabolites.42–44 FFAs and cholesterol, especially when accumulated in the mitochondria, are considered the “aggressive” lipids leading to TNFα-mediated liver damage and reactive oxygen species (ROS) formation.45,46 These lipids could also be present in a non-steatotic liver and act as early “inflammatory” hits, leading to the whole spectrum of NAFLD pathologies. The concept of lipotoxicity and involved lipid species has been introduced: abundant FAs cause lipotoxicity via the induction of ROS release, which causes inflammation, apoptosis, and thus, the progression to NASH and fibrogenesis.46–48
In summary, TG accumulation in the cytoplasm of hepatocytes, as the hallmark of NAFLD, arises from an imbalance between lipid acquisition (FA uptake and de novo lipogenesis) and removal (mitochondrial FA oxidation and export as a component of VLDL particles) and accompanies multiple pathophysiological mechanisms in NASH (Figure 1). In order to control the progression of NAFLD, it is important to understand the regulatory mechanisms of lipid accumulation in the human liver.
Figure 1.
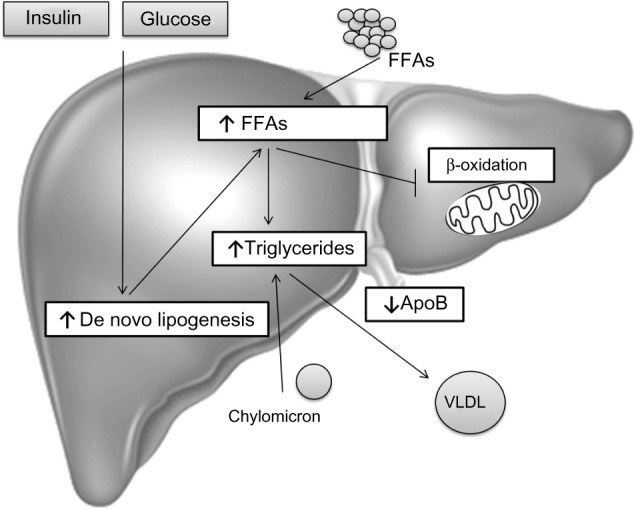
Hepatic steatosis.
Notes: The hallmark of NAFLD is triglyceride accumulation in the cytoplasm of hepatocytes as a result of an imbalance between lipid input and output: 1) an increase in FFAs uptake derived from the circulation due to increased lipolysis from adipose tissue and/or from the diet in the form of chylomicrons; 2) an increase in glucose and insulin levels in response to carbohydrate intake that promotes de novo lipogenesis; 3) a decrease in FA mitochondrial oxidation; 4) a decrease in triglyceride hepatic secretion by packaging with ApoB into VLDLs. In NAFLD patients, enhanced acquisition of FAs through uptake and de novo lipogenesis are not compensated by FA oxidation or production of VLDL particles.
Abbreviations: ApoB, apolipoprotein B; FFAs, free fatty acids; FA, fatty acid; NAFLD, non-alcoholic fatty liver disease; VLDL, very-low-density lipoprotein.
Hepatic FA uptake
One of the sources for hepatic FAs is FFA recruitment from the blood stream. FFAs are derived from lipolysis in adipocytes, which usually occurs in the fasting state, promoted by catecholamines, natriuretic peptides, and glucagon, and are usually repressed by insulin.49 However, the IR state (obesity, metabolic syndrome) goes along with increased adipocyte lipolysis, leading to abundant FFAs in the plasma pool independently from the nutritional status.50 FFAs are then taken up by the hepatocytes in a facilitated fashion rather than by passive processes.51 FA uptake into the liver contributes to the steady balance of hepatic TGs, as well as the pathogenesis of NAFLD. The rate of FA uptake from plasma into cells depends on the FA concentration in the plasma and the hepatocellular capacity for FA uptake, which also depends on the number and activity of transporter proteins on the sinusoidal plasma membrane of the hepatocyte. The main plasma membrane transporters for FFAs are FA transporter protein (FATP), caveolins, FA translocase (FAT)/CD36, and FA-binding protein (FABP).52–56
FATP
Six FATP isoforms have been identified in mammalian cells, which contain a common motif for FA uptake and fatty acyl-CoA synthetase function.57 Of these isoforms, FATP2 and FATP5 are highly expressed in the liver, and are utilized as major FATPs for the normal physiological context. In mouse hepatocytes, adenovirus-mediated knockdown of FATP2 or genetic deletion of FATP5 significantly decreases the rates of FA uptake.58 Indeed, FATP5 knockout mice have shown resistance to diet-induced obesity and hepatic TG accumulation.58 In humans, a promoter polymorphism in the liver-specific FATP5 is associated with features of the metabolic syndrome and steatosis.59
Caveolins
Caveolins consist of three protein family members termed caveolins 1, 2, and 3. They are found in the membrane structures called caveolae, which are important for protein trafficking and the formation of lipid droplets. Caveolin 1 knockout mice exhibited lower TG accumulation in the liver and showed resistance to diet-induced obesity, showing the importance of this protein in TG synthesis.60 Some authors suggest there is an involvement of caveolin 1 in abnormal lipogenesis and mitochondrial function typical of steatotic hepatocytes in NAFLD.61
FAT/CD36
It is well-known that FFAs are taken up into cells by passive diffusion and by protein-mediated mechanisms involving a number of FA transporters, of which FAT/CD36 is the best characterized. FAT/CD36 is expressed in a wide variety of cells including macrophages, adipocytes, myocytes, enterocytes, and hepatocytes. This transmembrane protein plays an important role in facilitating the uptake and intracellular trafficking of FFAs, as well as esterification into TGs in heart and skeletal muscle cells; this function is largely dependent on its translocation from intracellular depots to the plasma membrane. Insulin, muscular contractions, and the transcription factor Forkhead box protein O1 (FoxO1) induce FAT/CD36 translocation and enhance FFA uptake.62
Hepatic FAT/CD36 expression is normally weak, but its expression is enhanced in rodents with fatty liver.63 Moreover, some authors have demonstrated that FAT/CD36 mRNA levels increase concomitantly with hepatic TG content in different animal models of liver steatosis.64,65 Further studies have shown that FAT/CD36 is a common target gene of liver X receptor (LXR), pregnane X receptor, and PPARγ in promoting hepatic steatosis in a murine model.66 However, little is known about the significance of FAT/CD36 in human liver diseases. In morbidly obese patients with NAFLD, Greco et al showed that hepatic FAT/CD36 mRNA levels were positively related to liver fat content67 and Bechmann et al found a significant correlation between hepatic FAT/CD36 mRNA and apoptosis in patients with NASH.68 Other authors have described that hepatic FAT/CD36 upregulation is significantly associated with IR, hyperinsulinemia, and increased steatosis in patients with NASH.62
FABPs
The FABPs are a group of molecules that coordinate inflammatory and metabolic responses in cells.69 These proteins are a family of 14- to 15-kDa proteins that bind with high affinity to hydrophobic ligands such as saturated and unsaturated long-chain FAs (LCFAs).70 Two isoforms of FABPs, aP2 (FABP4) and mal1 (FABP5) are the isoforms coexpressed in adipocytes and macrophages.71 The expression of these FABP isoforms is controlled transcriptionally during adipocyte differentiation and is regulated by PPARγ agonists, insulin, and FAs. The functions of cytoplasmic FABPs include enhancement of FFA solubility and transport to specific enzymes and cellular compartments (to the mitochondria and peroxisomes for oxidation; to the endoplasmic reticulum [ER] for reesterification; into lipid droplets for storage; or to the nucleus for gene expression regulation).71,72 Disruption or pharmacological blockade of FABP4 protects mice from dyslipidemia, atherosclerosis, IR, and fatty liver in the context of either a high-fat diet or genetically induced obesity.73 The definitive biology and function of FABPs in human physiology and disease are still not fully clarified.69,73 Few studies have assessed the involvement of hepatic FABP4 expression in NAFLD. Greco et al and Taskinen et al have described FABP4 as being upregulated in subjects with high liver fat content.67,74 The expression of FABP4 and FABP5 in the liver was correlated with hepatic fatty infiltration in NAFLD patients.75
Recent studies have also suggested that hepatic FA uptake via FATPs can be a novel therapeutic strategy for NAFLD. Adenovirus-mediated knockdown of FATP2 or FATP5 reduced hepatic TG accumulation in high-fat fed mice.76,77 Moreover, both deoxycholic and ursodeoxycholic acid have shown promise as inhibitors of FATP5-mediated FA uptake, suggesting that they may improve hepatic steatosis in high-fat fed mice.78
PPARγ
PPARγ is a master transcriptional regulator of adipogenesis and plays an important role in the process of lipid storage.79 Thus, PPARα and PPARγ have opposing functions in the regulation of fat metabolism; PPARα promotes utilization, while activation of PPARγ promotes storage. Indeed, as increased PPARγ expression has been found in steatotic livers, it has been suggested that the role of PPARγ in the activation of lipogenic genes may contribute to the development of steatosis. Nevertheless, several studies have shown that PPARγ overexpression can prevent the progression of hepatic steatosis in murine models, and treatment with the PPARγ agonist rosiglitazone has been shown to have similar effects. The protective effects of PPARγ could be due to higher insulin sensitivity in adipose tissue and skeletal muscle leading to a reduction in FFA deposition in the liver. Adiponectin has also been shown to be increased by PPARγ, which also contributes to insulin sensitivity as well as upregulating PPARα expression, which leads to further hepatic FA oxidation. Furthermore, PPARγ expression has been shown to have anti-inflammatory and anti-fibrotic effects in stellate cells, macrophages, and epithelial cells. Westerbacka et al have described that PPARγ was overexpressed in the fatty liver of obese human subjects.75
Activation of PPARγ in adipose tissue has been proposed to promote the relocalization and storage of fat in adipose tissue, protecting peripheral tissues from lipotoxicity.
Regarding this, the TZDs have proven to be effective drugs for improving insulin sensitivity and treating type 2 diabetes. Moreover, pioglitazone and rosiglitazone are highly effective in improving NAFLD outcomes in patients.80 Unfortunately, the clinical use of TZDs against NAFLD has been hampered by side effects.
De novo lipogenesis
The process in which the liver synthesizes endogenous FAs is called de novo lipogenesis. This includes de novo synthesis of FAs through a complex cytosolic polymerization in which glucose is converted to acetyl-CoA by glycolysis and the oxidation of pyruvate. Acetyl-CoA carboxylase (ACC1) then converts acetyl-CoA into malonyl-CoA. Finally, FA synthase (FAS) catalyzes the formation of palmitic acid from malonyl-CoA and acetyl-CoA.81–83 Depending on the metabolic state, FAs are then processed to TGs and stored or rapidly metabolized.
Dietary fats are packed in chylomicrons and hydrolyzed, releasing FAs of which approximately 20% are delivered to the liver.7 In the fasting state, a decline of insulin levels stimulates adipocyte TG hydrolase, thereby releasing FFAs that are transported to the liver. In the liver, FFAs derived from peripheral tissue, endogenous synthesis, or diet, can be used for: 1) energy and ketone body production via mitochondrial β-oxidation; 2) sterified and stored as TGs in lipid droplets; or 3) packaged with apolipoprotein B into VLDL that is secreted into the circulation.83,84 In NAFLD patients, enhanced acquisition of FAs through uptake and de novo lipogenesis are not compensated by FA oxidation or production of VLDL particles (Figure 1).
The rate of de novo lipogenesis is regulated primarily at the transcriptional level. Several nuclear transcription factors are involved such as LxRα, sterol regulatory element-binding protein 1c (SREBP1c), carbohydrate-responsive element-binding protein (ChREBP), and farnesoid X receptor (FxR); and enzymes (ACC1, FAS, and steroyl CoA desaturase 1 [SCD1]). Postprandially, plasma glucose and insulin levels rise, which promote activation of lipogenesis through the activation of ChREBP and SREBP1c, respectively.34,85 In humans, NAFLD has been associated with increased hepatic expression of several genes involved in de novo lipogenesis.86,87
LXRs
LXRs are ligand-activated transcription factors that belong to the nuclear receptor (NR) superfamily.88 There are two LXR isoforms termed α and β. LxRα is mainly expressed in the liver, adipose tissue, and intestine, whereas LxRβ is ubiquitously expressed.89 In addition to modulating cholesterol metabolism, LXRs have been characterized as major regulators of hepatic FA biosynthesis.90 A major function of LxRα in the liver is the stimulation of de novo lipogenesis, through the induction of SREBP1c, ACC1, FAS, and SCD1 (Figure 2).91–93
Figure 2.
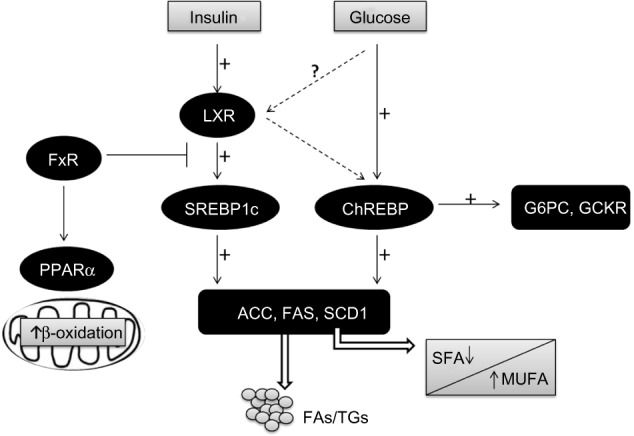
Transcriptional control of lipogenesis and glycolysis.
Notes: The conversion of glucose into FAs through de novo lipogenesis is nutritionally regulated by glucose and insulin signaling pathways, which induce the expression of glycolytic and lipogenic genes synergistically in response to dietary carbohydrates. Insulin activates the transcription factor SREBP1c, which induces lipogenic enzymes (ACC1, FAS, SCD1), while glucose activates the transcription factor ChREBP, which induces both lipogenic (ACCl, FAS, SCD1) and glycolytic (G6PC, GCKR) enzymes. ChREBP is also a direct target of LXRs, and modifies the ratio of MUFA/SFA in favor of MUFA by stimulating SCD1 activity. Recently, glucose was also identified as activating LXR’s genes. Hepatic FxR activation inhibits FA/TG synthesis by suppressing SREBP1c and LXRα activation, and inducing the expression of PPARα, which promotes mitochondrial oxidation of FAs.
Abbreviations: ACC, acetyl-CoA carboxylase; ChREBP, carbohydrate-responsive element-binding protein; FA, fatty acid; FAS, fatty acid synthase; FFAs, free fatty acids; FxR, farnesoid X receptor; G6PC, glucose 6-phosphatase; GCKR, glucokinase regulatory protein; LXR, liver X receptor; MUFA, monosaturated fatty acids; PPARα, peroxisomal proliferator-activated receptor alpha; SCD1, steroyl CoA desaturase 1; SFA, saturated fatty acids; SREBP1c, sterol regulatory element-binding protein 1c; TG, triglyceride.
Several authors have described an enhanced expression of LxRα and SREBP1c in NAFLD.87,94,95
SREBP1c
SREBPs are a family of membrane-bound transcription factors. SREBPs are synthesized as 125 kD precursors embedded in the ER. Proteolytic cleavage then allows the accumulation of active SREBP in the nucleus.
There are different SREBP isoforms: SREBP1c and SREBP2 are expressed in the liver, while SREBP1a is expressed only at very low levels in the liver of adult mice, rats, and humans.96 SREBP1c, the predominant isoform in the liver, preferentially affects the transcription of genes that regulate de novo lipid synthesis, although SREBP2 regulates genes involved in cholesterol biosynthesis and metabolism. The SREBP1a isoform, despite its very low levels in the liver, transactivates both lipogenic and cholesterol genes.97
To date, the main regulation demonstrated for SREBP1c is at the transcriptional level. SREBP1c transcription is induced by two quite disparate stimuli: insulin, a hormone released in response to carbohydrate intake and leading to a parallel increase in both the membrane-bound precursor and the mature nuclear form, and LxRα, a transcription factor that acts as a cholesterol sensor.92,93,98–100 In response to feeding, SREBP1c binds to its lipogenic genes, such as ACC1, FAS, and SCD1, and to its own gene, thereby stimulating hepatic lipogenesis (Figure 2).96,101–105
Different authors have described an enhanced expression of SREBP1c and LxRα in NAFLD.87,94,95 However, Nagaya et al demonstrated the downregulation of the hepatic SREBP1c-mediated lipogenic pathway in advanced NASH patients; SREBP1c mRNA levels were inversely correlated with the fibrosis stage.106 These discrepancies might be explained by differences in the cohort of studied patients. For example, Higuchi et al94 included normal weight patients with NAFLD and Lima-Cabello et al95 included patients with NAFLD and with steatosis related to chronic hepatitis C virus infection in mildly overweight men and women. Moreover, Higuchi et al did not evaluate either histological findings nor protein levels or intracellular localization of SREBP1c.94
ChREBP
De novo lipogenesis is regulated by glucose and insulin signaling pathways in response to dietary carbohydrate intake to induce glycolytic and lipogenic gene expression. SREBP1c has emerged as a major mediator of insulin action on lipogenic genes. However, SREBP1c activity alone is not sufficient for the stimulation of glycolytic and lipogenic gene expression.107,108 Over recent years, it has been reported that the liver transcription factor ChREBP is required for the induction of glycolytic gene expression by glucose and that it acts together with SREBP1c to stimulate lipogenic genes.109–111 Interestingly, ChREBP was also identified as a direct target of LXRs, which are an important regulator of the lipogenic pathway through the transcriptional control of SREBP1c, ACC1, FAS, and SCD1.112–117 Oxysterols are known as ligands of LXRs, but glucose was also shown to activate LXRs and to induce their target genes, including ChREBP (Figure 2).107,118
Postprandial hyperglycemia raises the hepatic concentrations of phosphorylated intermediates, causing activation of ChREBP, which binds to the promoter of its target genes as a heterotetramer with its ubiquitously expressed partner Max-like protein X (Mlx). ChREBP target genes include not only enzymes of glycolysis and lipogenesis that predispose to hepatic steatosis, but also glucose 6-phosphatase (G6PC), which catalyzes the final reaction in glucose production, and glucokinase regulatory protein (GCKR), which inhibits hepatic glucose uptake.119,120 Transcriptional induction of G6PC and GCKR manifests as hepatic glucose intolerance or IR.121 Studies using a dominant negative variant of Mlx identified target genes of ChREBP–Mlx that promote hepatic glucose intolerance when overexpressed.120
Study results of the role and impact of ChREBP in glucose and lipid metabolism have been confusing. Global ChREBP deficiency in C57BL/6J mice results in IR.122 On the other hand, ChREBP deficiency123,124 or expression of a dominant negative Mlx isoform125 in an obese ob/ob background decreases hepatic steatosis and other related metabolic alterations, including IR. Benhamed et al126 hypothesized that these opposite phenotypes in these two murine models may reside in the fact that ChREBP controls both glycolysis and lipogenesis, and that the beneficial effect of ChREBP deficiency may only be apparent in the context of lipid overload. The authors showed that mice overexpressing ChREBP, on a standard diet, remained insulin sensitive, despite increased lipogenesis resulting in hepatic steatosis. However, mice that overexpress ChREBP, on a high-fat diet, showed normal insulin levels and improved insulin signaling and glucose tolerance compared with controls, despite having greater hepatic steatosis. This effect seems to be mediated by the fact that ChREBP modifies the monounsaturated FAs/saturated FAs (MUFA/SFA) balance in favor of MUFA, by stimulating SCD1 activity. Taken together, these results demonstrated that increasing ChREBP, by buffering detrimental FAs and favoring lipid partitioning, can dissociate hepatic steatosis from IR, with beneficial effects on both glucose and lipid metabolism. Interestingly, ChREBP expression in liver biopsies from patients with NASH was higher when steatosis was greater than 50% and lower in the presence of severe IR,126 sup porting this conclusion.
Furthermore, because insulin induces enzymes of lipogenesis by activation of SREBP1c and represses G6PC through other transcriptional regulators, a mechanism of “selective IR” has been proposed to explain the simultaneous elevation of hepatic glucose production (or G6PC expression) and lipogenesis in human type 2 diabetes or models of IR.127
FxR
The FxR is a member of the NR superfamily and a receptor for bile acids. FxR activation leads to alterations in pathways involved in energy metabolism. It is mainly expressed in the liver, intestine, kidneys, and the adrenal glands, with less expression in adipose tissue and heart.128–130 FxR has emerged as a master regulator of lipid and glucose homeostasis in the liver and of inflammatory processes at hepatic and extrahepatic sites. Also, a number of synthetic FxR agonists are being used for the treatment of different hepatic and metabolic disorders, resulting in a lower inflammatory and fibrogenic process.131,132
The generation of FxR knockout mice showed a clear role for FxR as the master regulation of bile acid homeostasis.133,134 However, FxR knockout mice also revealed elevated levels of cholesterol and TGs in both the plasma and liver, suggesting a key role for FxR lipid metabolism as well. In fact, it was demonstrated that FxR needs to be activated in order to reduce the expression of SREBP1c.135 More recently, in addition to bile acid and lipid metabolism, it has been shown that FxR also plays an important role in glucose metabolism, improving insulin sensitivity and glucose tolerance in a diabetic mice model.136,137
Regarding its role in lipid metabolism, the majority of literature seems to point to the fact that FxR activation is beneficial in situations of excess, such as obesity and diabetes. FxR activation seems to reduce TGs levels by: 1) reducing FA synthesis in the liver, through the reduction of SREBP1c and LxR expression;138 2) inducing the expression of PPARα, which promotes FFA catabolism via β-oxidation; 3) increasing TG clearance; and 4) increasing adipose tissue storage and altering adipokine patterns (Figure 2).139,140
Another hepatic protective mechanism of FxR activation has been shown to be maintenance of gut integrity against gut-derived endotoxins through the induction of antibacterial factors such as angiogenin, inducible NO synthase, and interleukin-18 (IL18).131,132
Patients with NAFLD have lower protein and mRNA FxR levels, which has been attributed to higher TG synthesis and induced expression of SREBP1c and LxRα.138
ACC1 and FAS
In the process of FA synthesis, ACC1 converts acetyl-CoA, an essential substrate of FAs, to malonyl-CoA. FAS then utilizes both acetyl-CoA and malonyl-CoA to form palmitic acid (C16:0). Both are highly regulated by a transcriptional factor, SREBP1c, and play important roles in the energy metabolism of FAs. They are currently considered an attractive target for regulating the human diseases of obesity, diabetes, cancer, and cardiovascular complications. Dorn et al found that FAS expression was impaired in SS, while the absence of SS in hepatic inflammation did not affect FAS expression.141 In agreement with Dorn et al, several authors have described an enhanced expression of FAS in NAFLD.142 These authors have also described that ACC1 mRNA expression was higher in NAFLD. In support of increased FA synthesis in NAFLD, Morgan et al found that ACC1 and FAS mRNA expression were significantly higher in high-fat mice.143 All these findings suggest that ACC1 and FAS might be a new diagnostic marker or therapeutic target for NAFLD.
FoxO1
FoxO1 is a transcription factor with an important role not only in glycogenolysis and gluconeogenesis, but also in lipid metabolism.
With regard to lipid metabolism, liver-specific transgenic expression of active FoxO1 induces the expression of genes involved in lipid transport and decreases the expression of important genes for glycolysis and lipid/sterol synthesis, resulting in lower postprandial TG concentrations compared to in wild-type mice.144 However, using a similar murine model, another group observed enhanced lipogenesis and liver steatosis.145 Similarly, adenoviral delivery of an active FoxO1 variant to the liver results in lipogenesis, hepatic steatosis, and reduced FA oxidation. These increases in lipogenesis result from a feedback loop that enhances insulin signaling, thereby modulating lipid metabolism through SREBP1c in a FoxO1-independent manner.146
FoxO1 not only inhibits SREBP1c expression but also suppresses the expression of genes directly involved in FA synthesis, including FAS and adenosine triphosphate (ATP) citrate lyase.144
With regard to glucose metabolism, under fasting conditions, the liver provides energy by releasing glucose into the bloodstream. Initially, this results from the breakdown of liver glycogen stores (glycogenolysis), whereas with prolonged fasting, the primary source of glucose is gluconeogenesis. Studies with adenoviral vectors in isolated hepatocytes confirm that FoxO1 stimulates the expression of gluconeogenic genes and suppresses the expression of genes involved in glycolysis, the shunt pathway, and lipogenesis, including glucokinase and SREBP1c. Taken together, these results indicate that FoxO1 proteins promote hepatic glucose production through multiple mechanisms and contribute to the regulation of other important metabolic pathways in adapting to fasting and feeding in the liver, including glycolysis, the pentose phosphate shunt, and lipogenic and sterol synthetic pathways.144
Chronic expression of an active FoxO1 mutant in the liver leads to increased expression of genes involved in gluconeogenesis, resulting in elevated plasma glucose and insulin levels, which are not able to maintain normal glycemia.144 Valenti et al suggest that increased FoxO1 activity may play a role in the pathogenesis of hepatic IR associated with NAFLD.147
Reduction of FoxO1 in both liver and white adipose tissue using an antisense oligonucleotide-mediated approach improves glucose tolerance and both hepatic and peripheral insulin action in mice with diet-induced obesity.148 Consistent with these studies, FoxO1 haploinsufficiency is able to rescue the loss of insulin sensitivity in insulin receptor–haploinsufficient mice partly by reducing the hepatic expression of gluconeogenic genes.149
FA oxidation
Oxidation of FAs occurs within the mitochondria, peroxisomes, and the ER. It facilitates the degradation of activated FAs to acetyl-CoA. FAs are activated by acyl-CoA-synthetase to acyl-CoA in the cytosol, which is indispensable for enabling FAs to cross membranes and enter organelles. Short- and medium-chain FAs pass the mitochondrial membrane without activation. However, activated LCFAs are shuttled across the membrane via carnitine palmitoyltransferase-1 (CPT1). Malonyl-CoA, an early intermediate of de novo lipogenesis, is an inhibitor of CPT1. In the fed state, FA oxidation is inhibited and de novo lipogenesis promoted, allowing for storage and distribution of lipids. In general, short-, medium-, and long-chain FAs are oxidized within mitochondria (β-oxidation), while toxic, very-long-chain FAs are oxidized within peroxisomes. In diabetes or FA overload, cytochrome P450 (CYP4A)-dependent ω-oxidation of LCFAs occurs in the ER and induces ROS and lipid peroxidation. During the process of β-oxidation, electrons are indirectly donated to the electron transport chain to drive ATP synthesis. Acetyl-CoA can be further processed via the tricarboxylic acid cycle, or in the case of FA abundance, be converted into ketone bodies. PPARα and insulin signaling are again involved in the regulation of FA oxidation and the formation of ketone bodies via transcriptional regulation of mitochondrial 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase.82
PPARα
In the liver, PPARα plays a pivotal role in FA metabolism by upregulating the expression of numerous genes involved in mitochondrial FA and peroxisome FA oxidation, as well as numerous other aspects of FA metabolism in the cell.150 As a consequence, activation of PPARα can prevent and decrease hepatic fat storage.151–154 When PPARα sensing is inefficient, overnight or prolonged fasting leads to severe hepatic steatosis, as seen in PPAR-α−/− mice.155,156 PPARα−/− mice fail to upregulate FA oxidation systems in the liver and cannot oxidize the influxed FAs, and thus develop severe hepatic steatosis. PPARα−/− mice also develop severe steatohepatitis when maintained on a diet deficient in methionine and choline.153,157,158 Also of importance is that administering PPARα agonists to rats not only prevents the development of methionine- and choline-deficient diet-induced steatohepatitis by preventing intrahepatic lipid and lipoperoxide accumulation, but also reverses hepatic fibrosis by decreasing the expression of fibrotic markers and reducing the number of stellate cells.153,157–159 The efficacy of these agonists in the treatment of NAFLD in human subjects has not yet been studied in depth. From the available data, no definitive conclusion can be made on the efficacy of PPARα agonists on NAFLD due to study limitations, such as small sample size, incomplete data, and the use of agonists in combination with other strategies.160
Besides governing metabolic processes, PPARα also regulates inflammatory processes, mainly by inhibiting inflammatory gene expression. Hepatic PPARα activation has been repeatedly shown to reduce hepatic inflammation elicited by acute exposure to cytokines and other compounds.161–165
In conclusion, PPARα activation plays a role in the modulation of hepatic steatosis due to its effects: upregulation of FA oxidation systems and the ensuing burning of energy, reduction in the toxicity of FAs, and its anti-inflammatory effect (Figure 3).153,155,156,158
Figure 3.

Fatty acid oxidation.
Notes: In the liver, mitochondrial, peroxisomal, and microsomal FA oxidation are regulated by PPARα and metabolize energy. Increased sensing of PPARα results in energy burning and reduced fat storage. Decreased sensing of PPARα leads to a reduction in energy utilization and increased lipogenesis, resulting in steatosis and steatohepatitis.
Abbreviations: CPT1, carnitine palmitoytransferase-1; FA, fatty acid; PPARα, peroxisomal proliferator-activated receptor alpha.
CPT1
CPT1 is a regulatory enzyme in the mitochondria that transfers FAs from the cytosol to the mitochondria prior to β-oxidation (Figure 3). Inhibition of CPT1 has been shown to prevent IR induced by a high-fat diet, partly due to a reduction in some of the deleterious intermediates generated by incomplete FA oxidation and partly to a shift toward increased glucose oxidation for energy production.166 Kohjima et al showed that CPT1 expression in humans is reduced by 50% in NAFLD compared with that in the normal liver.142
Inflammation and NAFLD
It is well-known that the balance between pro- and anti-inflammatory acting cytokines is fundamental in the control of systemic and hepatic insulin action, and as a consequence, in the development of NAFLD. IR is an important feature of NAFLD and is caused by a variety of factors, including soluble mediators derived from adipose tissue and/or immune cells: the adipocytokines (Figure 4).167
Figure 4.

Cytokines and NAFLD.
Notes: The balance/imbalance of pro- and anti-inflammatory cytokines secreted by adipose may profoundly affect the liver. Hepatic adiponectin mRNA expression was lower in individuals with NAFLD. However, TNFα and IL6 mRNA expression were higher in these patients. NALFD is associated with more proinflammatory cytokines and with fewer anti-inflammatory cytokines.
Abbreviations: ADIPOR2, adiponectin receptor type 2; IL6, interleukin-6; NAFLD, non-alcoholic fatty liver disease; TNFα, tumor necrosis factor alpha; TNFR, tumor necrosis factor receptor; IL6R, interleukin-6 receptor.
Adiponectin
Adiponectin, one of the major products of adipocytes, is a prototypic anti-inflammatory and anti-diabetic adipocytokine, the actions of which are mainly exerted by the activation of adenosine monophosphate (AMP)-activated kinase and PPARα. Adiponectin has two specific receptors: adiponectin receptor type 1 and 2 (ADIPOR1 and 2). ADIPOR1 is widely expressed, whereas ADIPOR2 can be mainly observed in the liver.168 Serum levels of adiponectin are lower in individuals with obesity, type 2 diabetes, and in conditions of IR,169 whereas adiponectin synthesis is induced by weight loss and PPARγ activation by its ligands, TZDs.28 In general, studies have suggested that adiponectin exerts anti-inflammatory effects, stimulates secretion of anti-inflammatory cytokines such as IL10 or IL1 receptor antagonist (IL1Ra), blocks nuclear factor κB (NF-κB) activation, and inhibits the release of TNFα, IL6, and chemokines.170,171
The liver is not a relevant source of circulating adiponectin, but it is a major target organ for many of its effects. In mice with alcoholic and non-alcoholic fatty liver disease, administering recombinant adiponectin ameliorated necroinflammation and steatosis, partly via inhibition of the hepatic production of TNFα and the decrease in plasma concentration of this proinflammatory cytokine.172
In humans, adiponectin serum levels were lower in patients with NASH in comparison to matched controls and to patients with SS, independently of IR or the waist–hip ratio. IR and low adiponectin serum levels were associated with increased steatosis and necroinflammation, but not with severe fibrosis, which was predicted only by IR.173 Another study has shown that morbidly obese patients with IR undergoing bariatric surgery have lower mRNA adiponectin expression in adipose tissue and lower serum levels of adiponectin than those without IR. This low expression of adiponectin may predispose patients to the progressive form of NAFLD or to NASH.174
Low mRNA expression of adiponectin and ADIPOR2 was found in the liver of patients with NASH compared with those with SS. Moreover, ADIPOR2 expression was inversely related to alanine aminotransferase and the fibrosis stage.175 More recently, Moschen et al demonstrated in a prospective study that rapid weight loss after bariatric surgery results in a significant improvement of both histological and biochemical liver parameters, which is accompanied by an increase of adiponectin serum levels, as well as hepatic mRNA adiponectin expression.176
Altogether, there is now strong evidence that circulating adiponectin levels are lower in obesity and related human disorders, including NAFLD.176,177
TNFα and IL6
TNFα and IL6 are two important proinflammatory adipocytokines and the expression of both is hugely increased in the fat cells of obese human subjects and patients with IR.178,179 In patients with severe obesity, the mRNA expression of IL6 and TNFα is clearer in adipose compared to liver tissue.180
TNFα was identified more than two decades ago as the first inflammatory molecule linked with IR.181 Higher serum levels of TNFα and soluble TNFα receptor 2 (TNFR2) have been found in patients with NASH compared with healthy subjects and these differences were independent of higher IR. However, no significant differences in TNFα and TNFR2 were found between SS and NASH patients.173
Enhanced TNFα hepatic expression was recently demonstrated in a group of obese patients with NAFLD. Crespo et al reported increased hepatic expression of TNFα and TNFR2 in patients with NASH compared to patients with SS. In these patients, more advanced fibrosis was also accompanied by the increased hepatic expression of TNFα.182 In line with these results, TNFα plasma levels have been shown to correlate positively with the grade of liver fibrosis assessed by ultrasound-guided liver biopsy in patients with advanced stages of NAFLD.183
Furthermore, certain TNFα polygenetic polymorphisms have been found to have higher IR indices, a higher prevalence of impaired glucose tolerance, and higher susceptibility to the development of NAFLD and NASH.184,185
IL6 is a pleiotropic cytokine expressed in many inflammatory cells in response to different types of stimuli, regulating a number of biological processes including IR and the regulation of inflammation. It is known to be the main stimulating factor for hepatocyte synthesis and the secretion of C-reactive protein in humans,186 and for this reason, it has been proposed as a potential mediator leading to NAFLD. However, the true mechanisms driving IL6 induced NAFLD remain unclear.
Preliminary studies have found that IL6 plays a protective role in liver fibrosis by promoting hepatocyte proliferation and by protecting against oxidative stress and mitochondrial dysfunction.187 On the other hand, Wieckowska et al demonstrated markedly increased IL6 expression in the liver of patients with NASH compared to those with SS or normal liver. Hepatic IL6 expression also correlated positively with the severity of inflammation and fibrosis. IL6 plasma levels that were measured in parallel in this study correlated well with liver IL6 expression.6 In another study, IL6 was evaluated among several serum markers in NAFLD patients, and IL6 circulating levels were significantly increased in patients with NAFLD as compared to healthy controls, but not in NASH compared to SS.188
Weight loss resulted in a dramatic decrease of IL6 subcutaneous and hepatic expression with a subsequent reduction in expression of the hepatic suppressor of cytokine signaling 3 (SOCS3) and improved insulin sensitivity. On the other hand, TNFα expression after weight loss only decreased in adipose tissue, not in hepatic tissue.180 This would suggest that the liver might be a key organ for adipose tissue-derived IL6 and TNFα because continuous TNFα/IL6 exposure affects hepatic IR.189
Visfatin
Visfatin, also termed pre-B cell colony enhancing factor (PBEF) or nicotinamide phosphoribosyltransferase (NAMPT) was first identified in 1994 as a protein secreted by activated lymphocytes, synergizing with IL7 and stem cell factor in early B cell formation.190 Although the first discovery of this molecule suggested primarily a cytokine function, its rediscovery as the key enzyme in generating nicotinamide adenine dinucleotide has considerably widened its biological perspective.191 Its extracellular functions (cytokine-like) are mainly proinflammatory as it potently induces various other proinflammatory cytokines such TNFα and IL6. Its intracellular functions concentrate on regulating the activity of NAD-consuming enzymes such as various sirtuins, thereby also affecting TNFα biosynthesis, cell lifespan, and longevity.
Only a few reports have so far addressed the role of this adipocytokine in human NAFLD. In patients with NAFLD, visfatin shows higher serum concentrations and weight loss is associated with both a decrease in serum levels and a reduction in liver mRNA expression, suggesting that the fatty liver might indeed contribute to an observable increase in serum visfatin levels.176 In the same study, immunohistochemistry staining for visfatin was carried out in 18 paired liver biopsies. The staining showed that visfatin was abundantly expressed in hepatocytes; weight loss decreased this expression dramatically. Another report has demonstrated the correlation of visfatin serum levels with liver histology in NAFLD, and such high circulating levels could predict the presence of portal inflammation in NAFLD patients.192
A protective role of visfatin against hepatocyte inflammatory damage was suggested by Jarrar et al.193 In that study, serum visfatin circulating levels in NAFLD patients were higher than in healthy control individuals, both lean and obese without NAFLD. Furthermore, when NASH occurred, visfatin concentration decreased significantly compared with SS, but was still significantly higher than in obese or lean healthy subjects without NAFLD.
Our findings are in line with data reporting that circulating levels and hepatic expression of visfatin are significantly higher in a group of morbidly obese women compared to lean controls and morbidly obese women with normal liver histology. Moreover, serum visfatin correlated well with IL6 and C-reactive protein.194
All these findings suggest that visfatin is a molecule with an important role in the pathophysiology of NAFLD and indicate that the liver could be a major source for this cytokine.
PPARδ
The PPARs family consists of three members: namely, PPARα, PPARβ/δ, and PPARγ. These receptors act as FA sensors that control many metabolic programs that are essential for systematic energy homeostasis. Today, due to its ubiquitous profile, much less is known about PPARδ than the other two in relation to human obesity and NAFLD.195
Oliver et al showed that IR obese rhesus monkeys normalized fasting glucose and insulin, increased high-density lipoprotein cholesterol and reduced low-density lipoprotein (LDL) cholesterol after treatment with a potent and specific PPARδ agonist, the GW501516.196 Other studies in an animal model of adenovirus-mediated hepatic PPARδ overexpression showed that PPARδ regulates lipogenesis and glucose utilization for glycogen synthesis. These effects could result in hepatic protection from FFA-mediated damage, possibly due to the generation of protective MUFA and the lowering of lipotoxic SFA levels.197
Overweight and obese men subjected to the PPARδ agonists, GW501516 or MBX-8025, exhibited improved insulin sensitivity and decreased fasting plasma TGs, non-esterified FAs, apolipoprotein B-100, and LDL-cholesterol, with diminished liver fat content quantified by magnetic resonance imaging.198,199
However, the final mechanisms underlying PPARδ effects in the liver of NAFLD patients still need further study.
NAFLD and lipotoxicity
The pathogenesis of NAFLD is often interpreted by the “double-hit” hypothesis. The primary insult or the “first hit” is lipid accumulation in the liver,8,200 followed by a “second hit” in which proinflammatory mediators induce inflammation, hepatocellular injury, and fibrosis.201 This paradigm suggested TG accumulation to be the “first hit” that predisposes to further liver damage in the pathogenesis of NASH, but has recently been replaced by a more complex model as emerging evidence points to FAs and their metabolites as the true lipotoxic agents.202 Interestingly, lipid accumulation and altered composition of phospholipids within ER membranes further promotes ER stress and IR in obese mice.203 Cytosolic TGs are therefore now considered to be inert, and in fact, lipid droplet accumulation seems to be hepatoprotective.204 However, TG accumulation and lipid droplet formation go hand in hand with pathophysiological mechanisms in NASH. FAs, as well as acyl-CoA and acetyl-CoA, have been identified as potential causes of lipotoxicity.205 FAs have been found to initiate the extrinsic apoptosis cascade and also to interfere with NR signaling, which might influence the extent of hepatocyte damage and further promote IR and ER stress.206 Accordingly, β-oxidation of LCFA within peroxisomes and ω-oxidation within the ER are upregulated in NASH and contribute to lipotoxicity and ROS formation.142 This might be secondary to inhibition of mitochondrial β-oxidation due to an accumulation of malonyl-CoA and the inhibition of CPT1. In fact, recent studies indicate that activation of mitochondrial FA oxidation protects from steatosis and IR.207
It is known that FAs induce the production of TNFα. Hepatic TNF receptor expression correlates with the severity of NAFLD disease.182 Also, TNF receptor activation increases expression of SREBP1c, which induces hepatic lipogenesis and lipid accumulation.208 TNFα activation is further paralleled by death-receptor expression, which facilitates activation of the extrinsic apoptosis cascade. Apoptosis is indeed the predominant form of hepatocellular injury in NASH. In fact, apoptotic activity within the unhealthy liver correlates with disease severity, and thus, cleaved cytokeratin-18 fragments in the serum of NAFLD could effectively be utilized as surrogate markers for the progression of NAFLD.209 As previously mentioned, FA accumulation also leads to the induction of ER stress and ROS formation, which again promotes hepatic injury.210
On the other hand, other studies indicate that metabolic oxidative stress, autophagy, and inflammation are hallmarks of NASH progression. In this sense, CYP2E1, the principal isoform of the CYP450 enzyme, seems to be critically important in NASH development by promoting oxidative/nitrosative stress, protein modifications, inflammation, and IR.211,212 Moreover, Das et al suggest that purinergic receptor X7 (P2X7), upregulated by CYP2E1, might have a key role in autophagy induced by metabolic oxidative stress in NASH.213
In summary, while hepatic TG accumulation seems to be a benign symptom of hepatic steatosis, FA metabolites contribute to the progression of NAFLD to NASH. IR promotes the recruitment of FFAs from the serum pool as well as intrahepatic FA accumulation, which induces apoptosis and ROS formation. FAs themselves also promote hepatic IR via TNF receptor activation, indicating a vicious cycle of lipid accumulation (Figure 1D). Other mechanisms could also contribute to liver damage. Regarding that, some authors have even suggested a “multiple parallel hits hypothesis” to explain the pathophysiology of NAFLD.41
Cannabinoid receptors (CB1, CB2) in NAFLD
The endocannabinoid (EC) system consists of cannabinoid receptors, endogenous cannabinoid ligands, and their biosynthetic and degradative enzymes, and has recently emerged as a ubiquitous system with key functions in a variety of physiological settings. Over the last decade, the EC system has emerged as a pivotal mediator of acute and chronic liver injury. ECs regulate appetite behavior and are lipid mediators that produce similar effects to those of marijuana by acting on membrane-bound receptors.214 Cannabinoid receptors are localized mainly in the brain, but are also present in minor amounts in the liver and some other peripheral tissues (CB1) and in immune and hematopoietic cells (CB2).215,216
ECs may also regulate peripheral energy metabolism, as demonstrated by their CB1-mediated effect on lipoprotein lipase activity in adipocytes217 and their ability to stimulate lipogenesis in hepatocytes.218,219 Cannabinoids exert their effects through two different cannabinoid receptors: CB1 and CB2. Under physiological conditions, the EC system is silent, since CB1 and CB2 receptors are faintly expressed. In contrast, induction of CB receptors and/or increased levels of ECs are common features of liver injuries of diverse origins.220 Both receptors have been implicated in the development of liver fibrosis secondary to various etiologies. CB1-mediated EC tone is enhanced in experimental diet-induced or genetic models of NAFLD, and is characterized by upregulation of adipose tissue and hepatocyte CB1 receptors, and by increased liver synthesis of anandamide. The pathogenic role of CB1 receptors in NAFLD is supported by the resistance to steatosis of obese mice bearing a global or hepatocyte-specific CB1 deletion, or of rodents administered rimonabant or AM6545, a CB1 antagonist.221–223 Studies with cultured hepatocytes and liver slices further indicate that the steatogenic properties of CB1 arise from altered hepatic lipid metabolism, consisting of a combination of hepatocyte activation of SREBP1c-mediated lipogenesis, reduction of FA oxidation via inhibition of AMP kinase, and decreased release of TG-rich VLDL.221,222,224 In addition, the adipose tissue may largely contribute to the steatogenic process via CB1-induced release of FFAs by adipocytes (Figure 5).225
Figure 5.

Mechanisms of CB1 involved in hepatic lipid accumulation.
Notes: The activation of CB1 receptors in adipose tissue promotes LPL activity, which results in increased FFA release into the liver. The activation of hepatic CB1 receptors contributes to liver fat accumulation by increased de novo hepatic lipogenesis, decreased FA oxidation, and decreased secretion of TG-rich VLDL.
Abbreviations: ACC1, acetyl-CoA carboxylase; CB, cannabinoid; CPT1, carnitine palmitoyltransferase-1; FA, fatty acid; FAS, fatty acid synthase; FFA, free fatty acid; LPL, lipoprotein lipase; SREBP1c, sterol regulatory element-binding protein 1c; TG, triglyceride; VLDL, very-low-density lipoprotein.
Also, a potential impact of CB1 receptors on the inflammatory response associated with NASH has been suggested by experiments in obese rats showing that rimonabant reduces liver inflammation.222,223 Although the underlying mechanism remains to be delineated, in hepatocytes, CB1 receptors could contribute to the acute phase response, via activation of cAMP responsive element-binding protein (CREBH), a liver-specific transcription factor that upregulates acute phase response genes.226 In addition, fat CB1 receptors reduce the production of adiponectin, an adipokine which reduces hepatic inflammation.222,223
With regard to liver fibrosis, it has been shown that marijuana use may correlate with the progression of fibrosis in patients with hepatitis C.227 However, each receptor seems to have opposing roles in the liver. The CB2 receptor has been shown to be upregulated in the livers of cirrhosis patients and to ameliorate the progression of fibrosis.228 In contrast, CB1 receptor activation has been linked to the progression of fibrosis, and CB1 antagonists have been shown to inhibit the progression of fibrosis.229 Indeed, clinical trials with a CB1 receptor antagonist have shown that antagonism of CB1 can result in weight loss and improved metabolic and cardiac parameters in overweight and obese populations.230 In summary, enhanced CB1 tone promotes liver fibrogenesis and cardiovascular alterations associated with cirrhosis, and contributes to the pathogenesis of NAFLD. On the other hand, upregulated CB2 signaling displays hepatoprotective effects, reducing liver inflammation, and improving liver fibrogenesis. Antagonism of CB1 and agonism of CB2 receptors have been identified as promising therapeutic strategies for the management of liver diseases.
Conclusion
NAFLD is characterized by IR, which leads to the deposition of fat, predominantly TGs, in the liver. The steatotic liver exhibits low-grade liver injury. However, a number of patients develop progressive liver injury with hepatocyte apoptosis, greater oxidative stress, and liver inflammation. The factors that lead to the progression of steatosis to steatohepatitis, are likely to be multiple and complex. We proposed a model of hepatocyte injury in fatty liver: in the susceptible steatotic hepatocyte, circulating FFAs can activate ER stress and apoptosis. While hepatic TG accumulation seems to be a benign symptom of hepatic steatosis, FA metabolites might contribute to the progression of NAFLD to NASH. IR promotes the recruitment of FFAs from the serum pool as well as intrahepatic FA accumulation through altered hepatic lipid metabolism, which finally induces apoptosis and ROS formation. Better understanding of the molecular pathways of liver injury should promote the development of diagnostic and therapeutic interventions aimed at reducing the morbidity and mortality associated with NAFLD.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev. 2007;65(6 Pt 2):57–63. doi: 10.1111/j.1753-4887.2007.tb00329.x. [DOI] [PubMed] [Google Scholar]
- 2.Lazo M, Clark JM. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin Liver Dis. 2008;28(4):339–350. doi: 10.1055/s-0028-1091978. [DOI] [PubMed] [Google Scholar]
- 3.Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 4.Nakamuta M, Kohjima M, Morizono S, et al. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2005;16(4):631–635. [PubMed] [Google Scholar]
- 5.Machado M, Marques-Vidal P, Cortez-Pinto H. Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol. 2006;45(4):600–606. doi: 10.1016/j.jhep.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. Am J Gastroenterol. 2008;103(6):1372–1379. doi: 10.1111/j.1572-0241.2007.01774.x. [DOI] [PubMed] [Google Scholar]
- 7.Ekstedt M, Franzén LE, Mathiesen UL, et al. Long-Term Follow-up of Patients with NAFLD and Elevated Liver Enzymes. Hepatology. 2006;44(4):865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 8.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl 1):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 9.Dowman JK, Tomlinson JW, Newsome PN. Systematic review: the diagnosis and staging of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2011;33(5):525–540. doi: 10.1111/j.1365-2036.2010.04556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37(4):917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 11.Adams LA, Waters OR, Knuiman MW, Elliott RR, Olynyk JK. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. Am J Gastroenterol. 2009;104(4):861–867. doi: 10.1038/ajg.2009.67. [DOI] [PubMed] [Google Scholar]
- 12.Adams LA, Feldstein AE. Non-invasive diagnosis of nonalcoholic fatty liver and nonalcoholic steatohepatitis. J Dig Dis. 2011;12(1):10–16. doi: 10.1111/j.1751-2980.2010.00471.x. [DOI] [PubMed] [Google Scholar]
- 13.Yan E, Durazo F, Tong M, Hong K. Nonalcoholic fatty liver disease: pathogenesis, identification, progression, and management. Nutr Rev. 2007;65(8 Pt 1):376–384. doi: 10.1301/nr.2007.aug.376-384. [DOI] [PubMed] [Google Scholar]
- 14.Sorbi D, Boynton J, Lindor KD. The ratio of aspartate aminotransferase to alanine aminotransferase: potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol. 1999;94(4):1018–1022. doi: 10.1111/j.1572-0241.1999.01006.x. [DOI] [PubMed] [Google Scholar]
- 15.Adams LA, Angulo P, Lindor KD. Nonalcoholic fatty liver disease. CMAJ. 2005;172(7):899–905. doi: 10.1503/cmaj.045232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Festi D, Schiumerini R, Marzi L, et al. Review article: the diagnosis of non-alcoholic fatty liver disease – availability and accuracy of non-invasive methods. Aliment Pharmacol Ther. 2013;37(4):392–400. doi: 10.1111/apt.12186. [DOI] [PubMed] [Google Scholar]
- 17.Federico A, Trappoliere M, Loguercio C. Treatment of patients with non-alcoholic fatty liver disease: current views and perspectives. Dig Liver Dis. 2006;38(11):789–801. doi: 10.1016/j.dld.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Pacifico L, Celestre M, Anania C, Paolantonio P, Chiesa C, Laghi A. MRI and ultrasound for hepatic fat quantification: relationships to clinical and metabolic characteristics of pediatric nonalcoholic fatty liver disease. Acta Paediatr. 2007;96(4):542–547. doi: 10.1111/j.1651-2227.2007.00186.x. [DOI] [PubMed] [Google Scholar]
- 19.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Dig Dis Sci. 2010;55(3):560–578. doi: 10.1007/s10620-009-1081-0. [DOI] [PubMed] [Google Scholar]
- 20.Yilmaz Y, Ulukaya E, Dolar E. A “biomarker biopsy” for the diagnosis of NASH: promises from CK-18 fragments. Obes Surg. 2008;18(11):1507–1508. doi: 10.1007/s11695-008-9639-z. author reply 1509–1510. [DOI] [PubMed] [Google Scholar]
- 21.Schwenger KJ, Allard JP. Clinical approaches to non-alcoholic fatty liver disease. World J Gastroenterol. 2014;20(7):1712–1723. doi: 10.3748/wjg.v20.i7.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCarthy EM, Rinella ME. The role of diet and nutrient composition in nonalcoholic fatty liver disease. J Acad Nutr Diet. 2012;112(3):401–409. doi: 10.1016/j.jada.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51(1):121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.St George A, Bauman A, Johnston A, Farrell G, Chey T, George J. Effect of a lifestyle intervention in patients with abnormal liver enzymes and metabolic risk factors. J Gastroenterol Hepatol. 2009;24(3):399–407. doi: 10.1111/j.1440-1746.2008.05694.x. [DOI] [PubMed] [Google Scholar]
- 25.Haukeland JW, Konopski Z, Eggesbø HB, et al. Metformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trial. Scand J Gastroenterol. 2009;44(7):853–860. doi: 10.1080/00365520902845268. [DOI] [PubMed] [Google Scholar]
- 26.Oh MK, Winn J, Poordad F. Review article: diagnosis and treatment of non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;28(5):503–522. doi: 10.1111/j.1365-2036.2008.03752.x. [DOI] [PubMed] [Google Scholar]
- 27.Van Wagner LB, Rinella ME. The role of insulin-sensitizing agents in the treatment of nonalcoholic steatohepatitis. Therap Adv Gastroenterol. 2011;4(4):249–263. doi: 10.1177/1756283X11403809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutchman G, Modi A, Kleiner DE, et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology. 2007;46(2):424–429. doi: 10.1002/hep.21661. [DOI] [PubMed] [Google Scholar]
- 29.Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135(4):1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 30.Maroni L, Guasti L, Castiglioni L, et al. Lipid targets during statin treatment in dyslipidemic patients affected by nonalcoholic fatty liver disease. Am J Med Sci. 2011;342(5):383–387. doi: 10.1097/MAJ.0b013e318213e526. [DOI] [PubMed] [Google Scholar]
- 31.Ekstedt M, Franzén LE, Mathiesen UL, Holmqvist M, Bodemar G, Kechagias S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: a histopathological follow-up study. J Hepatol. 2007;47(1):135–141. doi: 10.1016/j.jhep.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Tandra S, Vuppalanchi R. Use of statins in patients with liver disease. Curr Treat Options Cardiovasc Med. 2009;11(4):272–278. doi: 10.1007/s11936-009-0028-2. [DOI] [PubMed] [Google Scholar]
- 33.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55(6):2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 34.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog Lipid Res. 2009;48(1):1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Petta S, Muratore C, Craxì A. Non-alcoholic fatty liver disease pathogenesis: the present and the future. Dig Liver Dis. 2009;41(9):615–625. doi: 10.1016/j.dld.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Utzschneider KM, Kahn SE. Review: the role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91(12):4753–4761. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 37.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 38.Savage DB, Semple RK. Recent insights into fatty liver, metabolic dyslipidaemia and their links to insulin resistance. Curr Opin Lipidol. 2010;21(4):329–336. doi: 10.1097/MOL.0b013e32833b7782. [DOI] [PubMed] [Google Scholar]
- 39.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118(3):829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23(2):201–229. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 41.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 42.Koliwad SK, Streeper RS, Monetti M, et al. DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J Clin Invest. 2010;120(3):756–767. doi: 10.1172/JCI36066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45(6):1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 44.Amaro A, Fabbrini E, Kars M, et al. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology. 2010;139(1):149–153. doi: 10.1053/j.gastro.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40(1):185–194. doi: 10.1002/hep.20283. [DOI] [PubMed] [Google Scholar]
- 46.Marí M, Caballero F, Colell A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4(3):185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 47.Cheung O, Sanyal AJ. Abnormalities of lipid metabolism in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28(4):351–359. doi: 10.1055/s-0028-1091979. [DOI] [PubMed] [Google Scholar]
- 48.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28(4):360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arner P. Human fat cell lipolysis: biochemistry, regulation and clinical role. Best Pract Res Clin Endocrinol Metab. 2005;19(4):471–482. doi: 10.1016/j.beem.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. 2007;10(2):142–148. doi: 10.1097/MCO.0b013e328042ba90. [DOI] [PubMed] [Google Scholar]
- 51.Berk PD. Regulatable fatty acid transport mechanisms are central to the pathophysiology of obesity, fatty liver, and metabolic syndrome. Hepatology. 2008;48(5):1362–1376. doi: 10.1002/hep.22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin G, Nemoto M, Gelman L, et al. The human fatty acid transport protein-1 (SLC27A1; FATP-1) cDNA and gene: organization, chromosomal localization, and expression. Genomics. 2000;66(3):296–304. doi: 10.1006/geno.2000.6191. [DOI] [PubMed] [Google Scholar]
- 53.Ge F, Zhou S, Hu C, Lobdell H, Berk PD. Insulin- and leptin-regulated fatty acid uptake plays a key causal role in hepatic steatosis in mice with intact leptin signaling but not in ob/ob or db/db mice. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G855–G866. doi: 10.1152/ajpgi.00434.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou SL, Stump D, Sorrentino D, Potter BJ, Berk PD. Adipocyte differentiation of 3T3-L1 cells involves augmented expression of a 43-kDa plasma membrane fatty acid-binding protein. J Biol Chem. 1992;267(20):14456–14461. [PubMed] [Google Scholar]
- 55.Zhou SL, Stump D, Kiang CL, Isola LM, Berk PD. Mitochondrial aspartate aminotransferase expressed on the surface of 3T3-L1 adipocytes mediates saturable fatty acid uptake. Proc Soc Exp Biol Med. 1995;208(3):263–270. doi: 10.3181/00379727-208-43854. [DOI] [PubMed] [Google Scholar]
- 56.Trigatti BL, Anderson RG, Gerber GE. Identification of caveolin-1 as a fatty acid binding protein. Biochem Biophys Res Commun. 1999;255(1):34–39. doi: 10.1006/bbrc.1998.0123. [DOI] [PubMed] [Google Scholar]
- 57.Doege H, Stahl A. Protein-mediated fatty acid uptake: novel insights from in vivo models. Physiology (Bethesda) 2006;21:259–268. doi: 10.1152/physiol.00014.2006. [DOI] [PubMed] [Google Scholar]
- 58.Doege H, Baillie RA, Ortegon AM, et al. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology. 2006;130(4):1245–1258. doi: 10.1053/j.gastro.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Auinger A, Valenti L, Pfeuffer M, et al. A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis. Horm Metab Res. 2010;42(12):854–849. doi: 10.1055/s-0030-1267186. [DOI] [PubMed] [Google Scholar]
- 60.Fernández MA, Albor C, Ingelmo-Torres M, et al. Caveolin-1 is essential for liver regeneration. Science. 2006;313(5793):1628–1632. doi: 10.1126/science.1130773. [DOI] [PubMed] [Google Scholar]
- 61.Mastrodonato M, Calamita G, Rossi R, et al. Altered distribution of caveolin-1 in early liver steatosis. Eur J Clin Invest. 2011;41(6):642–651. doi: 10.1111/j.1365-2362.2010.02459.x. [DOI] [PubMed] [Google Scholar]
- 62.Miquilena-Colina ME, Lima-Cabello E, Sánchez-Campos S, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60(10):1394–1402. doi: 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- 63.Inoue M, Ohtake T, Motomura W, et al. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336(1):215–222. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- 64.Buqué X, Martínez MJ, Cano A, et al. A subset of dysregulated metabolic and survival genes is associated with severity of hepatic steatosis in obese Zucker rats. J Lipid Res. 2010;51(3):500–513. doi: 10.1194/jlr.M001966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Degrace P, Moindrot B, Mohamed I, et al. Upregulation of liver VLDL receptor and FAT/CD36 expression in LDLR−/− apoB100/100 mice fed trans-10, cis-12 conjugated linoleic acid. J Lipid Res. 2006;47(12):2647–2655. doi: 10.1194/jlr.M600140-JLR200. [DOI] [PubMed] [Google Scholar]
- 66.Zhou J, Febbraio M, Wada T, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134(2):556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 67.Greco D, Kotronen A, Westerbacka J, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol. 2008;294(5):G1281–G1287. doi: 10.1152/ajpgi.00074.2008. [DOI] [PubMed] [Google Scholar]
- 68.Bechmann LP, Gieseler RK, Sowa J-P, et al. Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int. 2010;30(6):850–859. doi: 10.1111/j.1478-3231.2010.02248.x. [DOI] [PubMed] [Google Scholar]
- 69.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7(6):489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karakas SE, Almario RU, Kim K. Serum fatty acid binding protein 4, free fatty acids, and metabolic risk markers. Metabolism. 2009;58(7):1002–1007. doi: 10.1016/j.metabol.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zimmerman AW, Veerkamp JH. New insights into the structure and function of fatty acid-binding proteins. Cell Mol Life Sci. 2002;59(7):1096–1116. doi: 10.1007/s00018-002-8490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chmurzyńska A. The multigene family of fatty acid-binding proteins (FABPs): function, structure and polymorphism. J Appl Genet. 2006;47(1):39–48. doi: 10.1007/BF03194597. [DOI] [PubMed] [Google Scholar]
- 73.Krusinová E, Pelikánová T. Fatty acid binding proteins in adipose tissue: a promising link between metabolic syndrome and atherosclerosis? Diabetes Res Clin Pract. 2008;82(Suppl 2):S127–S134. doi: 10.1016/j.diabres.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 74.Taskinen MR, Adiels M, Westerbacka J, et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. 2011;31(9):2144–2150. doi: 10.1161/ATVBAHA.111.224808. [DOI] [PubMed] [Google Scholar]
- 75.Westerbacka J, Kolak M, Kiviluoto T, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007;56(11):2759–2765. doi: 10.2337/db07-0156. [DOI] [PubMed] [Google Scholar]
- 76.Falcon A, Doege H, Fluitt A, et al. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab. 2010;299(3):E384–E393. doi: 10.1152/ajpendo.00226.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doege H, Grimm D, Falcon A, et al. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem. 2008;283(32):22186–22192. doi: 10.1074/jbc.M803510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nie B, Park HM, Kazantzis M, et al. Specific bile acids inhibit hepatic fatty acid uptake in mice. Hepatology. 2012;56(4):1300–1310. doi: 10.1002/hep.25797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okamura M, Inagaki T, Tanaka T, Sakai J. Role of histone methylation and demethylation in adipogenesis and obesity. Organogenesis. 2010;6(1):24–32. doi: 10.4161/org.6.1.11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51(2):679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56(4):952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 83.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fuchs M. Non-alcoholic fatty liver disease: the bile acid-activated farnesoid x receptor as an emerging treatment target. J Lipids. 2012;2012:934396. doi: 10.1155/2012/934396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferré P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes Metab. 2010;12(Suppl 2):83–92. doi: 10.1111/j.1463-1326.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 86.Mitsuyoshi H, Yasui K, Harano Y, et al. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. 2009;39(4):366–373. doi: 10.1111/j.1872-034X.2008.00464.x. [DOI] [PubMed] [Google Scholar]
- 87.Kohjima M, Higuchi N, Kato M, et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. 2008;21(4):507–511. [PubMed] [Google Scholar]
- 88.Baranowski M. Biological role of liver X receptors. J Physiol Pharmacol. 2008;59(Suppl 7):31–55. [PubMed] [Google Scholar]
- 89.Faulds MH, Zhao C, Dahlman-Wright K. Molecular biology and functional genomics of liver X receptors (LXR) in relationship to metabolic diseases. Curr Opin Pharmacol. 2010;10(6):692–697. doi: 10.1016/j.coph.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 90.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277(11):9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 91.Peet DJ, Turley SD, Ma W, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93(5):693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 92.Repa JJ, Liang G, Ou J, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14(22):2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schultz JR, Tu H, Luk A, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14(22):2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Higuchi N, Kato M, Shundo Y, et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol Res. 2008;38(11):1122–1129. doi: 10.1111/j.1872-034X.2008.00382.x. [DOI] [PubMed] [Google Scholar]
- 95.Lima-Cabello E, Garcia-Mediavilla MV, Miquilena-Colina ME, et al. Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clin Sci (Lond) 2011;120(6):239–250. doi: 10.1042/CS20100387. [DOI] [PubMed] [Google Scholar]
- 96.Horton JD, Goldstein JL, Brown MS. Critical review SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99(5):838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96(24):13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Foretz M, Pacot C, Dugail I, et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19(5):3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Azzout-Marniche D, Bécard D, Guichard C, et al. Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem J. 2000;350(Pt 2):389–393. [PMC free article] [PubMed] [Google Scholar]
- 101.Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem. 2000;275(42):32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 102.Chakravarty K, Leahy P, Becard D, et al. Sterol regulatory element-binding protein-1c mimics the negative effect of insulin on phosphoenolpyruvate carboxykinase (GTP) gene transcription. J Biol Chem. 2001;276(37):34816–34823. doi: 10.1074/jbc.M103310200. [DOI] [PubMed] [Google Scholar]
- 103.Shimano H. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog Lipid Res. 2001;40(6):439–452. doi: 10.1016/s0163-7827(01)00010-8. [DOI] [PubMed] [Google Scholar]
- 104.Horton JD, Bashmakov Y, Shimomura I, Shimano H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci U S A. 1998;95(11):5987–5992. doi: 10.1073/pnas.95.11.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim JB, Sarraf P, Wright M, et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest. 1998;101(1):1–9. doi: 10.1172/JCI1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagaya T, Tanaka N, Suzuki T, et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J Hepatol. 2010;53(4):724–731. doi: 10.1016/j.jhep.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 107.Denechaud PD, Dentin R, Girard J, Postic C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Lett. 2008;582(1):68–73. doi: 10.1016/j.febslet.2007.07.084. [DOI] [PubMed] [Google Scholar]
- 108.Foufelle F, Ferré P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J. 2002;366(Pt 2):377–391. doi: 10.1042/BJ20020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dentin R, Pégorier JP, Benhamed F, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279(19):20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 110.Ishii S, Iizuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A. 2004;101(44):15597–15602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J Biol Chem. 2005;280(12):12019–12027. doi: 10.1074/jbc.M413063200. [DOI] [PubMed] [Google Scholar]
- 112.Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem. 2007;282(1):743–751. doi: 10.1074/jbc.M605023200. [DOI] [PubMed] [Google Scholar]
- 113.Ulven SM, Dalen KT, Gustafsson JA, Nebb HI. LXR is crucial in lipid metabolism. Prostaglandins Leukot Essent Fatty Acids. 2005;73(1):59–63. doi: 10.1016/j.plefa.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 114.Chen G, Liang G, Ou J, Goldstein JL, Brown MS. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc Natl Acad Sci U S A. 2004;101(31):11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Joseph SB, Laffitte B, Patel PH, et al. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem. 2002;277(13):11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 116.Zhang Y, Yin L, Hillgartner FB. SREBP-1 integrates the actions of thyroid hormone, insulin, cAMP, and medium-chain fatty acids on ACCalpha transcription in hepatocytes. J Lipid Res. 2003;44(2):356–368. doi: 10.1194/jlr.M200283-JLR200. [DOI] [PubMed] [Google Scholar]
- 117.Chu K, Miyazaki M, Man WC, Ntambi JM. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol Cell Biol. 2006;26(18):6786–6798. doi: 10.1128/MCB.00077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mitro N, Mak PA, Vargas L, et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445(7124):219–223. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 119.Arden C, Petrie JL, Tudhope SJ, et al. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes. 2011;60(12):3110–3120. doi: 10.2337/db11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ma L, Robinson LN, Towle HC. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem. 2006;281(39):28721–28730. doi: 10.1074/jbc.M601576200. [DOI] [PubMed] [Google Scholar]
- 121.de la Iglesia N, Mukhtar M, Seoane J, Guinovart JJ, Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem. 2000;275(14):10597–10603. doi: 10.1074/jbc.275.14.10597. [DOI] [PubMed] [Google Scholar]
- 122.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101(19):7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Iizuka K, Miller B, Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am J Physiol Endocrinol Metab. 2006;291(2):E358–E364. doi: 10.1152/ajpendo.00027.2006. [DOI] [PubMed] [Google Scholar]
- 124.Dentin R, Benhamed F, Hainault I, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55(8):2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 125.Iizuka K, Takeda J, Horikawa Y. Hepatic overexpression of dominant negative Mlx improves metabolic profile in diabetes-prone C57BL/6J mice. Biochem Biophys Res Commun. 2009;379(2):499–504. doi: 10.1016/j.bbrc.2008.12.100. [DOI] [PubMed] [Google Scholar]
- 126.Benhamed F, Denechaud P, Lemoine M, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122(6):2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7(2):95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 128.Forman BM, Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81(5):687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 129.Lu TT, Makishima M, Repa JJ, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6(3):507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem. 2003;278(1):104–110. doi: 10.1074/jbc.M209505200. [DOI] [PubMed] [Google Scholar]
- 131.Downes M, Verdecia MA, Roecker AJ, et al. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell. 2003;11(4):1079–1092. doi: 10.1016/s1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7(8):678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 133.Kok T, Hulzebos CV, Wolters H, et al. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J Biol Chem. 2003;278(43):41930–41937. doi: 10.1074/jbc.M306309200. [DOI] [PubMed] [Google Scholar]
- 134.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 135.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116(4):1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103(4):1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang ZX, Shen W, Sun H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol Int. 2010;4(4):741–748. doi: 10.1007/s12072-010-9202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Teodoro JS, Rolo AP, Palmeira CM. Hepatic FXR: key regulator of whole-body energy metabolism. Trends Endocrinol Metab. 2011;22(11):458–466. doi: 10.1016/j.tem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 140.Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal. 2010;8:e005. doi: 10.1621/nrs.08005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dorn C, Riener MO, Kirovski G, et al. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int J Clin Exp Pathol. 2010;3(5):505–514. [PMC free article] [PubMed] [Google Scholar]
- 142.Kohjima M, Enjoji M, Higuchi N, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20(3):351–358. [PubMed] [Google Scholar]
- 143.Morgan K, Uyuni A, Nandgiri G, et al. Altered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2008;20(9):843–854. doi: 10.1097/MEG.0b013e3282f9b203. [DOI] [PubMed] [Google Scholar]
- 144.Zhang W, Patil S, Chauhan B, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281(15):10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 145.Qu S, Altomonte J, Perdomo G, et al. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147(12):5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116(9):2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Valenti L, Rametta R, Dongiovanni P, et al. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes. 2008;57(5):1355–1362. doi: 10.2337/db07-0714. [DOI] [PubMed] [Google Scholar]
- 148.Samuel VT, Choi CS, Phillips TG, et al. Targeting foxo1 in mice using antisense oligonucleotide improves hepatic and peripheral insulin action. Diabetes. 2006;55(7):2042–2050. doi: 10.2337/db05-0705. [DOI] [PubMed] [Google Scholar]
- 149.Nakae J, Biggs WH, Kitamura T, et al. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32(2):245–253. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- 150.Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61(4):393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103(11):1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Harano Y, Yasui K, Toyama T, et al. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 2006;26(5):613–620. doi: 10.1111/j.1478-3231.2006.01265.x. [DOI] [PubMed] [Google Scholar]
- 153.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38(1):123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 154.Chou CJ, Haluzik M, Gregory C, et al. WY14,643, a peroxisome proliferator-activated receptor alpha (PPARalpha) agonist, improves hepatic and muscle steatosis and reverses insulin resistance in lipoatrophic A-ZIP/F-1 mice. J Biol Chem. 2002;277(27):24484–24489. doi: 10.1074/jbc.M202449200. [DOI] [PubMed] [Google Scholar]
- 155.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem. 2000;275(37):28918–28928. doi: 10.1074/jbc.M910350199. [DOI] [PubMed] [Google Scholar]
- 156.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 157.Kashireddy PV, Rao MS. Lack of peroxisome proliferator-activated receptor alpha in mice enhances methionine and choline deficient diet-induced steatohepatitis. Hepatol Res. 2004;30(2):104–110. doi: 10.1016/j.hepres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 158.Rao MS, Papreddy K, Musunuri S, Okonkwo A. Prevention/reversal of choline deficiency-induced steatohepatitis by a peroxisome proliferator-activated receptor alpha ligand in rats. In Vivo. 2002;16(2):145–152. [PubMed] [Google Scholar]
- 159.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39(5):1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 160.Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Triglyceride Metab Dis. 2012;1821(5):809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 161.Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. A paradigm for gene regulation: inflammation, NF-kappaB and PPAR. Adv Exp Med Biol. 2003;544:181–196. doi: 10.1007/978-1-4419-9072-3_22. [DOI] [PubMed] [Google Scholar]
- 162.Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappa B and AP-1. J Biol Chem. 1999;274(45):32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- 163.Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem. 2000;275(47):36703–36707. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 164.Gervois P, Vu-Dac N, Kleemann R, et al. Negative regulation of human fibrinogen gene expression by peroxisome proliferator-activated receptor alpha agonists via inhibition of CCAAT box/enhancer-binding protein beta. J Biol Chem. 2001;276(36):33471–33477. doi: 10.1074/jbc.M102839200. [DOI] [PubMed] [Google Scholar]
- 165.Gervois P, Kleemann R, Pilon A, et al. Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J Biol Chem. 2004;279(16):16154–16160. doi: 10.1074/jbc.M400346200. [DOI] [PubMed] [Google Scholar]
- 166.Huang YY, Gusdon AM, Qu S. Nonalcoholic fatty liver disease: molecular pathways and therapeutic strategies. Lipids Health Dis. 2013;12:171. doi: 10.1186/1476-511X-12-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008;14(3–4):222–231. doi: 10.2119/2007-00119.Tilg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tilg H. Adipocytokines in nonalcoholic fatty liver disease: key players regulating steatosis, inflammation and fibrosis. Curr Pharm Des. 2010;16(17):1893–1895. doi: 10.2174/138161210791208929. [DOI] [PubMed] [Google Scholar]
- 169.Maeda N, Takahashi M, Funahashi T, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50(9):2094–2099. doi: 10.2337/diabetes.50.9.2094. [DOI] [PubMed] [Google Scholar]
- 170.Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun. 2004;323(2):630–635. doi: 10.1016/j.bbrc.2004.08.145. [DOI] [PubMed] [Google Scholar]
- 171.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 172.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112(1):91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40(1):46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 174.Baranova A, Gowder SJ, Schlauch K, et al. Gene expression of leptin, resistin, and adiponectin in the white adipose tissue of obese patients with non-alcoholic fatty liver disease and insulin resistance. Obes Surg. 2006;16(9):1118–1125. doi: 10.1381/096089206778392149. [DOI] [PubMed] [Google Scholar]
- 175.Kaser S, Moschen A, Cayon A, et al. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut. 2005;54(1):117–121. doi: 10.1136/gut.2003.037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Moschen AR, Molnar C, Wolf AM, et al. Effects of weight loss induced by bariatric surgery on hepatic adipocytokine expression. J Hepatol. 2009;51(4):765–777. doi: 10.1016/j.jhep.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 177.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50(3):957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 178.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 179.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest. 1995;95(5):2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Moschen AR, Molnar C, Geiger S, et al. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut. 2010;59(9):1259–1264. doi: 10.1136/gut.2010.214577. [DOI] [PubMed] [Google Scholar]
- 181.Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology. 1992;130(1):43–52. doi: 10.1210/endo.130.1.1727716. [DOI] [PubMed] [Google Scholar]
- 182.Crespo J, Cayón A, Fernández-Gil P, et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34(6):1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 183.Lesmana CRA, Hasan I, Budihusodo U, et al. Diagnostic value of a group of biochemical markers of liver fibrosis in patients with non-alcoholic steatohepatitis. J Dig Dis. 2009;10(3):201–206. doi: 10.1111/j.1751-2980.2009.00386.x. [DOI] [PubMed] [Google Scholar]
- 184.Valenti L, Fracanzani AL, Dongiovanni P, et al. Tumor necrosis factor alpha promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology. 2002;122(2):274–280. doi: 10.1053/gast.2002.31065. [DOI] [PubMed] [Google Scholar]
- 185.Zhou YJ, Li YY, Nie YQ, et al. Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. J Gastroenterol Hepatol. 2010;25(4):772–777. doi: 10.1111/j.1440-1746.2009.06144.x. [DOI] [PubMed] [Google Scholar]
- 186.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340(6):448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 187.Cressman DE, Greenbaum LE, DeAngelis RA, et al. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274(5291):1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 188.Haukeland JW, Damås JK, Konopski Z, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44(6):1167–1174. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 189.Senn JJ, Klover PJ, Nowak IA, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. 2003;278(16):13740–13746. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- 190.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14(2):1431–1437. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Moschen AR, Gerner RR, Tilg H. Pre-B Cell colony enhancing factor/NAMPT/visfatin in inflammation and obesity-related disorders. Curr Pharm Des. 2010;16(17):1913–1920. doi: 10.2174/138161210791208947. [DOI] [PubMed] [Google Scholar]
- 192.Aller R, de Luis DA, Izaola O, et al. Influence of visfatin on histopathological changes of non-alcoholic fatty liver disease. Dig Dis Sci. 2009;54(8):1772–1777. doi: 10.1007/s10620-008-0539-9. [DOI] [PubMed] [Google Scholar]
- 193.Jarrar MH, Baranova A, Collantes R, et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;27(5):412–421. doi: 10.1111/j.1365-2036.2007.03586.x. [DOI] [PubMed] [Google Scholar]
- 194.Auguet T, Terra X, Porras JA, et al. Plasma visfatin levels and gene expression in morbidly obese women with associated fatty liver disease. Clin Biochem. 2013;46(3):202–208. doi: 10.1016/j.clinbiochem.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 195.Videla LA, Pettinelli P. Misregulation of PPAR functioning and its pathogenic consequences associated with nonalcoholic fatty liver disease in human obesity. PPAR Res. 2012;2012:107434. doi: 10.1155/2012/107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Oliver WR, Jr, Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98(9):5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Liu S, Hatano B, Zhao M, et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. J Biol Chem. 2011;286(2):1237–1247. doi: 10.1074/jbc.M110.138115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Risérus U, Sprecher D, Johnson T, et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta Promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57(2):332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- 199.Ooi EMM, Watts GF, Sprecher DL, Chan DC, Barrett PHR. Mechanism of action of a peroxisome proliferator-activated receptor (PPAR)-delta agonist on lipoprotein metabolism in dyslipidemic subjects with central obesity. J Clin Endocrinol Metab. 2011;96(10):E1568–E1576. doi: 10.1210/jc.2011-1131. [DOI] [PubMed] [Google Scholar]
- 200.Haider DG, Schindler K, Schaller G, Prager G, Wolzt M, Ludvik B. Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J Clin Endocrinol Metab. 2006;91(4):1578–1581. doi: 10.1210/jc.2005-2248. [DOI] [PubMed] [Google Scholar]
- 201.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36(1):4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 202.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52(2):774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 203.Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ricchi M, Odoardi MR, Carulli L, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol. 2009;24(5):830–840. doi: 10.1111/j.1440-1746.2008.05733.x. [DOI] [PubMed] [Google Scholar]
- 205.Han MS, Park SY, Shinzawa K, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res. 2008;49(1):84–97. doi: 10.1194/jlr.M700184-JLR200. [DOI] [PubMed] [Google Scholar]
- 206.Nolan CJ, Larter CZ. Lipotoxicity: why do saturated fatty acids cause and monounsaturates protect against it? J Gastroenterol Hepatol. 2009;24(5):703–706. doi: 10.1111/j.1440-1746.2009.05823.x. [DOI] [PubMed] [Google Scholar]
- 207.Orellana-Gavalda JM, Herrero L, Malandrino MI, et al. Molecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidation. Hepatology. 2011;53(3):821–832. doi: 10.1002/hep.24140. [DOI] [PubMed] [Google Scholar]
- 208.Endo M, Masaki T, Seike M, Yoshimatsu H. TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c) Exp Biol Med (Maywood) 2007;232(5):614–621. [PubMed] [Google Scholar]
- 209.Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50(4):1072–1078. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Feldstein AE. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin Liver Dis. 2010;30(4):391–401. doi: 10.1055/s-0030-1267539. [DOI] [PubMed] [Google Scholar]
- 211.Abdelmegeed MA, Banerjee A, Yoo SH, et al. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol. 2012;57(4):860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Seth RK, Das S, Kumar A, et al. CYP2E1-dependent and leptin-mediated hepatic CD57 expression on CD8+ T cells aid progression of environment-linked nonalcoholic steatohepatitis. Toxicol Appl Pharmacol. 2014;274(1):42–54. doi: 10.1016/j.taap.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Das S, Seth RK, Kumar A, et al. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2013;305(12):G950–G963. doi: 10.1152/ajpgi.00235.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Di Marzo V, Piscitelli F, Mechoulam R. Cannabinoids and endocannabinoids in metabolic disorders with focus on diabetes. Handb Exp Pharmacol. 2011;(203):75–104. doi: 10.1007/978-3-642-17214-4_4. [DOI] [PubMed] [Google Scholar]
- 215.Pacher P, Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50(2):193–211. doi: 10.1016/j.plipres.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58(3):389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cota D, Marsicano G, Tschap M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112(3):423–431. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Osei-Hyiaman D, DePetrillo M, Pacher P, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115(5):1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jeong W, Osei-Hyiaman D, Park O, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7(3):227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 220.Mallat A, Teixeira-Clerc F, Deveaux V, Manin S, Lotersztajn S. The endocannabinoid system as a key mediator during liver diseases: new insights and therapeutic openings. Br J Pharmacol. 2011;163(7):1432–1440. doi: 10.1111/j.1476-5381.2011.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Osei-Hyiaman D, Liu J, Zhou L, et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118(9):3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tam J, Vemuri VK, Liu J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120(8):2953–2966. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Gary-Bobo M, Elachouri G, Gallas JF, et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46(1):122–129. doi: 10.1002/hep.21641. [DOI] [PubMed] [Google Scholar]
- 224.Jourdan T, Demizieux L, Gresti J, et al. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: evidence from cultured explants. Hepatology. 2012;55(3):790–799. doi: 10.1002/hep.24733. [DOI] [PubMed] [Google Scholar]
- 225.Jourdan T, Djaouti L, Demizieux L, Gresti J, Vergès B, Degrace P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes. 2010;59(4):926–934. doi: 10.2337/db09-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chanda D, Kim DK, Li T, et al. Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J Biol Chem. 2011;286(32):27971–27979. doi: 10.1074/jbc.M111.224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Hézode C, Roudot-Thoraval F, Nguyen S, et al. Daily cannabis smoking as a risk factor for progression of fibrosis in chronic hepatitis C. Hepatology. 2005;42(1):63–71. doi: 10.1002/hep.20733. [DOI] [PubMed] [Google Scholar]
- 228.Muñoz-Luque J, Ros J, Fernández-Varo G, et al. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J Pharmacol Exp Ther. 2008;324(2):475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Teixeira-Clerc F, Julien B, Grenard P, et al. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12(6):671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 230.Van Gaal L, Pi-Sunyer X, Després JP, McCarthy C, Scheen A. Efficacy and safety of rimonabant for improvement of multiple cardiometabolic risk factors in overweight/obese patients: pooled 1-year data from the Rimonabant in Obesity (RIO) program. Diabetes Care. 2008;31(Suppl 2):S229–S240. doi: 10.2337/dc08-s258. [DOI] [PubMed] [Google Scholar]