

Abstract
Wernicke's encephalopathy (WE) is a severe neurological syndrome caused by thiamine (vitamin B1) deficiency and clinically characterized by the sudden onset of mental status changes, ocular abnormalities, and ataxia. Apart from chronic alcoholism, the most common cause of WE, a lot of other conditions causing malnutrition and decreasing thiamine absorption such as gastrointestinal surgical procedures and hyperemesis gravidarum must be considered as predisposing factors. Due to its low prevalence and clinical heterogeneity, WE is often misdiagnosed, leading to persistent dysfunctions and, in some cases, to death. Nowadays, MR imaging of the brain, showing T2 and FLAIR hyperintensities in typical (thalami, mammillary bodies, tectal plate, and periaqueductal area) and atypical areas (cerebellum, cranial nerve nuclei, and cerebral cortex), is surely the most important and effective tool in the diagnostic assessment of WE. The aim of this paper is to propose a state of the art of the role of MR imaging in the early diagnosis of this complex disease.
1. Introduction
Wernicke's encephalopathy (WE) is an uncommon but severe neurological syndrome, caused by thiamine (vitamin B1) deficiency. It is characterized by the sudden onset of altered consciousness, ophthalmoplegia, and ataxia [1]. This classic clinical triad, however, is present in only a minority of patients, making this condition often misdiagnosed and, consequently, life-threatening; its prognosis depends on prompt early intravenous administration of thiamine [2]. Magnetic resonance (MR) imaging, showing typical (thalami, mammillary bodies, tectal plate, and periaqueductal area) and atypical (cerebellum, cranial nerve nuclei, and cerebral cortex) signal-intensity alterations, is an essential tool to get the right diagnosis, especially when clinical presentation is incomplete [3]. The most common cause of thiamine deficiency is chronic alcohol abuse but a lot of other conditions such as gastrointestinal surgical procedures, hyperemesis gravidarum, and chemical therapy have been reported as predisposing factors [4].
We propose a review of the relevant literature focusing our attention on the role of MR imaging in the early diagnosis that, in recent years, has been fully confirmed.
2. Epidemiology
The prevalence of Wernicke's encephalopathy in the general population has been estimated from autopsy studies and varies from 0.4 to 2.8 [5, 6]; it seems to be much higher in alcoholics (AL) than in nonalcoholics (NA). These studies have revealed that the diagnosis of WE is often made only postmortem and less than 20% of the patients obtain a right diagnosis during life [6].
There is no racial predilection and the male sex is more affected than the female one (M : F ratio = 1.7 : 1) [7].
3. Aetiopathogenesis
Thiamine is a water-soluble vitamin involved in the maintenance of membrane integrity and osmotic gradients across cell membranes [8]. It is stored in body tissues, especially in the liver, predominantly as thiamine diphosphate (TDP) [9]. TDP plays an important role in the conversion of glucose into energy, acting as an essential cofactor for several enzymes in the Krebs cycle and in the pentose phosphate pathway. The Krebs cycle, also known as the tricarboxylic acid cycle (TCA cycle) or the citric acid cycle, is a crucial metabolic pathway that represents a key part of aerobic respiration in cells. It is constituted by a series of chemical reactions, occurring in the mitochondrion, that lead to the oxidation of acetate derived from carbohydrates, fatty acids, and amino acids into carbon dioxide, producing chemical energy in the form of adenosine triphosphate (ATP). In the citric acid cycle, TDP is an important coenzyme for the pyruvate dehydrogenate complex, which converts pyruvate, produced by glycolysis, into acetyl-CoA, and for the alpha-ketoglutarate dehydrogenase complex, that transforms alpha-ketoglutarate into succinil-CoA.
TDP also acts as cofactor of the enzyme transketolase, involved in the pentose phosphate pathway, a cytosolic process generating pentoses (5-carbon sugars), essential for nucleic acid synthesis [10]. A healthy adult requires approximately 1-2 mg of thiamine daily, depending on the carbohydrate intake. Body's reserves of thiamine are only 30–50 mg, so any malnutrition condition lasting more than 3-4 weeks can cause complete depletion of the vitamin's stores [3]. In case of thiamine deficiency, intracellular TDP is depleted leading to a series of metabolic alterations in the central nervous system. The decreased activities of the pyruvate dehydrogenate complex, the alpha-ketoglutarate dehydrogenase complex, and the transketolase, resulting in a reduction of Krebs cycle and pentose phosphate pathway efficiency, induce a cellular energy deficit due to reduced passage of pyruvate into the citric acid cycle, consequent intracellular accumulation of toxic intermediates such as lactate and alanine, reduction in PH, and cerebral lactic acidosis. Another effect is the intracellular accumulation of glutamate, derived from the transamination of alpha-ketoglutarate; glutamate limits the function of ATP-dependent cellular pumps, inducing failure to maintain cellular electrolyte homeostasis. Glutamate is consequently discharged in the extracellular space and, due to its function of excitatory neurotransmitter, neurons are exposed to excitotoxic damage. Excessive extracellular glutamate can bind to NMDA (N-methyl-D-aspartate) receptors, causing high calcium (Ca2+) concentration within the cells, leading to necrosis or apoptosis. Glial cells and neurons control of ionic gradients across the cell membrane is therefore impaired, causing cytotoxic edema. Thiamine deficiency also induces blood-brain barrier (BBB) dysfunctions with consequent vasogenic edema. Disruption of the BBB can be a result of physical processes such as the mechanical opening of endothelial tight junctions with increased vesicular transport across brain endothelial cells or of chemical-mediated alterations such as increased endothelial cell permeability due to the presence of amyloid precursor protein (APP) in susceptible brain areas. However, if thiamine is adequately administered before the occurrence of cellular death, the earliest alterations are reversible, creating the concept of “reversible biochemical lesion” [11]. Dysregulation of these thiamine dependent metabolic pathways represents the biochemical mechanism responsible for the signs and symptoms of WE.
The most common cause of thiamine deficiency is chronic alcohol abuse. Alcoholism is not directly responsible for vitamin B1 deficiency; its effects are related to the complications of liver cirrhosis such as problems to the gastrointestinal tract with low absorption rate at the mucosal level and consequent malnourishment [12]. Apart from alcohol, a lot of other conditions causing malnutrition and decreased thiamine absorption such as gastrointestinal surgical procedures (including gastric bypass surgery, gastrojejunostomy, gastrectomy, and colectomy) [13–18], therapy with intragastric balloon [19], hyperemesis gravidarum [20, 21], terminal tumor [22], chemical therapy [23, 24], allogenic stem cell transplantation [25], AIDS [26], anorexia nervosa [27], fasting [28], starvation [29], hemodialysis [30], pancreatitis [31], wrong formula feeding [32], parenteral nutrition, hyperalimentation [33, 34], and prolonged intravenous glucose infusion [35] have been reported as predisposing factors.
Besides Wernicke's encephalopathy, thiamine deficiency is the main cause of beriberi, a syndrome usually diagnosed in people whose diet consists mainly of polished white rice, which is very low in thiamine because the thiamin-bearing husk has been removed [36]. The clinical spectrum of this disease comprises cardiac beriberi, neuropathic beriberi, and gastrointestinal beriberi.
Cardiac beriberi is characterized by high-output cardiac failure with right-sided heart predominance [37]. Neuropathic beriberi is a painful sensorimotor peripheral neuropathy that typically affects the lower extremities [38]. In the recent literature, the term “dry beriberi” is usually used to describe neuropathic beriberi, while the term “wet beriberi” often refers to cardiac beriberi [39]. The idea of a primary gastrointestinal beriberi, characterized by abdominal pain, vomiting, and lactic acidosis, has been introduced recently but it might represent a consequence of cardiac beriberi [40].
4. Histologic Findings
In acute WE, pathologic findings comprise intra- and extracellular edema with swelling of astrocytes and oligodendrocytes and increased microglial cells, variable degrees of necrosis, demyelination, vascular proliferation, petechial hemorrhage, and disruption of brain-blood barrier [41–43]. These findings suggest that both vasogenic and cytotoxic edema are involved in the pathogenic mechanism of WE [8]; Liu et al. [44] suggested that vasogenic edema might precede cytotoxic edema. The most evident alterations are described at the level of the structures around the third ventricle such as the medial thalami, the periaqueductal grey matter, the mammillary bodies, and the tectal plate of the midbrain [41]. All these areas are considered typical sites of involvement. Because of their high oxidative metabolism, it has been suggested that these regions are particularly sensitive to thiamine deficiency [8]. The cerebellum, the dentate nuclei, the cranial nerve nuclei, the red nuclei, the caudate nuclei, the splenium, and the cerebral cortex are recognized as less commonly involved areas [3].
5. Clinical Manifestations and Diagnosis
Classically, WE is characterized by the sudden onset of a typical clinical triad: mental status changes, ophthalmoplegia, and ataxia; however, this complete triad can be seen in just one-third of patients. The most constant clinical finding is represented by mental status changes [45]. These changes comprise confusional state, spatial disorientation, dizziness, drowsiness, apathy, cognitive impairment with disturbance in memory and inability to concentrate, coma, and death; such symptoms derive from an involvement of thalamic nuclei or mammillary bodies [10, 46, 47].
Among ocular disorders, complete ophthalmoplegia occurs rarely, while the most common ocular abnormality is nystagmus, usually horizontal. Other ophthalmic alterations include bilateral decreased visual acuity, diplopia, palsy of both lateral recti or other ocular muscles and conjugate-gaze palsies resulting from lesions of the pontine tegmentum and of the abducens and oculomotor nuclei, torpid reaction of the pupils to light, retinal hemorrhage, papilledema, anisocoria, and ptosis [7, 10, 48, 49].
Equilibrium disorders comprehend gait ataxia, that can vary from mild gait disturbance to a complete inability to stand; it results from an involvement of cerebellar vermis and vestibular dysfunction. Some patients also experience polyneuropathy and dysarthria [7, 50].
The onset of the disease can be characterized by several other findings such as cardiac failure with hypotension and tachycardia, gastrointestinal symptoms like abdominal pain and nausea, hypothermia due to the involvement of the posterior hypothalamic regions, deafness due to thalamic involvement, and epileptic seizures in case of glutamatergic hyperactivity [51–55].
Because of this large variability in clinical presentation, in 1997, Caine et al. suggested that, in chronic alcoholics, a suspicion of Wernicke's encephalopathy should be based on two of the following four conditions: malnutrition, oculomotor abnormalities, cerebellar dysfunction, and an altered mental state [56]. In nonalcoholic patients, WE commonly presents with altered mental state without other symptoms and the diagnosis is often delayed or missed [4].
If left untreated, Wernicke's encephalopathy can lead to the Korsakoff syndrome, a form of memory disturbance characterized by anterograde and retrograde amnesia and confabulation, related to lesions in the thalamus and mammillary bodies [57].
The differential diagnosis should include stroke and intracranial hemorrhage, meningitis and encephalitis, brain tumors, cerebellar diseases, toxic ingestions, liver failure, Marchiafava-Bignami disease, and metronidazole-induced encephalopathy [58].
WE is essentially a clinical diagnosis; the determination of thiamine blood concentration and red blood cell transketolase activity can help to confirm it [59, 60]. However, these measurements are limited by a lack of specificity and technical difficulties. Computed tomography (CT) can show areas of reduced attenuation density at the level of the periaqueductal grey matter and the medial portion of the thalami but, in most cases, is negative in the acute phase of WE [3, 58]. Currently, MR imaging of the brain, with its high specificity, is surely the most important and effective tool to get the right diagnosis [61, 62].
6. MR Imaging Findings
On MR imaging, the pathologic alterations described above are typically seen as bilateral and symmetrical T2w and FLAIR (Fluid Attenuation Inversion Recovery) hyperintensities in the thalami (Figure 1), mammillary bodies (Figure 2), tectal plate, and periaqueductal area (Figure 3) [3, 58, 63, 64]. In these areas, the maintenance of cellular osmotic gradients is considered to be strictly related to thiamine levels.
Figure 1.

Forty-three-year-old man with a 10-year history of alcohol abuse. T2-weighted axial image showing bilateral and symmetric hyperintense signal alteration at the level of the medial portion of the thalami.
Figure 2.

Fifty-year-old man with a 7-year history of alcohol abuse. FLAIR coronal images showing bilateral and symmetric hyperintense signal alteration at the level of the mammillary bodies (a) and the medial portion of the thalami (b).
Figure 3.

Sixty-four-year-old man with a 15-year history of alcohol abuse. T2-weighted axial image showing hyperintense signal alteration at the level of periaqueductal gray matter.
Signal intensity alterations in the dorsal medulla and the pons [65], cerebellar dentate nuclei, red nuclei [66], the substantia nigra of the midbrain [67], cranial nerve nuclei, the vermis and the paravermian regions of the cerebellum [68], the corpus callosum [69], the fornices [70], the head of the caudate nucleus [71], and the frontal-parietal cortex (Figure 4) [72] represent atypical MRI findings; they are almost always found in association with the typical findings.
Figure 4.

Forty-five-year-old man, affected by acute lymphoblastic leukemia, under chemical therapy, experiencing nausea and oral pain with consequent anorexia. T2-weighted axial (a) and FLAIR coronal (b) images showing bilateral hyperintense signal alteration at the level of the fronto-parietal cortex.
Cerebellar signal intensity alterations can be observed in both AL and NA patients [73, 74]. Cerebellar involvement on imaging is rather rare but autopsy studies have demonstrated that the anterior-superior vermis or anterior hemisphere is affected in more than half of patients with WE [10]. The involvement of the caudate nuclei, in particular of the capita, may be due to their adjacent position to the lateral ventricles. Opdenakker et al. reported a case of hemorrhagic focus in the caput of the right nucleus caudatus of a patient with WE [75]. According to Zhong et al., the presence of lesions of the caudate nuclei, frequently observed in patients in comatose state, is a sign of pathologic evolution [76]. In accordance with the literature, cortical involvement indicates irreversible damage and poor prognosis [3, 4, 76]; recently, however, Cui et al. reported a case of nonalcoholic WE with cortical involvement in a patient that, after 45 days of vegetative state, regained consciousness thanks to parenteral thiamine administration [77].
The absence of MR signal-intensity alterations, however, does not exclude the diagnosis of WE. Gadolinium administration can be a useful tool to identify WE cases with negative MRI scan.
Contrast-enhanced T1-weighted images point out areas with disrupted blood-brain barrier and enhancement can be seen in about 50% of cases [43, 78, 79]. Strong enhancement of the mammillary bodies, for instance, can be the only sign of the disease [80, 81] and is more frequent in chronic alcoholics [2].
Important differences in MR imaging findings between AL and NA patients have been reported in the literature. First of all, in association with the typical alterations of WE, AL patients can also present atrophy of the mammillary bodies and the cerebellar vermis as a result of previous WE attacks. No atrophy, instead, is found in NA patients; in their case, signal intensity alterations represent the first result of thiamine-related metabolic pathway dysregulation [4, 41, 76].
Atypical findings, moreover, are found more frequently in NA patients. In 2009, Zuccoli et al. reviewed MR imaging findings and clinical records of fifty-six patients (24 AL and 32 NA) with WE and reported that signal-intensity alterations in areas considered atypical for the disease were noted only in the NA group and always in association with the typical findings; they explained these results speculating a possible protective effect of the alcohol on the brain areas that show atypical lesions in WE [82]. As an answer to this hypothesis, in 2010, Hygino da Cruz and his collaborators reported two cases of nonalcoholic WE demonstrating that both typical and atypical MR imaging findings may coexist [83]; in the same year, Sugai and Kikugawa added their experience describing atypical MRI findings in two patients with WE, both of whom were alcoholic [84]. In 2012, Ha et al. reported MR imaging findings of twenty-four patients (13 AL and 11 NA) with WE showing that the atypical MR imaging findings, including cerebral cortex and cranial nerve nuclei lesions, were present both in the AL group and in the NA group [85]. Few months later, Liou et al. described an interesting case of an AL patient affected by WE that showed lesions in olivary bodies, brain stem cranial nerve nuclei, and the dentate nuclei of cerebellum in absence of hyperintense lesions in the typically affected areas [86]. These findings suggest that further investigations are required before the relationship between alcohol and the brain lesions associated with WE can be understood.
In pediatric patients, signal-intensity alterations are often observed at the level of the basal ganglia with a characteristic involvement of the putamen, probably due to the high thiamine-dependent metabolism of these areas in children [87, 88].
As for the differential diagnosis on MR imaging, diseases showing symmetric signal-intensity alterations of the medial thalami should be considered: deep cerebral venous thrombosis [89, 90], paramedian thalamic syndrome [91], top-of-the-basilar syndrome (TOBS) [92], viral encephalitis [93], acute disseminated encephalomyelitis (ADEM) [94], atypical Creutzfeldt-Jakob disease [95, 96], primary cerebral lymphoma [97], influenza A virus infection [98], and West Nile virus meningoencephalitis [99]. All these diseases can be differentiated from WE thanks to their clinical characteristics and MR imaging findings; in particular, symmetric thalamic alterations in WE are usually associated with other characteristic neuroradiological signs such as symmetric alterations in the mammillary bodies, tectal plate, and periaqueductal area [63].
The differential diagnosis of symmetric signal-intensity alterations of the dentate nuclei, cranial nerves nuclei, abducens, red nuclei, and splenium should include metronidazole-induced encephalopathy (MIE) [3]. Metronidazole is believed to penetrate CSF and the central nervous system; it is safe but, at high dosages, can produce peripheral neuropathies and cerebellar dysfunction. At MR imaging, MIE can mimic WE presenting bilateral symmetric hyperintense lesions in cerebellar dentate nuclei, midbrain, dorsal pons, medulla, corpus callosum, and cerebral white matter; moreover, MIE can cause symptoms very similar to those produced by acute WE [100]. Metronidazole neurotoxicity may be mediated by the impairment of vitamin B1 action though its conversion to a thiamine analog and consequent vitamin B1 antagonism [101]. The differential diagnosis between metronidazole-induced encephalopathy and Wernicke's encephalopathy is even more complex, in patient with chronic gastrointestinal diseases under metronidazole treatment [82].
After an effective thiamine treatment, in conjunction with the resolution of clinical symptoms, MR imaging followup usually shows a gradual decrease and disappearance of signal-intensity alterations (Figure 5).
Figure 5.

Thirty-three-year-old woman, primipara, in her 16th week of gestation, with a 3-week history of severe persistent nausea and vomiting accompanied by a 6 kg weight loss. FLAIR coronal (a) and T2-weighted sagittal (b) images showing bilateral and symmetric hyperintense signal alteration at the level of the medial portion of the thalami and of the lower portion of the tectal plate. A 3-week posttherapy (thiamine administered 100 mg/day intravenously for 10 days then 300 mg/day orally) brain MRI showed partial resolution of the signal abnormalities previously observed (c-d).
7. Role of DWI
In the diagnosis of acute Wernicke's encephalopathy DWI (diffusion-weighted imaging), detecting changes in water diffusion associated with cellular dysfunction is surely a valuable additional imaging sequence. The lesions can show hyperintensity on DWI images and reduced, normal, or increased ADC (apparent diffusion coefficient) values [102]. According to the literature, areas with high signal changes on DWI and normal or increased ADC values indicate the presence of vasogenic edema (Figure 6), characterized by elevated diffusion due to a relative increase in water in the extracellular compartment, whereas DWI hyperintensities with either decreased ADC represent cytotoxic edema (Figure 7) of the neurons and glial cells, characterized by restricted diffusion [103–110]. High signal intensity on DWI and decreased ADC values in acute WE do not always correspond to irreversible tissue damage; as discussed previously, the initial cellular alterations can be reversed so, when intravenous administration of thiamine is sufficient, signal-intensities abnormalities may regress and represent reversible cytotoxic edema, probably due to the osmotic dysregulation induced by decreased cellular energy levels. According to Chu et al., hyperintense signal alterations on DWI do not always indicate irreversibility but the presence of tissue at risk, similar to the cells in the ischemic penumbra [111]. This heterogeneity may result from disease severity, acuteness, and timing of imaging.
Figure 6.
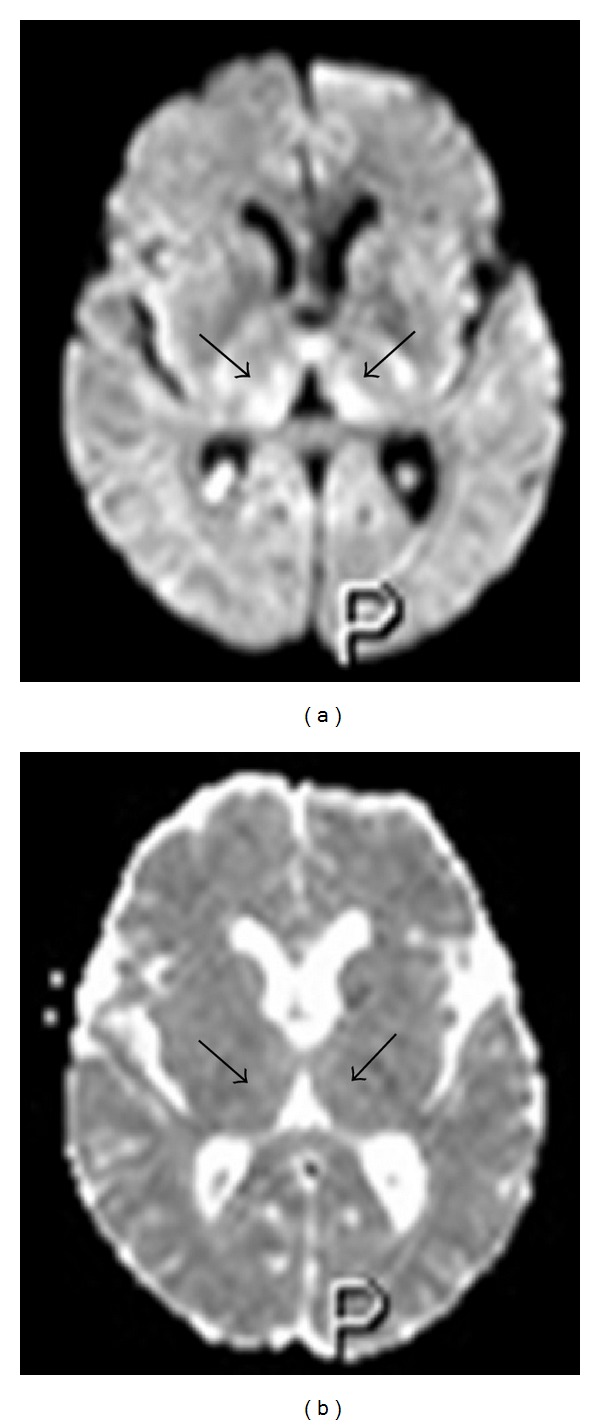
Fifty-three-year-old man in total parenteral nutrition. Axial diffusion-weighted image (a) showing bilateral and symmetric high signal at the level of the medial portion of the thalami with normal ADC values (b), indicating the presence of vasogenic edema.
Figure 7.

Forty-seven-year-old woman with a 16-year history of alcohol abuse. Axial diffusion-weighted image (a) showing bilateral and symmetric high signal at the level of the medial portion of the thalami with reduced ADC values (b), indicating the presence of cytotoxic edema.
8. Role of MR Spectroscopy
Proton MR spectroscopy is a powerful tool in the biochemical characterization of metabolic brain diseases in vivo. In the literature, there is a decent number of MR spectroscopy studies about thiamine deficiency but most of them have been performed on rats with very few studies applied to the investigation of WE in humans. In 2001, Murata et al. were the first to report proton MR spectroscopic findings in a patient with WE; they observed a decreased N-acetylaspartate (NAA)/creatine (Cr) ratio in the thalami and cerebellum and a lactate peak in the cerebellum (Figure 8). After thiamine administration, the NAA/Cr ratio increased at the level of the thalami but did not improve at the level of the cerebellum, suggesting irreversible tissue necrosis [112]. In 2002, Mascalchi et al. performed single-voxel MR spectroscopy in two patients with neurologic symptoms due to Wernicke's encephalopathy, before and after therapy, and placing their volume of interest in the thalami, in both cases, they observed a decreased NAA/Cr ratio without detectable lactate. They also reported an increased NAA/Cr ratio after thiamine therapy speculating it was presumably due to edema [113]. Few months later, in 2003, Rugilo and his collaborators reported proton MR spectroscopic findings in a patient with WE that showed a remarkable increase in lactate without decreased NAA/Cr ratio in the thalami. They hypothesized that the lactate peak was a result of the increased anaerobic oxidation of carbohydrates due to thiamine deficiency [114]. An analogous finding in a pediatric patient affected by WE has been recently reported by Rodan et al. Because of these few pieces of evidence, however, some other studies are required to assess the utility of MR spectroscopy as a potential diagnostic tool in Wernicke's encephalopathy [115].
Figure 8.

Forty-year-old man with a 10-year history of alcohol abuse. T2-weighted axial image (a) showing bilateral and symmetric hyperintense signal alteration at the level of the medial portion of the thalami and single-voxel MR spectroscopy image (b) demonstrating a decreased N-acetylaspartate (NAA)/creatine (Cr) ratio, a decreased N-acetylaspartate (NAA)/choline (Cho) ratio, and a lactate (Lac) peak, represented by a negative doublet (voxel taken from the right thalamus).
9. Treatment and Prognosis
Wernicke's encephalopathy is a medical emergency and any therapeutic delay may result in permanent neurological damage or death. The treatment of suspected or manifest WE is based on the administration of thiamine. To date, there is still no consensus on its optimal dose, modality of administration, and treatment time. The traditional recommendation is a parenteral dosage of at least 100 mg of thiamine per day [116]; recently, some authors have recommended that patients should be given 200 mg of thiamine three times a day [117]. It should be given before or concomitantly with any carbohydrates because glucose can precipitate the disorder [118]. Duration of treatment also remains an enigma; it should be continued until there is no further improvement in signs and symptoms. Except for a small risk of anaphylactic reactions, the overall safety of intravenous thiamine is good [116, 118]. Initial improvements in acute symptoms can be observed within the first week but they usually take 1–3 months to resolve. Nevertheless, persistent neurologic dysfunctions, such as nystagmus and gait ataxia, are common. If not treated or inappropriately treated with low doses of thiamine, WE can lead to irreversible brain damage that can cause death in about 20% of cases or Korsakoff syndrome in 85% of survivors. AL patients are more predisposed to develop Korsakoff syndrome than NA patients, probably due to the possible occurrence of multiple nervous lesions induced by repeated episodes of thiamine deficiency in alcoholics [118].
10. Conclusions
Due to its low prevalence in the general population and clinical heterogeneity, Wernicke's encephalopathy is often misdiagnosed leading to disabling persistent dysfunctions and, in 20% of cases, to death. Chronic alcoholism is surely the most common cause of WE but it is important to remember that a lot of other conditions responsible for thiamine deficiency must be considered. The diagnosis of this syndrome is essentially clinical but the complete triad (changes in consciousness, ophthalmoplegia, and ataxia) can be seen in just one-third of patients. These problems, in association with the technical difficulties of determination of thiamine blood concentration, make MR imaging of the brain the most important and effective tool in the diagnostic assessment of WE. Bilateral and symmetrical T2w and FLAIR hyperintensities in typical (thalami, mamillary bodies, tectal plate, and periaqueductal area) and atypical (cerebellum, cranial nerve nuclei, and cerebral cortex) areas are characteristic. Contrast-enhanced MRI, pointing out areas with disrupted blood-brain barrier, is necessary to identify WE cases with negative scan. Signal-intensity alterations in atypical sites are found more frequently in NA patients but can be also detected in alcoholics. The role of DWI with ADC is still not well defined but may help to distinguish between vasogenic and cytotoxic edema. As to MR spectroscopy utility, further studies on its application to humans affected by WE are required. In conclusion, a responsible awareness of both clinical and neuroradiological Wernicke's encephalopathy features is fundamental to recognize it and reduce its morbidity and mortality.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Wernicke C. Lehrbuch der Gehirnkrankheiten fur Aerzte und Studirende. Vol. 2. Kassel, Germany: Fisher; 1881. Die akute haemorrhagische polioencephalitis superior; pp. 229–242. [Google Scholar]
- 2.Zuccoli G, Gallucci M, Capellades J, et al. Wernicke encephalopathy: MR findings at clinical presentation in twenty-six alcoholic and nonalcoholic patients. American Journal of Neuroradiology. 2007;28(7):1328–1331. doi: 10.3174/ajnr.A0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. American Journal of Roentgenology. 2009;192(2):501–508. doi: 10.2214/AJR.07.3959. [DOI] [PubMed] [Google Scholar]
- 4.Fei GQ, Zhong C, Jin L, et al. Clinical characteristics and MR imaging features of nonalcoholic Wernicke encephalopathy. American Journal of Neuroradiology. 2008;29(1):164–169. doi: 10.3174/ajnr.A0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harper C, Fornes P, Duyckaerts C, Lecomte D, Hauw J-J. An international perspective on the prevalence of the Wernicke-Korsakoff syndrome. Metabolic Brain Disease. 1995;10(1):17–24. doi: 10.1007/BF01991779. [DOI] [PubMed] [Google Scholar]
- 6.Harper C. The incidence of Wernicke’s encephalopathy in Australia—a neuropathological study of 131 cases. Journal of Neurology Neurosurgery and Psychiatry. 1983;46(7):593–598. doi: 10.1136/jnnp.46.7.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Victor M. The Wernicke-Korsakoff syndrome. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. 28, part 2. Amsterdam, The Netherlands: North-Holland; 1976. pp. 243–270. [Google Scholar]
- 8.Harper C, Butterworth R. Nutritional and metabolic disorders. In: Graham DI, Lantos PL, editors. Greenfield’s Neuropathology. 6th edition. Vol. 1. London, UK: Hodder Arnold; 1997. pp. 601–652. [Google Scholar]
- 9.Gangolf M, Czerniecki J, Radermecker M, et al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013616.e13616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders due to Alcoholism and Malnutrition. 2nd ed edition. Philadelphia, Pa, USA: F.A. Davis; 1989. [Google Scholar]
- 11.Hazell AS, Todd KG, Butterworth RF. Mechanisms of neuronal cell death in Wernicke’s encephalopathy. Metabolic Brain Disease. 1998;13(2):97–122. doi: 10.1023/a:1020657129593. [DOI] [PubMed] [Google Scholar]
- 12.Thomson AD. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol and Alcoholism Supplement. 2000;35(1):2–7. doi: 10.1093/alcalc/35.supplement_1.2. [DOI] [PubMed] [Google Scholar]
- 13.Singh S, Kumar A. Wernicke encephalopathy after obesity surgery: a systematic review. Neurology. 2007;68(11):807–811. doi: 10.1212/01.wnl.0000256812.29648.86. [DOI] [PubMed] [Google Scholar]
- 14.Loh Y, Watson WD, Verma A, Chang ST, Stocker DJ, Labutta RJ. Acute Wernicke’s encephalopathy following bariatric surgery: clinical course and MRI correlation. Obesity Surgery. 2004;14(1):129–132. doi: 10.1381/096089204772787437. [DOI] [PubMed] [Google Scholar]
- 15.Becker DA, Ingala EE, Martinez-Lage M, Price RS, Galetta SL. Dry Beriberi and Wernicke’s encephalopathy following gastric lap band surgery. Journal of Clinical Neuroscience. 2012;19(7):1050–1052. doi: 10.1016/j.jocn.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Velasco MV, Casanova I, Sanchez-Pernaute A, et al. Unusual late-onset Wernicke’s encephalopathy following vertical banded gastroplasty. Obesity Surgery. 2009;19(7):937–940. doi: 10.1007/s11695-009-9841-7. [DOI] [PubMed] [Google Scholar]
- 17.D’Abbicco D, Praino S, Amoruso M, Notarnicola A, Margari A. Syndrome in syndrome: Wernicke syndrome due to afferent loop syndrome. Case report and review of the literature. Il Giornale di Chirurgia. 2011;32(11-12):479–482. [PubMed] [Google Scholar]
- 18.Nolli M, Barbier A, Pinna C, Pasetto A, Nicosia F. Wernicke’s encephalopathy in a malnourished surgical patient: clinical features and magnetic resonance imaging. Acta Anaesthesiologica Scandinavica. 2005;49(10):1566–1567. doi: 10.1111/j.1399-6576.2005.00879.x. [DOI] [PubMed] [Google Scholar]
- 19.Chaves LC, Faintuch J, Kahwage S, de Assis Alencar F. A cluster of polyneuropathy and Wernicke-Korsakoff syndrome in a bariatric unit. Obesity Surgery. 2002;12(3):328–334. doi: 10.1381/096089202321088093. [DOI] [PubMed] [Google Scholar]
- 20.Gárdián G, Vörös E, Járdánházy T, Ungureán A, Vécsei L. Wernicke’s encephalopathy induced by hyperemesis gravidarum. Acta Neurologica Scandinavica. 1999;99(3):196–198. doi: 10.1111/j.1600-0404.1999.tb07344.x. [DOI] [PubMed] [Google Scholar]
- 21.Zara G, Codemo V, Palmieri A, et al. Neurological complications in hyperemesis gravidarum. Neurological Sciences. 2012;33(1):133–135. doi: 10.1007/s10072-011-0660-y. [DOI] [PubMed] [Google Scholar]
- 22.Yae S, Okuno S, Onishi H, Kawanishi C. Development of Wernicke encephalopathy in a terminally ill cancer patient consuming an adequate diet: a case report and review of the literature. Palliative & Supportive Care. 2005;3(4):333–335. doi: 10.1017/s1478951505050509. [DOI] [PubMed] [Google Scholar]
- 23.Cho IJ, Chang HJ, Lee KE, et al. A case of Wernicke’s encephalopathy following fluorouracil-based chemotherapy. Journal of Korean Medical Science. 2009;24(4):747–750. doi: 10.3346/jkms.2009.24.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner JE, Alley JG, Sharpless NE. Medical problems in patients with malignancy: case 2. Wernicke’s encephalopathy: an unusual acute neurologic complication of lymphoma and its therapy. Journal of Clinical Oncology. 2004;22(19):4020–4022. doi: 10.1200/JCO.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 25.Baek JH, Sohn SK, Kim DH, et al. Wernicke’s encephalopathy after allogeneic stem cell transplantation. Bone Marrow Transplantation. 2005;35(8):829–830. doi: 10.1038/sj.bmt.1704893. [DOI] [PubMed] [Google Scholar]
- 26.Montine TJ, Fuller GN. Wernicke’s encephalopathy in a patient with AIDS. AIDS. 1993;7(5):744–745. [PubMed] [Google Scholar]
- 27.Saad L, Silva LF, Banzato CE, Dantas C, Garcia C. Anorexia nervosa and Wernicke-Korsakoff syndrome: a case report. Journal of Medical Case Reports. 2010;4, article 217 doi: 10.1186/1752-1947-4-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unlu E, Cakir B, Asil T. MRI findings of Wernicke encephalopathy revisited due to hunger strike. European Journal of Radiology. 2006;57(1):43–53. doi: 10.1016/j.ejrad.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Tsai HY, Yeh TL, Sheei-Meei W, Chen PS, Yang YK. Starvation-induced Wernicke’s encephalopathy in schizophrenia. Psychiatry and Clinical Neurosciences. 2004;58(3):338–339. doi: 10.1111/j.1440-1819.2004.01242.x. [DOI] [PubMed] [Google Scholar]
- 30.Ueda K, Takada D, Mii A, et al. Severe thiamine deficiency resulted in Wernicke’s encephalopathy in a chronic dialysis patient. Clinical and Experimental Nephrology. 2006;10(4):290–293. doi: 10.1007/s10157-006-0440-9. [DOI] [PubMed] [Google Scholar]
- 31.Arana-Guajardo AC, Cámara-Lemarroy CR, Rendón-Ramírez EJ, et al. Wernicke encephalopathy presenting in a patient with severe acute pancreatitis. Journal of Pancreas. 2012;13(1):104–107. [PubMed] [Google Scholar]
- 32.Fattal-Valevski A, Kesler A, Sela BA, et al. Outbreak of life-threatening thiamine deficiency in infants in Israel caused by a defective soy-based formula. Pediatrics. 2005;115(2):e233–e238. doi: 10.1542/peds.2004-1255. [DOI] [PubMed] [Google Scholar]
- 33.Francini-Pesenti F, Brocadello F, Manara R, Santelli L, Laroni A, Caregaro L. Wernicke’s syndrome during parenteral feeding: not an unusual complication. Nutrition. 2009;25(2):142–146. doi: 10.1016/j.nut.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Jeffrey K, James AG. Wernicke’s encephalopathy: complication of intravenous hyperalimentation. The Journal of the American Medical Association. 1977;238(20):2176–2177. doi: 10.1001/jama.238.20.2176. [DOI] [PubMed] [Google Scholar]
- 35.Koguchi K, Nakatsuji Y, Abe K, Sakoda S. Wernicke’s encephalopathy after glucose infusion. Neurology. 2004;62(3, article 512) doi: 10.1212/01.wnl.0000099189.56741.a7. [DOI] [PubMed] [Google Scholar]
- 36.Arnold D. British India and the “beriberi problem”, 1798–1942. Medical History. 2010;54(3):295–314. doi: 10.1017/s0025727300004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao SN, Chandak GR. Cardiac beriberi: often a missed diagnosis. Journal of Tropical Pediatrics. 2009;56(4):284–285. doi: 10.1093/tropej/fmp108. [DOI] [PubMed] [Google Scholar]
- 38.Howard AJ, Kulkarni O, Lekwuwa G, Emsley HC. Rapidly progressive polyneuropathy due to dry beriberi in a man: a case report. Journal of Medical Case Reports. 2010;4, article 409 doi: 10.1186/1752-1947-4-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Betrosian AP, Thireos E, Toutouzas K, Zabaras P, Papadimitriou K, Sevastos N. Occidental beriberi and sudden death. The American Journal of the Medical Sciences. 2004;327(5):250–252. doi: 10.1097/00000441-200405000-00020. [DOI] [PubMed] [Google Scholar]
- 40.Donnino M. Gastrointestinal beriberi: a previously unrecognized syndrome. Annals of Internal Medicine. 2004;141(11):898–899. doi: 10.7326/0003-4819-141-11-200412070-00035. [DOI] [PubMed] [Google Scholar]
- 41.Park SH, Kim M, Na DL, Jeon BS. Magnetic resonance reflects the pathological evolution of Wernicke encephalopathy. Journal of Neuroimaging. 2001;11(4):406–411. doi: 10.1111/j.1552-6569.2001.tb00070.x. [DOI] [PubMed] [Google Scholar]
- 42.Gui QP, Zhao WQ, Wang LN. Wernicke’s encephalopathy in nonalcoholic patients: clinical and pathologic features of three cases and literature reviewed. Neuropathology. 2006;26(3):231–235. doi: 10.1111/j.1440-1789.2006.00665.x. [DOI] [PubMed] [Google Scholar]
- 43.Schroth G, Wichmann W, Valavanis A. Blood-brain-barrier disruption in acute Wernicke encephalopathy: MR findings. Journal of Computer Assisted Tomography. 1991;15(6):1059–1061. doi: 10.1097/00004728-199111000-00034. [DOI] [PubMed] [Google Scholar]
- 44.Liu YT, Fuh JL, Lirng JF, DM H, Wang SJ. Correlation of magnetic resonance images with neuropathology in acute Wernicke’s encephalopathy. Clinical Neurology and Neurosurgery. 2006;108(7):682–687. doi: 10.1016/j.clineuro.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. Journal of Neurology, Neurosurgery and Psychiatry. 1986;49(4):341–345. doi: 10.1136/jnnp.49.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Keyser J, Deleu D, Solheid C, Ebinger G. Coma as presenting manifestation of Wernicke’s encephalopathy. Journal of Emergency Medicine. 1985;3(5):361–363. doi: 10.1016/0736-4679(85)90320-8. [DOI] [PubMed] [Google Scholar]
- 47.Lana-Peixoto MA, Dos Santos EC, Pittella JE. Coma and death in unrecognized Wernicke’s encephalopathy. An autopsy study. Arquivos de Neuro-Psiquiatria. 1992;50(3):329–333. doi: 10.1590/s0004-282x1992000300012. [DOI] [PubMed] [Google Scholar]
- 48.Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome. A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemporary Neurology Series. 1971;7:1–206. [PubMed] [Google Scholar]
- 49.Kulkarni S, Lee AG, Holstein SA, Warner JE. You are what you eat. Survey of Ophthalmology. 2005;50(4):389–393. doi: 10.1016/j.survophthal.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 50.Ghez C. Vestibular paresis: a clinical feature of Wernicke’s disease. Journal of Neurology, Neurosurgery and Psychiatry. 1969;32(2):134–139. doi: 10.1136/jnnp.32.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ackerman WJ. Stupor, bradycardia, hypotension and hypothermia. A presentation of Wernicke’s encephalopathy with rapid response to thiamine. Western Journal of Medicine. 1974;121(5):428–429. [PMC free article] [PubMed] [Google Scholar]
- 52.Wang S, Hou X, Ding S, et al. Refractory hypotension in a patient with Wernicke’s encephalopathy. Alcohol and Alcoholism. 2012;47(1):48–51. doi: 10.1093/alcalc/agr077. [DOI] [PubMed] [Google Scholar]
- 53.Reuler JB, Girard DE, Cooney TG. Wernicke’s encephalopathy. The New England Journal of Medicine. 1985;312(16):1035–1039. doi: 10.1056/NEJM198504183121606. [DOI] [PubMed] [Google Scholar]
- 54.Doss A, Mahad D, Romanowski CA. Wernicke’s encephalopathy: unusual findings in nonalcoholic patients. Journal of Computer Assisted Tomography. 2003;27(2):235–240. doi: 10.1097/00004728-200303000-00022. [DOI] [PubMed] [Google Scholar]
- 55.Zhang SQ, Guan YT. Acute bilateral deafness as the first symptom of Wernicke encephalopathy. American Journal of Neuroradiology. 2012;33(3):E44–E45. doi: 10.3174/ajnr.A3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caine D, Halliday GM, Kril JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. Journal of Neurology Neurosurgery and Psychiatry. 1997;62(1):51–60. doi: 10.1136/jnnp.62.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol and Alcoholism. 2009;44(2):148–154. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- 58.Cerase A, Rubenni E, Rufa A, et al. CT and MRI of Wernicke’s encephalopathy. La Radiologia Medica. 2011;116(2):319–333. doi: 10.1007/s11547-011-0618-x. [DOI] [PubMed] [Google Scholar]
- 59.Dreyfus PM. Clinical application of blood transketolase determinations. The New England Journal of Medicine. 1962;267:596–598. doi: 10.1056/NEJM196209202671204. [DOI] [PubMed] [Google Scholar]
- 60.Baker H, Frank O, Fennelly JJ, Leevy CM. A method for assaying thiamine status in man and animals. The American Journal of Clinical Nutrition. 1964;14(4):197–201. doi: 10.1093/ajcn/14.4.197. [DOI] [PubMed] [Google Scholar]
- 61.Antunez E, Estruch R, Cardenal C, Nicolas JM, Fernandez-Sola J, Urbano-Marquez A. Usefulness of CT and, MR imaging in the diagnosis of acute Wernicke’s encephalopathy. American Journal of Roentgenology. 1998;171(4):1131–1137. doi: 10.2214/ajr.171.4.9763009. [DOI] [PubMed] [Google Scholar]
- 62.Chung SP, Kim SW, Yoo IS, Lim YS, Lee G. Magnetic resonance imaging as a diagnostic adjunct to Wernicke’s encephalopathy in the ED. The American Journal of Emergency Medicine. 2003;21(6):497–502. doi: 10.1016/s0735-6757(03)00094-9. [DOI] [PubMed] [Google Scholar]
- 63.Gallucci M, Bozzao A, Splendiani A, Masciocchi C, Passariello R. Wernicke encephalopathy: MR findings in five patients. American Journal of Roentgenology. 1990;155(6):1309–1314. doi: 10.2214/ajr.155.6.2122685. [DOI] [PubMed] [Google Scholar]
- 64.Sullivan EV, Pfefferbaum A. Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol and Alcoholism. 2009;44(2):155–165. doi: 10.1093/alcalc/agn103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nardone R, Venturi A, Golaszewski S, Caleri F, Tezzon F, Ladurner G. MR atypical Wernicke encephalopathy showing extensive brain stem and diencephalic involvement. Journal of Neuroimaging. 2010;20(2):204–207. doi: 10.1111/j.1552-6569.2009.00374.x. [DOI] [PubMed] [Google Scholar]
- 66.Kang SY, Kang JH, Choi JC, Choi G. Wernicke’s encephalopathy: unusual manifestation on MRI. Journal of Neurology. 2005;252(12):1550–1552. doi: 10.1007/s00415-005-0886-9. [DOI] [PubMed] [Google Scholar]
- 67.Kalidass B, Sunnathkal R, Rangashamanna DV, Paraswani R. Atypical Wernicke’s encephalopathy showing involvement of substantia nigra. Journal of Neuroimaging. 2012;22(2):204–207. doi: 10.1111/j.1552-6569.2010.00545.x. [DOI] [PubMed] [Google Scholar]
- 68.Zuccoli G, Motti L. Atypical Wernicke’s encephalopathy showing lesions in the cranial nerve nuclei and cerebellum. Journal of Neuroimaging. 2008;18(2):194–197. doi: 10.1111/j.1552-6569.2007.00188.x. [DOI] [PubMed] [Google Scholar]
- 69.Lee ST, Jung YM, Na DL, Park SH, Kim M. Corpus callosum atrophy in Wernicke’s encephalopathy. Journal of Neuroimaging. 2005;15(4):367–372. doi: 10.1177/1051228405278352. [DOI] [PubMed] [Google Scholar]
- 70.Thomas AG, Koumellis P, Dineen RA. The fornix in health and disease: an imaging review. Radiographics. 2011;31(4):1107–1121. doi: 10.1148/rg.314105729. [DOI] [PubMed] [Google Scholar]
- 71.Nishida M, Sato H, Kobayashi N, Morimoto M, Hamaoka K. Wernicke’s encephalopathy in a patient with nephrotic syndrome. European Journal of Pediatrics. 2009;168(6):731–734. doi: 10.1007/s00431-008-0833-8. [DOI] [PubMed] [Google Scholar]
- 72.Pereira DB, Pereira ML, Gasparetto EL. Nonalcoholic Wernicke encephalopathy with extensive cortical involvement: cortical laminar necrosis and hemorrhage demonstrated with susceptibility-weighted MR phase images. American Journal of Neuroradiology. 2011;32(2):E37–E38. doi: 10.3174/ajnr.A2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim HA, Lee H. Atypical Wernicke’s encephalopathy with remarkable cerebellar lesions on diffusion-weighted MRI. European Neurology. 2007;58(1):51–53. doi: 10.1159/000102168. [DOI] [PubMed] [Google Scholar]
- 74.Lapergue B, Klein I, Olivot JM, Amarenco P. Diffusion weighted imaging of cerebellar lesions in Wernicke’s encephalopathy. Journal of Neuroradiology. 2006;33(2):126–128. doi: 10.1016/s0150-9861(06)77243-1. [DOI] [PubMed] [Google Scholar]
- 75.Opdenakker G, Gelin G, de Surgeloose D, Palmers Y. Wernicke encephalopathy: MR findings in two patients. European Radiology. 1999;9(8):1620–1624. doi: 10.1007/s003300050896. [DOI] [PubMed] [Google Scholar]
- 76.Zhong C, Jin L, Fei G. MR imaging of nonalcoholic Wernicke encephalopathy: a follow-up study. American Journal of Neuroradiology. 2005;26(9):2301–2305. [PMC free article] [PubMed] [Google Scholar]
- 77.Cui HW, Zhang BA, Peng T, Liu YR, Liu Y. Wernicke’s encephalopathy in a patient with acute pancreatitis: unusual cortical involvement and marvelous prognosis. Neurological Sciences. 2012;33(3):615–618. doi: 10.1007/s10072-011-0771-5. [DOI] [PubMed] [Google Scholar]
- 78.D’Aprile P, Gentile MA, Carella A. Enhanced MR in the acute phase of Wernicke encephalopathy. American Journal of Neuroradiology. 1994;15(3):591–593. [PMC free article] [PubMed] [Google Scholar]
- 79.Mascalchi M, Simonelli P, Tessa C, et al. Do acute lesions of Wernicke’s encephalopathy show contrast enhancement? Report of three cases and review of the literature. Neuroradiology. 1999;41(4):249–254. doi: 10.1007/s002340050741. [DOI] [PubMed] [Google Scholar]
- 80.Shogry ME, Curnes JT. Mamillary body enhancement on MR as the only sign of acute Wernicke encephalopathy. American Journal of Neuroradiology. 1994;15(1):172–174. [PMC free article] [PubMed] [Google Scholar]
- 81.Kavuk I, Agelink MW, Gaertner T, et al. Wernicke’s encephalopathy: unusual contrast enhancement revealed by magnetic resonance imaging. European Journal of Medical Research. 2003;8(11):492–494. [PubMed] [Google Scholar]
- 82.Zuccoli G, Santa Cruz DS, Bertolini M, et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. American Journal of Neuroradiology. 2009;30(1):171–176. doi: 10.3174/ajnr.A1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hygino da Cruz LC, Jr., Domingues RC, Vilanova I, Gasparetto EL. MR imaging findings in Wernicke encephalopathy: nonalcoholics may be similar to alcoholics. American Journal of Neuroradiology. 2010;31(6):E54–E55. doi: 10.3174/ajnr.A2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sugai A, Kikugawa K. Atypical MRI findings of Wernicke encephalopathy in alcoholic patients. American Journal of Roentgenology. 2010;195(5):W372–W374. doi: 10.2214/AJR.10.4539. [DOI] [PubMed] [Google Scholar]
- 85.Ha ND, Weon YC, Jang JC, Kang BS, Choi SH. Spectrum of MR imaging findings in Wernicke encephalopathy: are atypical areas of involvement only present in nonalcoholic patients? American Journal of Neuroradiology. 2012;33(7):1398–1402. doi: 10.3174/ajnr.A2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liou KC, Kuo SF, Chen LA. Wernicke encephalopathy with atypical magnetic resonance imaging. The American Journal of Emergency Medicine. 2012;30(9):2086.e1–2086.e3. doi: 10.1016/j.ajem.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 87.Kornreich L, Bron-Harlev E, Hoffmann C, et al. Thiamine deficiency in infants: MR findings in the brain. American Journal of Neuroradiology. 2005;26(7):1668–1674. [PMC free article] [PubMed] [Google Scholar]
- 88.Zuccoli G, Siddiqui N, Bailey A, Bartoletti SC. Neuroimaging findings in pediatric Wernicke encephalopathy: a review. Neuroradiology. 2010;52(6):523–529. doi: 10.1007/s00234-009-0604-x. [DOI] [PubMed] [Google Scholar]
- 89.Matheus MG, Castillo M. Imaging of acute bilateral paramedian thalamic and mesencephalic infarcts. American Journal of Neuroradiology. 2003;24(10):2005–2008. [PMC free article] [PubMed] [Google Scholar]
- 90.Forsting M, Krieger D, Seier U, Hacke W. Reversible bilateral thalamic lesions caused by primary internal cerebral vein thrombosis: a case report. Journal of Neurology. 1989;236(8):484–486. doi: 10.1007/BF00328513. [DOI] [PubMed] [Google Scholar]
- 91.Kumral E, Evyapan D, Balkir K, Kutluhan S. Bilateral thalamic infarction. Clinical, etiological and MRI correlates. Acta Neurologica Scandinavica. 2001;103(1):35–42. doi: 10.1034/j.1600-0404.2001.00141.x. [DOI] [PubMed] [Google Scholar]
- 92.Schmahmann JD. Vascular syndromes of the thalamus. Stroke. 2003;34(9):2264–2278. doi: 10.1161/01.STR.0000087786.38997.9E. [DOI] [PubMed] [Google Scholar]
- 93.Pilleri G, Pietrini V, Tagliavini F, Trabattoni G, Lechi A. “Thalamic” dementia in herpes encephalitis: clinico-pathological report. Acta Neuropathologica Supplement. 1981;7:156–159. doi: 10.1007/978-3-642-81553-9_47. [DOI] [PubMed] [Google Scholar]
- 94.Masson C, Colombani JM. Primary acute disseminated encephalomyelitis in the adult: study of 6 year follow-up of two patients. La Presse Médicale. 2002;31(37, part 1):1739–1745. [PubMed] [Google Scholar]
- 95.Carota A, Pizzolato GP, Gailloud P, et al. A panencephalopathic type of Creutzfeldt-Jakob disease with selective lesions of the thalamic nuclei in 2 Swiss patients. Clinical Neuropathology. 1996;15(3):125–134. [PubMed] [Google Scholar]
- 96.Zeidler M, Sellar RJ, Collie DA, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease . The Lancet. 2000;356(9213):1412–1418. doi: 10.1016/s0140-6736(00)02140-1. Erratum in: “The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease”, The Lancet, vol. 356, no. 9224, pp. 170, 2000. [DOI] [PubMed] [Google Scholar]
- 97.Brechteisbauer DL, Urbach H, Sommer T, Blümcke I, Woitas R, Solymosi L. Cytomegalovirus encephalitis and primary cerebral lymphoma mimicking Wernicke’s encephalopathy. Neuroradiology. 1997;39(1):19–22. doi: 10.1007/s002340050359. [DOI] [PubMed] [Google Scholar]
- 98.Nagai T, Yagishita A, Tsuchiya Y, Asamura S, Kurokawa H, Matsuo N. Symmetrical thalamic lesions on CT in influenza A virus infection presenting with or without Reye syndrome. Brain and Development. 1993;15(1):67–73. doi: 10.1016/0387-7604(93)90009-w. [DOI] [PubMed] [Google Scholar]
- 99.Ali M, Safriel Y, Sohi J, Llave A, Weathers S. West Nile virus infection: MR imaging findings in the nervous system. American Journal of Neuroradiology. 2005;26(2):289–297. [PMC free article] [PubMed] [Google Scholar]
- 100.Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. American Journal of Neuroradiology. 2007;28(9):1652–1658. doi: 10.3174/ajnr.A0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zuccoli G, Pipitone N, Santa Cruz D. Metronidazole-induced and Wernicke encephalopathy: two different entities sharing the same metabolic pathway? American Journal of Neuroradiology. 2008;29(9):p. E85. doi: 10.3174/ajnr.A1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lapergue B, Klein I, Olivot JM, Amarenco P. Diffusion weighted imaging of cerebellar lesions in Wernicke’s encephalopathy. Journal of Neuroradiology. 2006;33(2):126–128. doi: 10.1016/s0150-9861(06)77243-1. [DOI] [PubMed] [Google Scholar]
- 103.Hong KS, Kang DW, Cho YJ, Hwang YJ, Hur G. Diffusion-weighted magnetic resonance imaging in Wernicke’s encephalopathy. Acta Neurologica Scandinavica. 2002;105(2):132–134. doi: 10.1034/j.1600-0404.2002.1c205.x. [DOI] [PubMed] [Google Scholar]
- 104.Doherty MJ, Watson NF, Uchino K, Hallam DK, Cramer SC. Diffusion abnormalities in patients with Wernicke encephalopathy. Neurology. 2002;58(4):655–657. doi: 10.1212/wnl.58.4.655. [DOI] [PubMed] [Google Scholar]
- 105.Chung TI, Kim JS, Park SK, Kim B, Ahn K, Yang D. Diffusion weighted MR imaging of acute Wernicke’s encephalopathy. European Journal of Radiology. 2003;45(3):256–258. doi: 10.1016/s0720-048x(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 106.Halavaara J, Brander A, Lyytinen J, Setälä K, Kallela M. Wernicke’s encephalopathy: is diffusion-weighted MRI useful? Neuroradiology. 2003;45(8):519–523. doi: 10.1007/s00234-003-1043-8. [DOI] [PubMed] [Google Scholar]
- 107.Loh Y, Watson WD, Verma A, Krapiva P. Restricted diffusion of the splenium in acute Wernicke’s encephalopathy. Journal of Neuroimaging. 2005;15(4):373–375. doi: 10.1177/1051228405279037. [DOI] [PubMed] [Google Scholar]
- 108.Kim JE, Kim TH, Yu IK, Lee BR, Lee SJ, Oh GS. Diffusion-weighted MRI in recurrent Wernicke’s encephalopathy: a remarkable cerebellar lesion. The Journal of Clinical Neurology. 2006;2(2):141–145. doi: 10.3988/jcn.2006.2.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yucebilgin S, Cirpan T, Sanhal CY, Ozan E, Acar T, Ozsener S. Wernicke’s encephalopathy: a case report and MRI findings. Journal Belge de Radiologie: Belgisch Tijdschrift voor Radiologie. 2010;94(1):24–25. doi: 10.5334/jbr-btr.480. [DOI] [PubMed] [Google Scholar]
- 110.Moritani T, Smoker WR, Sato Y, Numaguchi Y, Westesson PA. Diffusion-weighted imaging of acute excitotoxic brain injury. American Journal of Neuroradiology. 2005;26(2):216–228. [PMC free article] [PubMed] [Google Scholar]
- 111.Chu K, Kang DW, Kim HJ, Lee Y, Park S. Diffusion-weighted imaging abnormalities in Wernicke encephalopathy: reversible cytotoxic edema? Archives of Neurology. 2002;59(1):123–127. doi: 10.1001/archneur.59.1.123. [DOI] [PubMed] [Google Scholar]
- 112.Murata T, Fujito T, Kimura H, Omori M, Itoh H, Wada Y. Serial MRI and 1H-MRS of Wernicke’s encephalopathy: report of a case with remarkable cerebellar lesions on MRI. Psychiatry Research. 2001;108(1):49–55. doi: 10.1016/s0925-4927(01)00304-3. [DOI] [PubMed] [Google Scholar]
- 113.Mascalchi M, Belli G, Guerrini L, Nistri M, Del Seppia I, Villari N. Proton MR spectroscopy of Wernicke encephalopathy. American Journal of Neuroradiology. 2002;23(10):1803–1806. [PMC free article] [PubMed] [Google Scholar]
- 114.Rugilo CA, Uribe Roca MC, Zurru MC, Capizzano AA, Pontello GA, Gatto EM. Proton MR spectroscopy in Wernicke encephalopathy. American Journal of Neuroradiology. 2003;24(5):952–955. [PMC free article] [PubMed] [Google Scholar]
- 115.Rodan LH, Mishra N, Tein I. MR spectroscopy in pediatric Wernicke encephalopathy. Neurology. 2013;80(10):p. 969. doi: 10.1212/WNL.0b013e3182840cd9. [DOI] [PubMed] [Google Scholar]
- 116.Donnino MW, Vega J, Miller J, Walsh M. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Annals of Emergency Medicine. 2007;50(6):715–721. doi: 10.1016/j.annemergmed.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 117.Galvin R, Bråthen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. European Journal of Neurology. 2010;17(12):1408–1418. doi: 10.1111/j.1468-1331.2010.03153.x. [DOI] [PubMed] [Google Scholar]
- 118.Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. The Lancet Neurology. 2007;6(5):442–455. doi: 10.1016/S1474-4422(07)70104-7. [DOI] [PubMed] [Google Scholar]